Article Text

Abstract
Background Li-Fraumeni syndrome (LFS) is characterized as an autosomal dominant cancer predisposition disorder caused by germline TP53 gene mutations. Both primary and therapy-related hematopoietic malignancies with LFS are associated with dismal outcomes with standard therapies and even allogenic stem cell transplantation (SCT).
Case presentation We reported a relapsed/refractory acute B-cell lymphoblastic lymphoma (B-LBL) patient in the context of LFS. He was identified to harbor a TP53 c.818G>A (p.R273H) germline mutation, and his family history was significant for rectal carcinoma in his father, an unknown cancer in his sister and acute lymphoblastic leukemia in his brother and one of his sons. The patient received murine monoclonal anti-CD19 and anti-CD22 chimeric antigen receptor (CAR) T-cell “cocktail” therapy and achieved complete remission with negative minimal residual disease (MRD), as assessed by morphology and multiparameter flow cytometry. Fifteen months after murine monoclonal CAR T-cell “cocktail” therapy, the patient’s B-LBL recurred. Fortunately, a round of fully human monoclonal anti-CD22 CAR T-cell therapy was still effective in this patient, and he achieved CR again and continued to be followed. Each time after infusion, the CAR T-cells underwent extremely rapid exponential expansion, which may be due to the disruption of TP53, a gene that can functionally control cell cycle arrest. Grade 4 and grade 1 cytokine release syndrome occurred after the first and second rounds of CAR T-cell therapy, respectively.
Conclusions This case provides the first report of the use of CAR T-cell therapy in a hematologic malignancy patient with LFS. As traditional chemotherapy and allogenic SCT are not effective therapy strategies for patients with hematologic malignancies and LFS, CAR T-cell therapy may be an alternate choice.
ChiCTR-OPN-16008526 and ChiCTR1900023922.
- haematology
- immunotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Li-Fraumeni syndrome (LFS) is a prototypical cancer predisposition syndrome caused by germline TP53 gene mutations and was first described in 1969 by Drs Frederick Li and Joseph Fraumeni.1–4 Patients with LFS have a predisposition to multiorgan tumorigenesis and are at increased risk of cancer-related morbidity. The LFS primary tumor spectrum includes soft tissue sarcomas, breast cancers, brain tumors, osteosarcomas and adrenocortical carcinomas.5 Other cancers associated with LFS include ovarian, gastrointestinal, pancreatic, genitourinary, skin, renal, thyroid, prostate and lung cancers, as well as hematopoietic malignancies including leukemia, lymphoma and MDS. The incidence of hematopoietic malignancies in LFS is ~4%.6 In addition, therapy-related hematopoietic malignancies are common in patients with LFS.7
Although patients with TP53 germline mutations are being identified in a rapidly increasing number because of improved genetic screening techniques and the increased acceptability of clinical testing for germline mutations, there are more patients with hereditary cancer predisposition syndromes than we thought, especially children.6 8 In a large genomic profiling study of 124 patients with childhood hypodiploid ALL, TP53 alterations were observed in 91.2% of low-hypodiploid patients. Moreover, in 43.3% of the TP53 mutation-carrying patients, the same mutations were also present in their non-tumor cells, suggesting that these were germline alterations and that this portion of patients had LFS.9 In adult low-hypodiploid acute lymphoblastic leukemia (ALL) patients, the incidence of germline TP53 mutations is not as high.10 The obvious disparity in TP53 germline mutation incidence between children and adult ALL patients suggests an early onset of disease, reflecting a high risk of cancer development.
In regard to treatment, Pepper et al found that TP53-mutated lymphocytes in LFS patients had an intrinsic resistance to conventional chemotherapeutic drugs.11 Moreover, a recent case series study showed that both primary and therapy-related hematopoietic malignancies with LFS were associated with dismal outcomes with standard therapies and even allogenic stem cell transplantation (SCT).5 The cohort of patients in that study included five patients with a history of solid tumors and a known LFS diagnosis who developed therapy-related acute myeloid leukemia (AML) or MDS during follow-up surveillance, as well as two patients who were diagnosed with de novo acute leukemia with an LFS diagnosis established during leukemia therapy. They had no responses or very short responses to induction therapy. Five patients received allogenic SCT after chemotherapy but did not achieve long-term remission. All of them had extremely poor outcomes. In summary, standard therapy and allogenic SCT do not provide long-term control of primary or therapy-related hematopoietic malignancies that are associated with LFS, and more efficient treatment methods are needed for these situations. Here, we report a relapsed/refractory acute B-cell lymphoblastic lymphoma patient in the context of LFS who was identified to harbor a TP53 c.818G>A (p.R273H) germline mutation. After failure of several lines of chemotherapy, this patient received murine monoclonal anti-CD19 and anti-CD22 chimeric antigen receptor (CAR) T-cell “cocktail” therapy and achieved complete remission (CR). Fifteen months after murine monoclonal CAR T-cell “cocktail” therapy, the patient’s B-cell lymphoblastic lymphoma (B-LBL) recurred. Fortunately, a round of fully human monoclonal anti-CD22 CAR T-cell therapy was still effective in this patient, and he achieved CR again. Before this study, immunotherapeutic strategies had not been studied in LFS. The present study showed a potential therapeutic strategy for LFS with hematologic malignancies.
Case report
A 38-year-old man presented to a local hospital in April 2017 with pain throughout his body that had been occurring for a month. He was diagnosed with diffuse large B-cell lymphoma (DLBCL) at stage IV and had an international prognostic index (IPI) score of 3. The patient was started on induction chemotherapy with four courses of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and carboplatin) and then attained partial remission by positron emission tomography-CT. He then underwent two courses of R-ICE (rituximab, ifosfamide, carboplatin, and etoposide), two courses of DHAP (dexamethasone, high-dose-Ara-C, and platinol) and one course of R-MA (rituximab,methotrexate,and Ara-C). He did not achieve CR.
To pursue CAR T-cell therapy, the patient was referred to our hospital in January 2018. The multiparameter flow cytometry results showed that 2.9% of nucleated cells expressed surface CD20, CD19, CD10, CD38, and CD22, as well as intracellular CD79a and TdT (terminal deoxynucleotidyl transferase), while Igκ and Igλ were negative. The immunophenotypic analysis is partially illustrated in figure 1A a–d. In addition, immunohistochemistry staining also showed that the bone marrow sections were TdT positive (figure 1B). According to these phenotypes, the patient was newly diagnosed with relapsed/refractory acute B-LBL.12 13

Immune and cytological analysis. (A) Phenotypic analysis of the bone marrow aspirate at diagnosis (a–d) and after fully human CAR T-cell infusion (e–h). Red dots represent CD19+ cells; green dots represent mature lymphocytes; blue dots represent progenitor B-cells; gray dots represent all the other cells. (B) Histological analysis of bone marrow sections by hematoxylin and eosin (H&E) and anti-TdT staining at diagnosis. (C) H&E staining of bone marrow aspirate slides at diagnosis (left) and at CR (right). CAR, chimeric antigen receptor; TdT, terminal deoxynucleotidyl transferase.
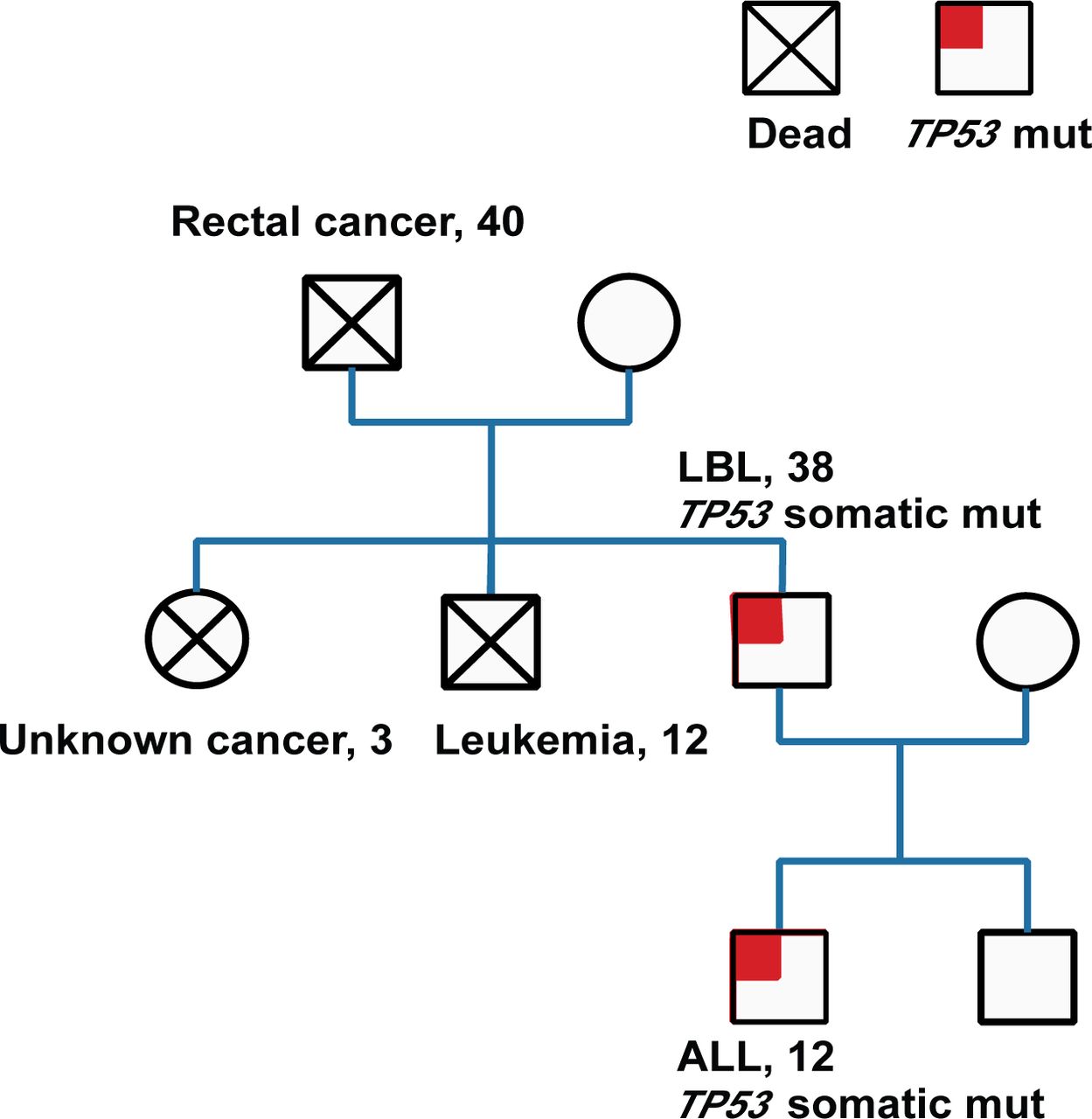
Hematological malignancy-targeted deep sequencing was performed on his plasma cell-free DNA and revealed a TP53 c.818G>A (p.R273H) mutation with a mutation allele fraction (MAF) of 88.8% by droplet digital PCR.online supplementary material To test whether the patient harbored a germline TP53 mutation, we analyzed his saliva samples and cells obtained by buccal swabbing. The TP53 germline analysis revealed the same mutation. Family studies were initiated, and the patient’s father, sister and brother had died from rectal carcinoma, an unknown tumor and ALL, respectively. Additionally, the same TP53 variant was also detected in one of the patient’s sons, who suffered from ALL 18 months later. Given these results, the patient was considered to have LFS (figure 2).
Supplemental material

Family history studies. Pedigree charts. The number indicates the age at which the symptomatic tumor was detected. ALL, acute lymphoblastic leukemia; LBL, lymphoblastic lymphoma.
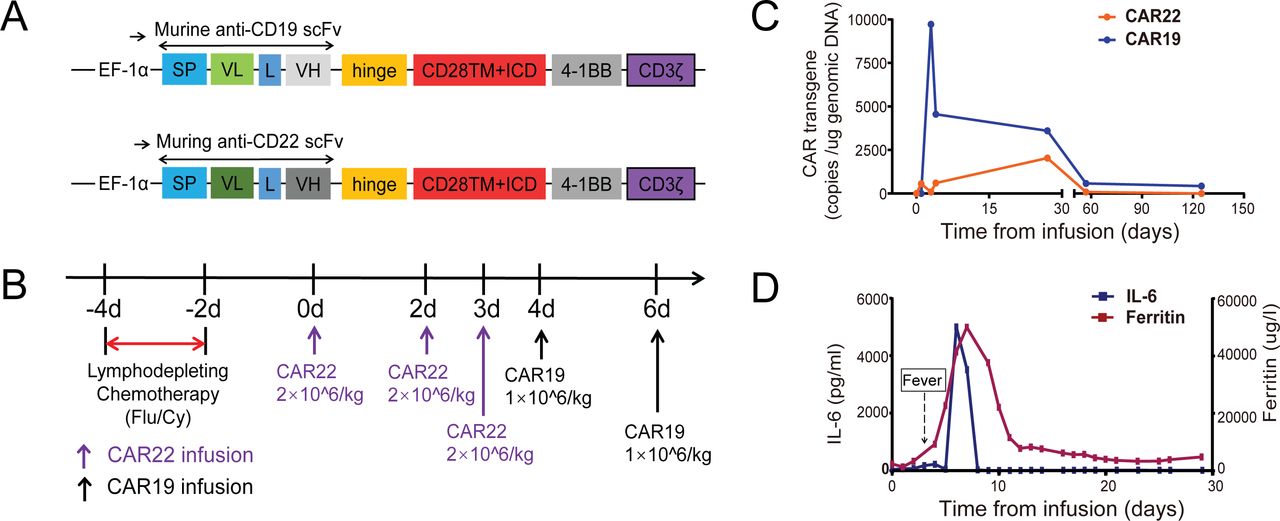
To stop tumor progression, the patient was given decitabine (25 mg/m2) and liposomal doxorubicin before CAR T-cell therapy. Then, he received lymphodepleting chemotherapy with fludarabine (25 mg/m2) and cyclophosphamide (300 mg/m2) for 3 days (days −4 to −2). Subsequently, he received murine monoclonal anti-CD19 and anti-CD22 CAR T-cell “cocktail” therapy (figure 3).14 Autologous CD22-targeted CAR T-cells (CAR22) (2*106 cells/kg) were infused on day 0, day +1, and day +2, followed by CD19-targeted CAR T-cells (CAR19) (1*106 cells/kg) on day +3 and day +5 (figure 3B). After day +5, he suffered a serious case of cytokine release syndrome (CRS) of grade 4 according to the grading system published by Lee et al, which lasted for 4 days and was characterized by a sustained high fever at 39°C to 40°C and hypoxia.15 At the same time, he also developed headache, dizziness, aphasia, dyscalculia and delirium lasting for a week, which fit the criteria for immune effector cell-associated neurotoxicity, grade 3.16 17 These symptoms were attenuated after plasmapheresis and steroid treatment. Both the CAR19 and CAR22 transgene copy numbers were tracked (figure 3C). At day +27, the patient achieved CR with negative MRD, as assessed by morphology and multiparameter flow cytometry of bone marrow aspirate samples.

The protocol and response for murine monoclonal anti-CD19 and anti-CD22 CAR T-cell “cocktail” therapy. (A) Schematic diagram of murine anti-CD19 and anti-CD22 CAR vectors. SP, signal peptide; VH, variable H chain; L, linker; VL, variable L chain. (B) The protocol of murine CAR22 and CAR19 “cocktail” infusion in combination with chemotherapy. Chemotherapy included fludarabine and cyclophosphamide. (C) Murine CAR22 and CAR19 transgene copy numbers detected by ddPCR. (D) Levels of IL-6 and ferritin after murine CAR22 and CAR19 infusion. CAR, chimeric antigen receptor; ddPCR, droplet digital PCR.
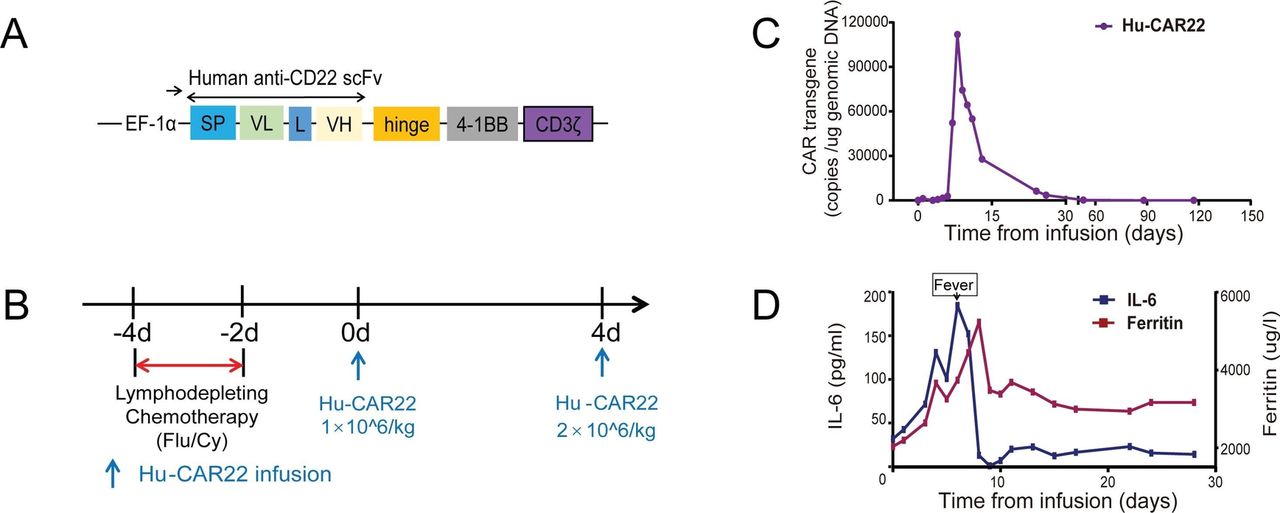
Fifteen months after CAR T-cell “cocktail” therapy, the patient’s B-LBL unfortunately recurred. In June 2019, he received fully human anti-CD22 CAR T-cell therapy after lymphodepleting chemotherapy with fludarabine and cyclophosphamide (figure 4). The cells carrying the fully human CAR transgene exhibited efficient expansion. The Cmax (maximum expansion) value reached 1.1*105 copies/µg genomic DNA at day +8 (figure 4C). At day +30, the patient achieved CR again (figure 1A e–h, C). He is currently in remission 4 months after fully human CAR T-cell therapy and continues to be followed.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The protocol and response for the fully human monoclonal anti-CD22 CAR T-cell therapy. (A) Schematic diagram of fully human anti-CD22 CAR vectors. SP, signal peptide; VH, variable H chain; L, linker; VL, variable L chain. (B) The protocol of Hu-CAR22 infusion in combination with chemotherapy. Chemotherapy included fludarabine and cyclophosphamide. (C) Hu-CAR22 transgene copy numbers detected by ddPCR. (D) Levels of IL-6 and ferritin after Hu-CAR22 infusion. CAR, chimeric antigen receptor; ddPCR, droplet digital PCR.
Discussion
An international consensus on the components of a cancer screening program for TP53 mutation carriers is lacking, and therefore, the personalized guidance for TP53 mutation carriers varies across institutions, regions, and countries. However, this situation has improved in recent years. In 2015, the MD Anderson Cancer Research Center announced the beginning of a program named LEAD (Li-Fraumeni Syndrome Education and Early Detection) for patients with LFS, which offers a comprehensive screening plan for LFS patients to detect cancers as early as possible. Furthermore, several surveillance protocols for TP53 mutation carriers have been proposed in Australia, the USA, and Canada.4 18–20 Nevertheless, in most countries, effective risk management approaches in carriers of TP53 germline mutations remain to be established, particularly in children.
In the present patient, screening for TP53 germline mutations was not performed, and LFS was not established during diagnosis and the beginning of treatment. The presence of his TP53 germline mutation was considered when the MAF of the TP53 c.818G>A (p.R273H) mutation detected in his plasma circulating tumor DNA was consistently above 50%, even when he achieved complete remission by multicolor flow cytometry. Thus, lipid biopsy may be applied as a method to discover germline mutations.
Patients with DLBCL that is refractory to chemotherapies or that has relapsed after SCT have a poor prognosis. Two CAR T-cell products were approved for use in DLBCL. Axi-cel is the first CAR T-cell product to be FDA approved for DLBCL. The overall response rate was 83%, with 58% of patients having complete responses.21 The second anti-CD19 CAR T-cell product to receive FDA approval for DLBCL is tisa-cel. The overall response rate to tisa-cel in adult relapsed or refractory DLBCL was 52%; 40% of subjects had complete responses.22 CAR T-cell therapy also showed promise in children and adults with refractory and relapsed B-cell ALL. In a reported trial, after receiving tisa-cel, 90% of patients achieved CR with 6-month event-free survival and OS values of 67% and 78%, respectively.23
Antigen escape-related relapse is a major challenge for long-term disease control in CAR T-cell therapy. Our group previously reported that treatment of relapsed/refractory (r/r) ALL patients with a cocktail of both CAR19 and CAR22 prevented antigen escape of CD19-CD22+blasts0.24 In this case, we first administered sequential CAR T-cell infusions targeting CD19 and CD22 to prevent antigen escape. This CAR T-cell “cocktail” was efficient, and the patient achieved CR with negative MRD, which lasted for 15 months. Furthermore, this study also indicated that a fully human CAR may still work even if the patient relapses after infusion of T-cells carrying a murine monoclonal antibody-based CAR.
This case provides evidence for the use of CAR T-cell therapy in hematologic malignancy patients with germline TP53 mutations. Each time after infusion, the CAR T-cells underwent extremely rapid exponential expansion. A recent study reported that TET2-disrupted anti-CD19 CAR T-cells exhibited growth advantages and displayed a central memory phenotype, which suggested that some key gene mutations may alter the function of CAR T-cells.25 In this case, we supposed that the extremely rapid exponential expansion was due to the disruption of TP53, a gene that can functionally control cell cycle arrest.
In conclusion, this is the first time that immunotherapeutic strategies have been used in LFS. Since LFS is associated with dismal outcomes with standard therapies and even allogenic SCT, CAR T-cell immunotherapy may be a promising treatment method. We suggest that CAR T-cell therapy be considered in LFS patients with r/r B-cell lymphoma or leukemia. As TP53 mutations will be present in CAR T-cells derived from LFS patients, attention should be paid to potential extremely rapid exponential expansion and CRS.
Acknowledgments
The authors would like to thank all members of the study team, the patient, and his family. We would also like to thank the wonderful work of Bio-Raid Company and Laso Biotherapeutics Company for the preparation of CAR T-cells.
References
Footnotes
LC, BX and XL contributed equally.
Contributors LC, XL, YL, JG, YW, XM, JW, HA, and MX performed the experiments, analyzed the data; BX, DW, YC, CL, GW, NW, YX, and JZ took care of the patient, provided clinical information. LC wrote the manuscript. YX and JZ directed the research.
Funding This work was supported in part by the National Natural Science Foundation of China (No. 81700160 to LC, No. 81873444 to YX and No. 81830008 to JZ).
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval This study was approved by the Medical ethics committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology (TJ-IRB20160310 and TJ-IRB20190609). Informed consent was obtained from the patient in strict accordance with the principles in Declaration of Helsinki.
Provenance and peer review Not commissioned; externally peer reviewed.