Article Text

Abstract
Immunotherapy is a promising new therapeutic field that has demonstrated significant benefits in many solid-tumor malignancies, such as metastatic melanoma and non-small cell lung cancer. However, only a subset of these patients responds to treatment. Glioblastoma (GBM) is the most common malignant primary brain tumor with a poor prognosis of 14.6 months and few treatment advancements over the last 10 years. There are many clinical trials testing immune therapies in GBM, but patient responses in these studies have been highly variable and a definitive benefit has yet to be identified. Biomarkers are used to quantify normal physiology and physiological response to therapies. When extensively characterized and vigorously validated, they have the potential to delineate responders from non-responders for patients treated with immunotherapy in malignancies outside of the central nervous system (CNS) as well as GBM. Due to the challenges of current modalities of radiographic diagnosis and disease monitoring, identification of new predictive and prognostic biomarkers to gauge response to immune therapy for patients with GBM will be critical in the precise treatment of this highly heterogenous disease. This review will explore the current and future strategies for the identification of potential biomarkers in the field of immunotherapy for GBM, as well as highlight major challenges of adapting immune therapy for CNS malignancies.
- neuroimmunology
- neuropathy
- neurosurgery
- tissue typing
- tumors
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Immune-based therapies harness a patient’s immune system to specifically target and destroy cancer cells.1 The field of immunotherapy has already made significant developments in the treatment of several aggressive cancer types, including melanoma,2 non-small cell lung cancer (NSCLC)3 and lymphomas.4 5 However, despite advances in immune therapy, there is an accompanying need for the development of biomarkers that accurately measure a patient’s response to therapy.
Biomarkers are quantifiable factors that gauge normal physiology, pathology, treatment response or biological responses to intervention.6 Relevant biomarkers can be used to determine which patients have effectively responded to immune therapy, identify risks and recurrences or even predict which patients are most likely to benefit from a particular treatment regimen.7
While immunotherapy offers some hope in the management of other cancers, it has not yet demonstrated improved overall survival (OS) for patients with glioblastoma (GBM).8–10 GBM, the most common malignant primary brain tumor in adults,11 12 is known for its poor median survival, high recurrence rate and limited treatment advancements.13 The intratumoral molecular heterogeneity and immunosuppressive tumor microenvironment of GBM are partly responsible for its therapeutic resistance and recurrence.14–18 Key dysregulations of the immune system within the tumor microenvironment, and this cancer’s anatomical location within the central nervous system (CNS) make the discovery of biomarkers to measure response to immune therapy particularly important.
Even in immune therapy trials reporting positive results, such as for untreated melanoma,2 only a subset of patients receives benefit from treatment. Similar mixed results have been reported in most immune therapy trials for various malignancies. High variability in patients’ clinical responses highlights the need for biomarkers which could guide clinicians to more precise treatment options based on each patient’s molecular and immune-related profiles.19
This review will examine current strategies and future directions in the discovery of GBM-specific biomarkers for immune therapy, major challenges that limit the ability to effectively adapt immunotherapy treatment in the CNS and highlight the potential role that genetic sequencing and other innovative technologies will have in discovering GBM-specific biomarkers.
Biomarkers in non-CNS cancers and the determination of responses to immune therapy
Several biomarkers, such as the presence of a pro-inflammatory T cell phenotype and molecular alterations in the tumor microenvironment, have made progress in helping to determine how patients will respond to checkpoint inhibition therapy in other cancers.20 21 However, most solid tumors exhibit molecular and immunological heterogeneity,22 which creates challenges in identifying biomarkers that determine which patients will benefit from immunotherapy. Tissue sampling, particularly of tumor tissue, remains the gold standard of pathological diagnosis and is critical for the evaluation and corroboration of most biomarkers under investigation. Tissue sampling is particularly important after treatment, as radiographic evidence of progression may not be reflected on tissue analysis.23
Immunophenotyping in non-CNS malignancy
Immunophenotyping, the characterization of the immune system components within tissue, has been a common technique for identifying potential biomarkers. This methodology focuses on cell type identification and analysis via antibody targeting such as immunohistochemistry (IHC). It is widely used in preclinical and clinical studies to investigate the immune system’s relationship with malignancy.
Lymphocytes, particularly T cells, were identified in tumors, blood and lymph nodes of patients with melanoma decades ago.24 Since this discovery, tumor infiltrating lymphocytes (TILs) and tumor-associated macrophages (TAMs) have been associated with disease prognosis in many solid tumors.25–28 In particular, CD103+ lymphocytes, most of which are CD8+ T cells, have been reported as a predictive biomarker of improved response with checkpoint inhibition therapy.29–31 Conversely, the presence of immunosuppressive cell types, such as regulatory T cells (Tregs), has been described as a negative prognostic biomarker in a variety of malignancies.32–34 Other groups report that the ratio of immunosuppressive and inflammatory cell types are prognostic biomarkers, with an increased ratio of Tregs cells to CD8+ cytotoxic T lymphocytes (CTLs) portending a poor prognosis.35 36
Studies with available pre-treatment and post-treatment tissue samples have helped to describe the effects of immune therapeutics on the tumor immune microenvironment of many cancers. These tissue-based pathological responses can then be related to survival and progression data from clinical trials to search for clinical correlation.
As study results of immunotherapies are released and recurrences are analyzed, there has been more data regarding pathological response following immune therapy. A phase II trial investigating imiquimod, an immunotherapy that activates toll-like receptor 7 on macrophages to increase the secretion of pro-inflammatory cytokines, in vulvar intraepithelial neoplasia demonstrated that the frequency of intratumoral CD4+ and CD8+ cells increased in response to treatment in responders, while non-responders demonstrated elevated Tregs.37 38 Notably, there was no significant difference in CD4 and CD8 levels in pre-treatment biopsy that would act as a predictive marker.
Lui et al reported improved survival in a subset of B16F10 melanoma-bearing mice with anti-PD-1 (programmed death-1) immunotherapy. On stratifying their population into responders and non-responders based on survival differences, they noticed the interferon (IFN)-γ secretion kinetics of peripheral lymphocytes could be used as an accurate predictive biomarker of response to this treatment.22
Chen et al performed a longitudinal study of patients with melanoma with multiple biopsies dictated by treatment response after serial checkpoint inhibitors of cytotoxic T lymphocyte associated protein-4 (CTLA-4) followed by PD-1 blockade.19 Patients underwent biopsy prior to initiation of anti-CTLA-4 therapy. Non-responders were re-biopsied prior to initiation of anti-PD-1 therapy. On-treatment biopsies were also obtained. Pre-treatment biopsies did not show any immune cell population differences between responders and non-responders, but early on-treatment biopsies showed higher CD8+ cell populations in responders to anti-CTLA-4. Pre-treatment biopsies prior to PD-1 blockade showed mostly higher CD3+, CD8+ and CD45RO+ cells in subsequent treatment responders. Early on-treatment biopsies of these patients’ tumors showed large increases in CD3, CD4, CD8, PD-1, PD-L1, LAG-3 and FoxP3 in responders. This study provided a rare view of predictive biomarkers and potential biomarkers of treatment response in longitudinal samples.
Programmed death-ligand 1 as an immune biomarker in non-CNS malignancy
Program death ligand-1 (PD-L1) is the ligand for PD-1, an immunosuppressive immune checkpoint that has been successfully targeted by multiple checkpoint inhibitors.39 Several clinical trials have analyzed the expression of PD-L1 in the tumor microenvironment as both a marker of prognosis for the tumors as well as to predict response to anti-PD-1/PD-L1 checkpoint inhibition.40–43 In a multicenter phase II, single-arm clinical trial, 270 patients with metastatic urothelial carcinoma were administered nivolumab, an anti-PD-1 antibody, to assess its safety and efficacy.44 They discovered that objective response was achieved in 28.4% of patients with PD-L1 expression >5%, in 23.8% of patients with PD-L1 expression >1% and in only 16.1% in patients with PD-L1 expression <1%. Conversely, in a randomized, phase III study designed to evaluate the safety and efficacy of nivolumab in 272 patients with advanced squamous cell NSCLC,45 they did not find any prognostic or predictive value of PD-L1 expression with nivolumab treatment. Due to these conflicting results, PD-L1 is not routinely used as a predictive biomarker in these patients.
Genetic biomarkers in non-CNS malignancy
Genetic profiling has demonstrated potential in discovering biomarkers in cancer. Das et al used genetic profiling to examine the immune response of tumors to checkpoint inhibition.46 When analyzing blood and tumor tissue from 45 patients undergoing checkpoint blockade, they found that combination therapy with antibodies targeting PD-1 and CTLA-4 led to an increase in T cell genes differentially expressed and a robust upregulation of IFN-γ. IFN-γ has been validated by numerous studies to be a good predictive biomarker in many solid tumors.47–49 Furthermore, Das et al showed that anti-PD-1 therapy induced cell lysis and expression of natural killer (NK) genes on T cells. A similar finding was reported in patients with melanoma.50 Gao et al showed that loss of IFN-γ in mice with melanoma was associated with poorer therapy response, further supporting the role of IFN-γ in antitumor response and long-term survival.51
Aside from identification of specific mutations, mutational burden determined by somatic genomic sequencing has been investigated in various malignancies to determine whether this can predict a response to checkpoint inhibition.52 High mutational burden has been associated with greater therapeutic response to immune therapy in non-CNS malignancies, particularly NSCLC and melanoma, among others.53 54 Mutational burden has been particularly supported as a predictive biomarker of clinical benefit for patients with NSCLC on immune therapy,55 56 despite it not being prognostic of survival for patients not on immune therapy.57
Goodman et al assessed the effects of tumor mutational burden (TMB) and clinical outcomes following immunotherapy in a retrospective review of 1638 patients with cancer, and reported a positive correlation between higher TMB and responsiveness to anti-PD-1/PD-L1 therapy in melanoma, NSCLC and many other tumors.58 This similar finding was reported in patients with colorectal cancer.59 Thus, clinical trials60 have sought to identify molecular alterations in cancers from patients who are classified as exceptional responders.61 Recent technological advancements have allowed the use of targeted sequencing of genes associated with increased mutational burden which could streamline the process of using mutational burden as a biomarker of response.62
Immunoscore in non-CNS malignancies
The Immunoscore is a newly developed pathological prognostic indicator which may develop into a predictive biomarker for immune therapy. Galon et al examined the density and location of specific infiltrative immune cells in patients with colorectal cancer and discovered that these factors were better predictors of OS than traditional histopathological methods.63 These findings have led to an additional system of cancer staging known as the Immunoscore. This system combines the traditional metrics used in the tumor, node, metatasis classification system, which primarily looks at the anatomical extent of disease for cancer staging,64 and a new component that evaluates the density of immune cells in the infiltrating tumor margin and the tumor core.65 This system has been used extensively in hepatocellular carcinoma, melanoma, colorectal, high-grade serous ovarian, invasive breast and gastric cancer.66–76 By routinely calculating an Immunoscore at the time of diagnosis prior to treatment with immune therapy, we may find that the Immunoscore can predict responses to checkpoint inhibition.
Biomarkers of response in metastatic CNS malignancies
Recent developments in checkpoint blockade have highlighted potential differences in response to immunotherapy between primary GBM and metastatic brain tumors. These clinical trials have demonstrated survival benefits for patients with tumors that frequently metastasize to the brain such as melanoma and NSCLC.77 78 The CheckMate-204 trial, combining nivolumab and ipilimumab, reported significant intracranial response rates of 46% in melanoma brain metastases.79 Predictive biomarkers in metastatic CNS malignancies have also been investigated by researchers. Capone et al looked at baseline neutrophil-to-lymphocyte ratio (NLR) as a predictive biomarker of response to nivolumab treatment in patients with advanced melanoma.80 Their group examined 97 patients with stage IV melanoma and discovered that absolute neutrophil count, NLR, derived neutrophil-to-lymphocyte ratio (dNLR) and lactate dehydrogenase were significantly associated with survival. More importantly, however, a subgroup analysis of 27 patients with brain metastases showed that NLR <4.7 and dNLR <3.8 were associated with extended OS and progression-free survival (PFS). A study by Berghoff et al used Immunoscore to evaluate the TILs in 116 brain metastases samples from melanoma, renal cell carcinoma and lung cancer.81 This study showed a strong positive correlation between median OS and CD3+, CD8+ and CD45RO+ TIL density.
Biomarkers in glioblastoma
A primary obstacle to the development of immunotherapy for GBM is the lack of accurate measures of treatment response. The Immunotherapy Response Assessment for Neuro-Oncology (iRANO), criteria used to determine if patients respond to immune therapy, relies on MRI to differentiate responders from non-responders.82 However, imaging alone cannot distinguish tumor progression from immunotherapy-induced inflammatory changes.83 84 Although the use of tissue sampling of a potential recurrence to guide treatment poses risks to patients, it remains the gold standard for the diagnosis of progression versus pseudoprogression. Moreover, there are relatively few published trial results of pathological changes in checkpoint inhibition-treated GBMs. Nonetheless, the development of new biomarkers could both help identify future responders and obviate the ongoing need for tissue confirmation of progression.
Immunophenotyping to determine biomarkers
As in other malignancies, immunophenotyping has been widely used in preclinical and clinical studies of GBM. In an immune-competent murine model of GBM, Zeng et al found that the combination of anti-PD-1 therapy and radiation increased the CD8+ T cell-to-Treg ratio in responders.85 Similarly, Fecci et al demonstrated that CTLA-4 blockade enhanced CD4+ T cell proliferative capacity and mitigated Treg-meditated suppression of these cells in murine models.86 Conversely, temozolomide increases the proportion of exhausted T cells in mice with intracranially implanted gliomas, and this reduces their response to checkpoint inhibition.87 This finding suggests that pre-existing T cell exhaustion may be a negative predictive biomarker of response to checkpoint inhibition.
Many groups have attempted to immunophenotype patients with glioma to better understand the baseline immune microenvironment of these tumors. In this way, the response to therapy in patients can be better defined. Immune infiltration varies widely between different grades of glioma.88–90 TAMs have been found to be one of the predominant immune cells found in patients with GBM.91 92 Tregs are also one of the predominant immune cell types in the immune microenvironment and may contribute to immune suppression.93 94 The increase in Tregs may be attributable to the secretion of the chemokine CCL2 by glioma tumors, which induces Treg migration into the tumor microenvironment.95 Single cell analyses of myeloid cells in the GBM microenvironment revealed that there is a high degree of intertumor heterogeneity between different patients and intratumor heterogeneity in comparison to healthy brain of the same patient.96 One group recently evaluated the T cells of patients with glioma in comparison to healthy controls and found that GBM tumors cause infiltrating T cell exhaustion and dysfunction, in spite of presence of recruited T cells.97 A study led by Kimiecik et al assessed the relationship between the frequency of lymphocyte infiltration and OS of patients with glioma treated with chemotherapy and radiation.98 They noticed extended survival in these patients was associated with increased CD3+ and CD8+ immune cell infiltrates. However, the formal Immunoscore, as reported by Galon et al, has not been applied to GBM patient databases. Such investigations could show that the Immunoscore is an important prognostic variable in patients with glioma, leading to its inclusion in patient stratification for glioma clinical trials.
One ex vivo study of TILs isolated from GBM tissue demonstrated a high percentage of exhausted T cells that overexpress multiple checkpoint molecule receptors.99 This group found that ex vivo treatment of these TILs with checkpoint inhibitors caused a subpopulation of T cells with a less differentiated phenotype to be reinvigorated.
It is currently unclear how standard therapy impacts the GBM immune microenvironment. A recent study described the immune infiltration of gliomas using IHC and found no significant difference in primary and recurrent GBMs, but this study was limited by a low number of matched samples.100 Another study suggests that an increase in Tregs in the perivascular space correlates to a decreased time to recurrence following standard treatments.101
Such immune phenotyping has been applied to clinical trial biomarker investigations. A pilot study investigating autologous cancer cell vaccinations using anti-CD3-stimulated lymphocytes in patients with recurrent grade III or IV astrocytoma found that the CD4/CD8 ratio of infused cells correlated with clinical outcomes, along with tumor grade and age.102 Additionally, patients with perivascular lymphocytic infiltration in their glioma tissue had a 4-month increase in survival compared with those with no lymphocyte infiltration.
Similarly, O’Rourke et al compared preinfusion and postinfusion EGFRvIII expression levels and immunosuppressive molecules in the tumor microenvironment while investigating peripherally infused EGFRvIII CAR T cell therapy for patients with recurrent GBM.103 They found a decrease in EGFRvIII expression in five of the seven patients who underwent postinfusion surgical resection and an increase in FOXP3+ cell frequency, IDO1, IL-10, PD-L1 and TGF-β. Interestingly, an increase in CD8+ T cell proliferation was also noted in three of the five patients evaluated.
Researchers have also investigated how immune phenotyping of peripheral immune cells may yield useful biomarkers in the future. This technique is easily performed, relatively inexpensive and less invasive than GBM tissue sampling. A phase I study assessed the use of VXMO1, a plasmid containing an attenuated Salmonella typhi, TY21a that encodes vascular endothelial growth factor receptor-2 (VEGFR-2), in patients with progressive GBM.104 This vaccine recruits VEGFR-2-targeting T cells and stimulates a systemic immune response. They performed T cell immune monitoring in the peripheral blood and IHC on brain tissue and discovered that a higher CD8-to-Treg ratio was associated with increased survival in patients with primary tumors. This ratio was also increased by VXM01 treatment. Additionally, a decrease in intratumoral PD-L1 expression correlated with increased survival, suggesting the benefit of anti-PD-L1 checkpoint inhibition in combination with VXM01.
A recent clinical trial performed a subset analysis of adjuvant intralesional autologous lymphokine-activated killer cell therapy for patients with primary GBM to differentiate responders versus non-responders.105 The researchers obtained autologous peripheral lymphokine-activated killer cells and incubated them in IL-2 prior to infusion into the tumor. They discovered that patients with more frequent CD3+CD16+CD56+ lymphokine-activated killer cells were associated with improved survival. Notably, this finding was more frequent in patients who did not receive corticosteroids in the month prior to leukapheresis.
A phase I clinical trial for recurrent malignant glioma evaluated the safety and efficacy of DNX-240, a tumor-selective replication-competent oncolytic adenovirus, and measured pre-treatment and post-treatment levels of checkpoint proteins and immune cell density in biopsy tissue.106 Comparing pre-treatment and post-treatment samples, they observed a decrease in TIM-3 levels following DNX-2401 administration, suggesting TIM-3 as a potential biomarker of response.
Cytokine levels as biomarkers
Beyond characterizing the presence of peripheral or tumor-infiltrating immune cells, measuring cytokine levels provides data regarding immune signaling and function. This may provide valuable insight into the complex relationship between pro-inflammatory and immunosuppressive signals in GBM and therapeutic response. Preclinical studies have suggested the potential utility of cytokine levels as biomarkers. For example, Wu et al found that GL261-Luc+-implanted mice treated with combination anti-PD-1 and anti-CXCR4 immunotherapy showed decreased production of pro-inflammatory cytokines including tumor necrosis factor-α and IFN-γ compared with controls.107
Clinical trials have reported cytokines as potential biomarkers for GBM, but their significance and utility remain unclear. Inogés et al conducted a phase II study of autologous DC vaccination in newly diagnosed patients with GBM.108 They found an increase in tumor-specific immune cell response after vaccination in 11 out of the 27 patients by measuring proliferation and cytokine production. However, they did not see any correlation between this immune response and survival.
Nonetheless, cytokine measurement is being increasingly incorporated into prospective study design. A phase I clinical trial studying the safety of administering vaccine therapy made from survivin peptide in conjugation with sargramostim, a granulocyte macrophage colony-stimulating factor (GM-CSF), for patients with GBM are defining treatment responders by measuring the IFN-γ levels for up to 6 months post-treatment.109
Tumor cell antigens as biomarkers
Outside of immune-specific markers, many tumor cell surface molecules have been identified in many malignancies, including GBM. These tumor-specific antigens are useful as targets in vaccine development and can also give insight into treatment response. Sampson et al conducted a phase II multicenter clinical trial to determine the immunogenicity of an EGFRvIII vaccine for patients with newly diagnosed EGFRvIII+ GBM.110 They discovered that delayed hypersensitivity responses to EGFRvIII, as well as the development of specific antibodies to EGFRvIII, significantly affected OS. Additionally, they noted that 82% of the patients who had recurrence had lost EGFRvIII expression in their tumors. The phase III multicentre trial of rindopepiput, an EGFRvIII vaccine, with temozolomide (TMZ) in newly diagnosed GBM cases reported negative outcomes and attributed their results to the differences in EGFRvIII expression in their patient cohort.111 These findings conflicted with the positive improvement in median OS (mOS) following vaccine treatment in the preceding phase II clinical trials.112
Tumor antigens may also act as biomarkers even when those antigens are not specifically targeted. A phase I clinical trial evaluating the immune response to a multiepitope-pulsed autologous dendritic cell vaccine in newly diagnosed patients with GBM showed that the expression of MAGE1 and AIM-2 on glioma cells was associated with significantly increased PFS.113 Additionally, they showed the expression levels of AIM-2 and MAGE1 correlated with both PFS and OS. For the five patients who underwent a second resection, there was a trend for increased survival with patients having tumors expressing gp100 and HER2 antigens, along with a decrease in CD133 expression levels.
PD-L1 as a biomarker in glioblastoma
PD-L1 expression has been widely investigated and reported as a potential biomarker within tumors and peripherally. An early analysis of PD-L1 expression on 135 specimens with GBM from both newly diagnosed and recurrent patients showed no clinical predictive or prognostic value.114 Contrary to these findings, Nduom et al found prognostic value for PD-L1, showing that high expression levels were associated with worse clinical outcomes.115 Furthermore, Pratt et al found varied PD-L1 expression, with high expression limited to a minority of patients.116
The clinical trial Checkmate 143 investigated the use of a PD-1 monoclonal antibody to improve median survival in recurrent patients with GBM in comparison to an anti-VEGF antibody, bevacizumab.8 117 They also retroactively measured PD-L1 expression to determine if the variability in expression could be used to distinguish responders from non-responders. However, this treatment did not increase mOS when compared with the control arm.
A phase II clinical trial using an autologous heat shock protein peptide vaccine, in combination with standard treatment, demonstrated decreased survival for newly diagnosed patients with GBM with high levels of PD-L1 on circulating myeloid cells, and elevated systemic immunosuppression.118 This association of PD-L1 expression and survival was not dependent on methylation status and was highly predictive of patient response to vaccine treatment.
Genetic biomarkers
Genetic and epigenetic aberrations are frequent in malignancy, including GBM. Further investigation of these features may reveal markers of response to immune therapy.
O6-methylguanine-DNA methyltransferase (MGMT) methylation is a predictive biomarker for response to standard chemoradiation.119 MGMT methylation status may play a similar role in immunotherapy. MGMT-methylation is associated with significantly improved survival compared with unmethylated MGMT patients in GBM vaccine therapy trials.12 120 Liau et al investigated autologous dendritic cell vaccine in newly diagnosed patients with GBM in a phase III clinical trial.121 They demonstrated that patients with a methylated MGMT had a better mOS of 34.7 months compared with 21.2 months in historic controls.
Mutational status in the tumor suppressor gene, phosphatase and tensin homolog (PTEN), may also be a predictive biomarker. A study conducted by Zhao et al evaluated the immune response of anti-PD-1 treatment in patients with GBM using genomic and transcriptomic analyzes.122 They discovered that the non-responders had PTEN mutations associated with immunosuppression, while responders had enriched MAPK pathways.
Isocitrate dehydrogenase (IDH) mutational status has been demonstrated as a prognostic factor in primary GBM,123 but this may not be a predictive marker for immune therapy. Desjardins et al evaluated the use of recombinant poliovirus in recurrent patients with GBM.124 In a retrospective analysis of their patient population, they did not find a survival advantage for patients who harbored the IDH1 mutation.
Recently, advances in genomic sequencing have allowed for more comprehensive analysis of newly discovered genes found in tumors, including in GBM. These genes can potentially act as biomarkers in GBM and its treatment with immunotherapy. Sequencing has given insight into GBM mutagenesis and oncogenesis, with groups reporting many GBMs demonstrating DNA damage repair deficiencies,125 extensive cell-to-cell heterogeneity from chromosomal instability126 and marked intratumoral genetic heterogeneity.127–129
Peng et al used sequencing to identify markers of genetic instability in long-term survivors of GBM following standard treatment.130 This group suggested that patients who do not fall within this category may be ideal targets for non-standard therapies such as immune therapy. Feng et al identified a series of immune-related genes associated with GBM prognosis and found that the heterogeneity within the lymphocyte population is associated with a greater degree of immune infiltration into the tumor.131 This gene panel could serve as a predictive biomarker for response to immune therapy.
Mutational burden
The relationship of mutational burden with immune therapy response in GBM remains nebulous. One study reported dramatic responses to anti-PD-1 checkpoint inhibition in two patients with the DNA mismatch repair deficiency known as Lynch disease.132 However, relatively few GBMs have high mutational burden as compared with other malignancies.133
Delayed-type hypersensitivity reactions
Delayed-type hypersensitivity (DTH) skin reactions, a measure of an inflammatory response to a specific foreign antigen, have also been used as a marker of immune response in clinical trials.134 A phase II multicenter trial exploring the immunogenicity and PFS of an EGFRvIII vaccine for newly diagnosed patients with GBM used patient serum and a DTH skin test with PEPvIII peptide to gauge the vaccine-induced immune response,135 elucidating additional clinical methods to identify minimally invasive biomarkers.
Similarly, a clinical trial conducted by Schneider et al used tumor cells modified with Newcastle Disease Virus (NDV) for 11 patients with GBM.136 Autologous tumor cells were extracted from the patients and incubated with NDV, then re-injected subcutaneously. They postulated that the vaccine would stimulate the immune system and show a local inflammatory response in skin with a corresponding relationship between OS and the rate of onset of the reaction. There was no significant correlation between area of skin inflammation and rate of systemic response or OS. However, the tumor samples of patients who underwent a second surgery after immunotherapy demonstrated a higher CD4+/CD8+ T cell infiltrating ratio following immunotherapy.
Multimodal biomarkers
Due to the complexity of the immune system in the disease state and in response to treatment, the simultaneous use of multiple biomarkers may best inform researchers in trial design and subsequent analysis. Additionally, it allows for more data and potential discovery to occur from fewer studies.
The ongoing phase I/II clinical trial for the IMA950 multipeptide vaccine in combination with poly-ICLC for GBM is seeking to determine if there is a correlation between clinical response and immunological response.137 They sample blood to measure cytokine secretion and proliferation of antigen-specific CD8+ and CD4+ T cells, frequency of myeloid cells and Tregs, and activation marker expression on tetramer positive cells. This study may show effective-response monitoring using only peripheral blood samples, benefiting patients and clinicians due to its relative non-invasiveness and ease of future implementation.
The randomized multi-institutional clinical trial conducted by the Ivy Foundation Early Phase Clinical Trials Consortium used gene analysis when aiming to demarcate immune therapy responders from non-responders.138 The study examined the immune response of 35 recurrent patients with GBM receiving neoadjuvant and/or adjuvant anti-PD-1 therapy. They noted that the use of neoadjuvant anti-PD-1 correlated with an upregulation of IFN-γ-related gene expression, while downregulating cell cycle-related genes in the tumor when compared with the adjuvant anti-PD-1 group. Additionally, they showed that this neoadjuvant treatment increased clonal T cell expansion, attenuated PD-L1 expression on peripheral blood T cells and decreased monocyte frequency. Neoadjuvant anti-PD-1 immune response was also characterized in a phase II single-arm study that compared pretumor and post-tumor tissue samples after treatment.139 They showed that neoadjuvant anti-PD-1 increased immune cell infiltration, T cell receptor clonal diversity among infiltrating T cells and enhanced expression of chemokine transcripts.
Future directions for biomarker identification and quantification
Given the need for rigorous biomarker collection in future clinical trials, novel techniques of biomarker quantification are vital. Despite the current standard of treatment for patients with GBM (figure 1A), biomarker collection should rely on repeat tissue sampling (figure 1B,C), as immunotherapy efficacy is difficult to interpret using MR imaging alone.140 Stereotactic biopsy has a low side-effect profile and provides pathological diagnosis and potential biomarkers that may be critical for effective stratification of patients to particular existing therapy or enrollment in a variety of active clinical trials (figure 2). While future trials may obviate the need for such tissue collection, we believe it is critical that tissue sampling and window of opportunity trials continue in patients with GBM until such less invasive biomarkers are developed (figure 3).

Different biomarker acquisition timepoints. (A) Standard. The current standard treatment course for patients with glioblastoma (GBM) used by practicing physicians. With this standard approach, biomarkers are only acquired from the primary resection tumor sample and usually without additional tissue samples taken during any subsequent trials. (B) Randomized neoadjuvant vs adjuvant immune therapy at recurrence. This schematic describes a more innovative approach to clinical trial design practiced by a few physicians allowing for more timepoints to acquire biomarkers. Following standard treatment, recurrence is suspected from MR imaging and patients can be enrolled on a trial to be treated with neoadjuvant immunotherapy prior to resection of the recurrent tumor or adjuvant immunotherapy after the second resection. Here, biomarkers can be collected from the primary tumor (I) and from recurrent tumor tissue following ±neoadjuvant immunotherapy (II). Additionally, non-surgical biomarkers can be collected during follow-up adjuvant immunotherapy visits (III). (C) Proposed trial scheme to maximize biomarker identification. The proposed ideal approach to designing clinical trials to ensure physicians obtain biomarkers at all critical timepoints. First, biomarkers are collected from the primary tumor sample from the initial biopsy and/or resection (I). On suspected recurrence on MRI, a second biopsy will be performed to confirm recurrence vs pseudoprogression and to collect biomarkers of the recurrent tumor prior to any immunotherapy treatment (II). Third, biomarkers will be collected from resected tumor tissue after neoadjuvant administration of immunotherapy (III). Fourth, during adjuvant immunotherapy treatment, non-surgical biomarkers can be acquired with patient follow ups (IV). Lastly, should a patient present with progression vs pseudoprogression again, biomarkers can be collected during a therapeutic surgical intervention (V).

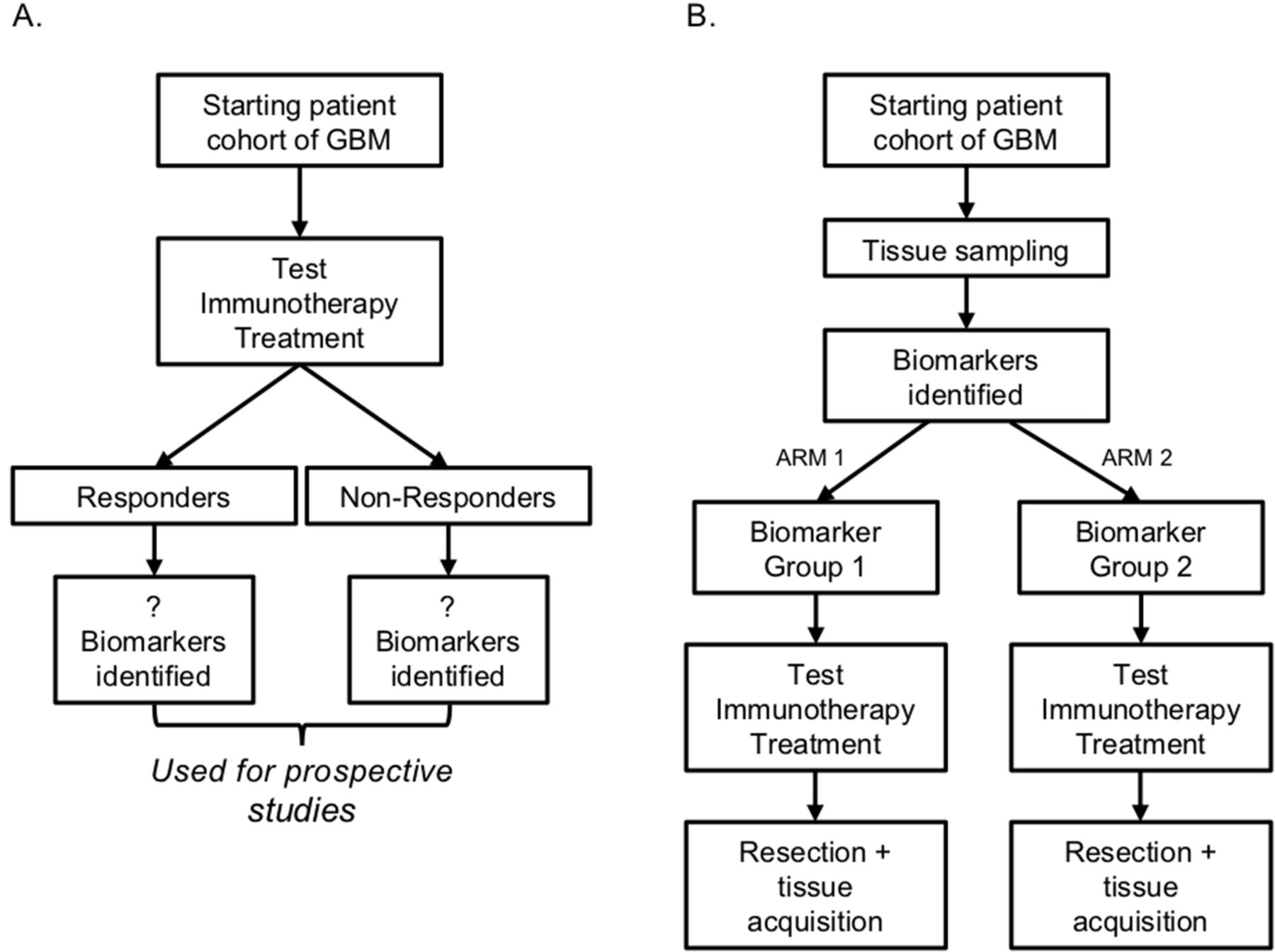
Immunotherapy trial design to find new predictive biomarkers. (A) A flow chart detailing how retrospective clinical trials can identify predictive biomarkers. As patients enroll in immunotherapy clinical trials, researchers can retrospectively delineate the responders to treatment from the non-responders and determine biomarker differences (mutation vs wild type, presence vs absence, high vs low expression). (B) Description of how identified predictive biomarkers in retrospective studies can be rigorously tested in prospective studies. On enrollment, patients will have their tissue sampled to identify changes in biomarkers identified in the cognate retrospective study. Then they will be stratified according to these criteria and administered treatment. Response from both groups will be compared to determine if the identified biomarkers are truly predictive of response to treatment.

Trial scheme for the use of previously identified predictive biomarkers. A flow chart depicting how predictive biomarkers can be used in current clinical practices. When patients are initially diagnosed with glioblastoma (GBM), their tissue will be sampled to determine the presence of predictive biomarkers. Depending on the biomarkers present, patients will be enrolled in clinical trials that have shown response to treatment when that biomarker is present. If the patient is responding to treatment they will remain on the clinical trial, but if not, they can be switched to a different clinical trial to continue a different immune therapy treatment.
Clinical trial design
Moving forward, there are many ways biomarkers can be used to improve immunotherapy for patients with GBM. Clinical trials can be designed both retrospectively and prospectively to look at responders and non-responders to immunotherapy. By stratifying patients by response, researchers can identify which biomarkers can be used to differentiate these two groups (figure 2A). Subsequent trials can determine if these biomarkers have predictive value by prospectively stratifying their patient cohort based on the biomarkers and comparing their treatment response with controls (figure 2B). This allows systematic, targeted immunotherapy for patients with GBM that will most likely benefit from a particular treatment regimen (figure 3).
Microdialysis catheters
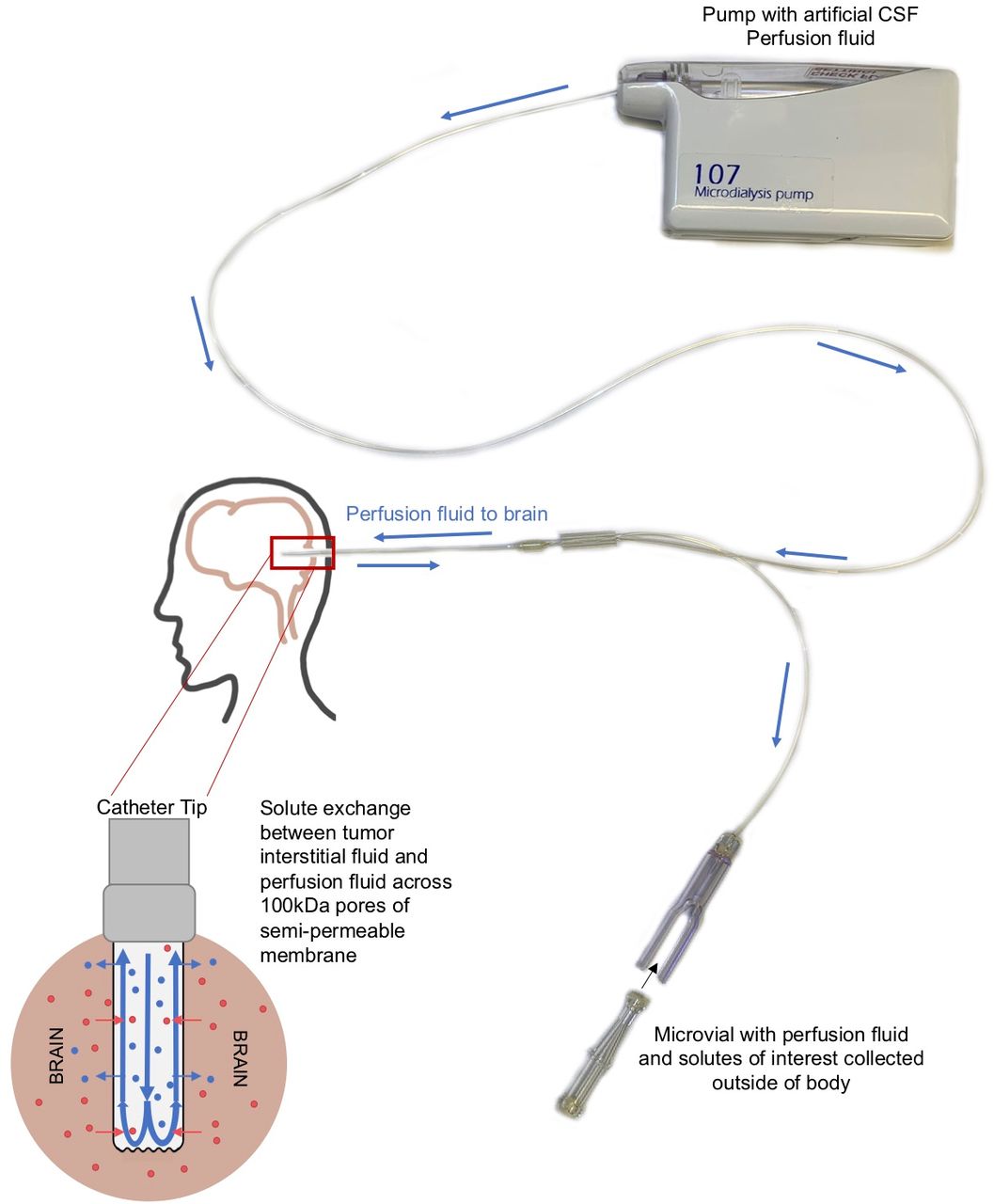
Cerebral microdialysis samples interstitial fluid of the brain using catheter placement141 (figure 4). This technology has been demonstrated to be safe to patients and is able to measure immune biomarkers in the context of cerebrovascular injury142 and traumatic brain injury.143 The use of microdialysis catheters for biomarker acquisition can provide clinicians with direct access to the tumor microenvironment by sampling the interstitial fluid of the brain and/or tumor. This may obviate the concern that biomarkers of response collected from other fluid compartments of the body, such as the blood or cerebrospinal fluid (CSF), may be diluted as compared with the interstitial fluid.144–146 Small early phase trials demonstrated the potential utility of cerebral microdialysis for cytokines in neuro-oncology. Portnow et al reported changes in 17 cytokines following chemotherapy for various intracerebral malignancies including GBM via microdialysis monitoring over 96 hours following craniotomy.147 Similarly, this technique has demonstrated immune response in the GBM peritumoral region following radiation therapy.148 The ongoing study ‘Cytokine Microdialysis for Immune Monitoring in Recurrent Glioblastoma Patients Undergoing Checkpoint Blockade’ uses microdialysis catheters to sample the tumor and normal brain microenvironments while concurrently collecting peripheral blood mononuclear cells and CSF of patients undergoing neoadjuvant anti-PD-1 immunotherapy prior to secondary surgical resection with continued infusions of combination of anti-PD-1 and anti-LAG-3 immunotherapy postoperatively.149 This comprehensive approach may identify new biomarkers for response, or lack thereof, in checkpoint inhibition for GBM as well as correlate between markers in different physiological spaces (figure 1C).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cytokine microdialysis catheter mechanism. A diagram describing the mechanism of the microdialysis catheter. Artificial cerebrospinal fluid (CSF) perfusion fluid is pushed through the catheter using a microdialysis pump. When the fluid reaches the catheter tip, solutes are exchanged between the artificial CSF perfusion fluid and the interstitial fluid of the brain through a semi-permeable membrane. The resulting mixture is then pushed into a microvial where it can be collected and analyzed.
Decreased invasive sampling
Recently, less invasive techniques to collect biomarkers to monitor response to treatment have been explored. Liquid biopsies have also been proposed as an alternative to conventional tissue biopsies in evaluation of treatment response.150 These biopsies typically involve the use of peripheral blood to identify the presence of intact circulating tumor cells, circulating tumor DNA (ctDNA) and other tumor-derived molecules that have entered into the bloodstream or CSF from primary or secondary tumors.127 Since liquid biopsies examine cancer-related factors present in the peripheral blood or CSF, they provide clinicians with a less invasive approach than typical tissue biopsies to acquire biomarkers. Potential biomarkers, including ctDNA, peripheral blood mononuclear cells and microRNA, have been found to be differentially present within serum, urine, stool and/or saliva samples after immune therapy.151 Kitahara et al used plasma ctDNA levels to monitor response to a peptide vaccine in patients with colorectal cancer.152 Cabel et al used ctDNA to distinguish tumor progression from pseudoprogression in patients with melanoma following checkpoint inhibition therapy and found that loss of detectable ctDNA at 8 weeks after treatment correlated with durable clinical response and improvement survival.153 Another study of 200 patients with breast, lung, colorectal or ovarian cancer showed a high concordance of mutations in ctDNA and tumor tissue.154 Additionally, an analysis of the colorectal patient cohort revealed that high levels of ctDNA was associated with lower OS and disease recurrence.
Interestingly, microbiota within stools samples have also been shown to play a potential role in immune therapy response. Matson et al analyzed baseline stool samples from patients with multiple melanoma before immunotherapy treatment and observed a significant correlation between commensal microbial composition and clinical response, finding Bifidobacterium longum, Collinsella aerofaciens and Enterococcus faecium to be more abundant in responders.155 Overall, although this field is relatively new, non-invasive strategies for biomarker collection may prove to be beneficial in stratifying patients and treatment-response monitoring for future immune therapy trials.
Non-invasive imaging development
Discerning pseudoprogression from progression of the disease based on imaging is a notoriously difficult problem because the inflammatory response can often mimic non-enhancing tumor progression.140 New imaging modalities are required that can either more accurately detect immune infiltration or specifically identify tumor progression separate from inflammation.
Antonios et al developed a non-invasive combination imaging technique to differentiate immune inflammatory changes from tumor progression in intracranial murine gliomas and in patients with GBM after dendritic cell vaccination and/or PD-1 inhibition.156 Their combination of MRI and PET imaging with a probe for deoxycytidine kinase (dCK), a protein marker overexpressed in immune cells, is a new technique that was able to effectively image immune response in intracranial tumors in preclinical murine models and in patients. Similarly, Rashidian et al use PET imaging to determine infiltration of CD8+, CD11b+ and CD45+ immune cells in tumors in response to anti-PD-1 treatment in a murine model.157 These modalities may provide clinicians a non-invasive way to monitor response of immunotherapy in GBM tumors, a critical necessity in measuring efficacy in future clinical trials.
Conclusion
As immune therapy continues to expand in response to promising results for malignancies with poor prognosis, the demand for effective biomarkers will only continue to increase. The discovery and development of effective techniques to predict therapeutic efficacy and measure patient response have the potential to significantly improve clinical decision-making. By enabling clinicians to treat patients with immune therapeutics that have the highest chance of evoking an objective response, biomarker development will improve the practice of immune therapeutics. The future of biomarker development will maximize positive outcomes to patients while minimizing exposure to ineffective therapy with potential risk.
References
Footnotes
Twitter @eknduom
JPL and AKN contributed equally.
Contributors JL, AN, HS, EKN conceptualized, wrote, designed the figures, and edited the paper. VS, KS, OA, GD conceptualized, wrote and edited the paper. All authors read and approved the final manuscript.
Funding This research was supported by the Intramural Research Program of the NINDS at the National Institutes of Health.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.