Article Text

Abstract
Background CTLA-4 blockade with ipilimumab is Food and Drug Administration-approved for melanoma as a monotherapy and has been shown to modulate the circulating T-cell repertoire. We have previously reported clinical trials combining CTLA-4 blockade with granulocyte-macrophage colony-stimulating factor (GM-CSF) in metastatic melanoma patients and in metastatic castration resistant prostate cancer (mCRPC) patients. Here, we investigate the effect that cancer type has on circulating T cells in metastatic melanoma and mCRPC patients, treated with ipilimumab and GM-CSF.
Methods We used next-generation sequencing of T-cell receptors (TCR) to compare the circulating T cells of melanoma and mCRPC patients receiving the same treatment with ipilimumab and GM-CSF by Wilcoxon rank sum test. Flow cytometry was utilized to investigate specific T-cell populations. TCR sequencing results were correlated with each T-cell subpopulation by Spearman’s rank correlation coefficient. Of note, 14 metastatic melanoma patients had samples available for TCR sequencing and 21 had samples available for flow cytometry analysis; 37 mCRPC patients had samples available for sequencing of whom 22 have TCR data available at both timepoints; 20 of these patients had samples available for flow cytometry analysis and 16 had data available at both timepoints.
Results While melanoma and mCRPC patients had similar pretreatment circulating T-cell counts, treatment induces greater expansion of circulating T cells in melanoma patients. Metastatic melanoma patients have a higher proportion of clones that increased more than fourfold after the treatment compared with mCRPC patients (18.9% vs 11.0%, p=0.017). Additionally, melanoma patients compared with mCRPC patients had a higher ratio of convergent frequency (1.22 vs 0.60, p=0.012). Decreases in clonality induced by treatment are associated with baseline CD8+ T-cell counts in both patient groups, but are more pronounced in the melanoma patients (r=−0.81, p<0.001 vs r=−0.59, p=0.02).
Trial registration numbers NCT00064129; NCT01363206.
- oncology
- immunology
- medicine
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Ipilimumab (Bristol-Myers Squibb) is a monoclonal, fully humanized IgG1 antibody against the T-cell coinhibitory receptor CTLA-4 that is the first agent to have shown improved survival in a randomized trial for patients with metastatic melanoma.1 Since the Food and Drug Administration approval of ipilimumab in 2011 for the treatment of unresectable or metastatic melanoma, there have been studies aimed at combining other therapies to improve on these outcome results and to gain better insight into the immunologic mechanism of these immunotherapies.2 One such combination is ipilimumab with granulocyte-macrophage colony-stimulating factor (GM-CSF). GM-CSF is a hematopoietic growth factor that stimulates the differentiation and proliferation of progenitor cells such as neutrophils, monocytes, macrophages, and myeloid-derived dendritic cells.3 Dendritic cells are antigen-presenting cells involved with primary and secondary T-cell immune response. Because GM-CSF also acts as a mediator of proliferation, maturation, and migration of dendritic cells, it is not surprising that adjuvant therapy of melanoma with GM-CSF has been reported to improve overall survival.4–6
Prostate cancer is less responsive to immunotherapies compared with melanoma.7 CTLA-4 blocking monoclonal antibodies has been studied extensively in patients with malignant melanoma. CTLA-4 blockade by ipilimumab results in long-term disease control in about 20% of metastatic melanoma patients.8 However, two phase III studies of ipilimumab in prostate cancer failed to show improvement in overall survival.9 10 There are many potential reasons for the difference in immunotherapy benefit between metastatic castration resistant prostate cancer (mCRPC) and melanoma patients including time of diagnosis and location of metastatic disease. One prominent theory is that melanoma is considered among the more immunogenic cancers. Genomic sequencing of tumor genetic material from melanoma patients reveals an increased rate of mutation especially UV-induced mutations.11 12 Exome sequencing of mCRPC patients, however, shows an overall low rate of mutation.13 Differences in response to CTLA-4 blockade between melanoma and prostate cancer could reflect the higher mutation frequency in melanoma that would result in a larger pool of neoantigens.14
We have previously reported response and outcome rates of a phase II clinical trial combination of ipilimumab and GM-CSF, which suggested that the combination could be more efficacious than ipilimumab monotherapy in patients with unresectable or metastatic melanoma.5 A randomized phase II clinical trial of ipilimumab ±GM-CSF has confirmed improved clinical outcomes for the combination.6 For mCRPC patients, we have found in a phase I trial that patients treated with ipilimumab and GM-CSF can result in clinical responses as well.15
CTLA-4 blockade has been shown to modulate the circulating T-cell repertoire. T cells recognize specific antigens via their T-cell receptors (TCR), which are made up of an α and β chain.16 The diversity of the T-cell repertoire is generated through somatic recombination of the V, D and J segments. Stochastic nucleotide addition and deletions in the junctions further diversifies the highly variable complementary determining region 3 (CDR3).17 In prior work, we have utilized next-generation sequencing of the TCR β chain to show that cancer patients treated with CTLA-4 blockade undergo active remodeling of their T-cell repertoire. Maintenance of high frequency clones and clonotype stability was associated with improved survival.18 Here, we compare the effect that two cancer types have on patient’s circulating T cells when treated with a combination of ipilimumab and GM-CSF.
Materials and methods
Study design
The ipi+GM-CSF mCRPC study is a phase I/II clinical trial where 42 mCRPC patients were treated with anti-CTLA-4 (ipilimumab; Bristol-Myers Squibb) and GM-CSF (sargramostim; Sanofi) as described previously.15 19 Study subjects had histologically confirmed metastatic prostate cancer with disease evident on CT, MRI, and/or bone scans. Subjects had disease progression CRPC as defined by the (prostate specific antigen) PSA Working Group Consensus Criteria.20 Serial cryopreserved peripheral blood mononuclear cells (PBMCs) obtained at baseline and six on-treatment timepoints from 37 patients. Of these patients, 20 patients had fluorescence activated cell sorting (FACS) data and 22 had TCR sequencing data at pretreatment (week 0); 16 patients had FACS data and 35 patients had TCR sequencing data at on-treatment (week 2) (see online supplementary figure 2 and table 1). The ipi+GM-CSF melanoma study is a phase II clinical trial of anti-CTLA-4 antibody (ipilimumab) and GM-CSF in 21 patients with metastatic melanoma and determined clinical outcomes and immunologic responses.5 Study subjects had histologically confirmed unresectable metastatic melanoma and at least one measureable lesion according to Immune-Related Response Criteria.21 GM-CSF was administered at 125 µg/m2 for 14 days beginning on the day of the ipilimumab infusion. Blood samples for determination of immune subsets were obtained before treatment, at week 3 (end of cycle 1) and at week 6 (end of cycle 2); 21 patients had FACS data and TCR sequencing data at pretreatment (week 0). Of these 21 patients, 14 patients also had available FACS data and TCR sequence at on-treatment (week 3) (see online supplementary figure 2 and table 2).
Supplemental material
Supplemental material
Supplemental material
Flow cytometry
PBMCs were thawed into complete media (10% heat-inactivated FBS, 1% non-essential amino acid, 2 mM L-Glu, 0.11 mg/mL sodium pyruvate, 25 mM hepes buffer, 2 g/L NaHC03, 2 g/L glucose) and washed twice with FACS buffer (PBS with 2% heat inactivated fetal bovis serum (FBS) 2 mM EDTA). Cell surface staining was performed in FACS buffer for 30 min at 4°C. Intracellular FoxP3 was performed using the FoxP3 fix/perm buffer set (Biolegend) according to the manufacturer’s protocol. The following antihuman antibodies were used: Alexa Fluor 700-CD3 (clone HIT3a), Brilliant violet 570-CD4 (clone RPA-T4), Brilliant violet 650-CD25 (clone BC96), Alexa Fluor 647-CD127 (clone A019D5), Alexa Fluor 488-FoxP3 (clone 206D), PECy7-CD69 (clone FN50), and Brilliant violet 421-PD-1 (clone EH12.2H7). Stained cells were fixed with Fluorofix buffer (Biolegend) according to manufacturer’s instructions and acquired with an LSRII (BD Biosciences) flow cytometer. Data analysis was performed with Flowjo software (Flowjo, LLC). Gating strategy is shown in online supplementary figure 1. CD8+ T cells were defined as CD4−CD3+; CD4 Teff cells were defined as CD4+CD3+FoxP3−; and Tregs were defined as CD4+CD3+FoxP3+. CD25, CD69, and PD-1 positive cells were gated off CD4 Teff cells, Tregs, CD8 T cells using isotype controls. CD127 positive cells were gated off CD4 Teff cells, Tregs, and CD8 T cells without an isotype control as the positive population was distinct from the negative population. Boolean analysis by Flowjo software was carried out to determine the different combinations of subsets. Percentage of positive cells were calculated back to the same parent lymphocyte gate. Frequency of total CD3+ and progeny subsets (in table 1) were obtained by adding the frequency of each subset from CD4 Teff cells, Tregs, and CD8 T cells. This was carried out in order to calculate correlation with TCR analysis as TCR sequencing was carried out from bulk PBMC. Absolute counts (per person of blood) for each immune subset were calculated by multiplying the percentage of lymphocyte gate with the absolute lymphocyte count quantitated on the day of blood drawn. Relative or ratio of counts was calculated as the counts in the on-treatment immune subset divided by the counts at baseline.
Supplemental material
Summary of change in circulating T-cell populations between mCRPC and metastatic melanoma patients
TCRβ amplification and sequencing, clonotype identification, and counting
The amplification and sequencing of TCRβ repertoire from RNA, read mapping to clonotypes via identification of V and J segments, and counting of the number of unique clonotypes have been previously described in detail.18 22 Of note, after filtering for read quality, reads were mapped to a clonotype if at least two identical reads were found in a given sample. Clonotype frequencies were calculated as the number of sequencing reads for each clonotype divided by the total number of passed reads in each sample.
Statistical methods
In general, frequency distribution and percentages were used to summarize categorical variables, and median with range were used to describe continuous variables with boxplots. Comparison of continuous variables between two groups was performed using Wilcoxon rank sum test. Pearson’s χ2 test was applied to determine statistical association between two categorical variables. The correlation between TCR results (diversity/dynamic indices) and FACS data was assessed by Spearman’s rank correlation coefficient. Statistical significance was declared at p-value <0.05 and no multiple testing adjustment was done. All statistical analysis was done with the statistical computing software R (https://www.r-project.org/).
TCR data assessment
TCR data analysis was performed following the analysis pipeline developed previously by our group.20 To determine the relative change in diversity over time, relative clonality was calculated as the ratio of the clonality at on-treatment versus the baseline clonality, where clonality is formulated as  , with
, with  being the frequency of clonotype i for a sample with n unique clonotypes.23 24 Morisita’s distance, a distance measurement from 0 to 1, maximally dissimilar to minimally dissimilar, respectively, was applied to examine the dynamic change in TCR repertoire from baseline to on-treatment for each subject.18 In addition, each T-cell clone was categorized as “increased” if fold change (FC) is ≥4, as “decreased” if FC is ≤0.25, and as “unchanged” if 0.25 <FC<4, where FC was defined as the clone frequency at on-treatment divided by the frequency at week 0. For each subject, the percentage of TCR sequences falling into each change category was computed. Ratio of TCR convergent frequency was calculated as the ratio of the TCR convergent frequency at on-treatment versus the baseline. TCR convergent frequency was calculated as the aggregate frequency of clonotypes sharing an amino acid sequence with at least one other clonotype. The comparison of dynamic indices between patient groups was done by Wilcoxon rank sum test.
being the frequency of clonotype i for a sample with n unique clonotypes.23 24 Morisita’s distance, a distance measurement from 0 to 1, maximally dissimilar to minimally dissimilar, respectively, was applied to examine the dynamic change in TCR repertoire from baseline to on-treatment for each subject.18 In addition, each T-cell clone was categorized as “increased” if fold change (FC) is ≥4, as “decreased” if FC is ≤0.25, and as “unchanged” if 0.25 <FC<4, where FC was defined as the clone frequency at on-treatment divided by the frequency at week 0. For each subject, the percentage of TCR sequences falling into each change category was computed. Ratio of TCR convergent frequency was calculated as the ratio of the TCR convergent frequency at on-treatment versus the baseline. TCR convergent frequency was calculated as the aggregate frequency of clonotypes sharing an amino acid sequence with at least one other clonotype. The comparison of dynamic indices between patient groups was done by Wilcoxon rank sum test.
Furthermore, for the top 100 clonotypes that were identified based on the clonal abundance at baseline, the standard deviation (SD) of the rank changes from baseline to on-treatment was calculated. The rank change is defined as log10 of the ratio of rank at on-treatment versus rank at pretreatment. The smaller the SD, the more consistent the rank order is across time.
TCR sequencing cannot be directly compared between different patients because individuals do not share exact TCR nucleotide sequencing matches. However, V and J gene usage can be used to compare across different patients. Gene usage is defined as the number of clonotypes that use a particular combination of V and J genes normalized by the total number of unique clones. Random forest method was used to identify the genes whose relative frequencies from baseline to on-treatment were significantly different between melanoma and mCRPC patients.25 Unsupervised hierarchical clustering and heatmaps were used to further illustrate the results.
Results
Treatment induces greater changes in circulating T cells in metastatic melanoma compared with mCRPC
At baseline, the CD4+ and CD8+ T cell counts were not significantly different between melanoma and mCRPC patients (0.68 vs 0.64, p=0.267 and 0.21 vs 0.28, p=0.31, respectively). However, melanoma patients had greater change in the CD3+CD4+, CD3+CD8+, total CD3+CD69+, total CD3+PD1+, and total CD3+CD25 CD69+CD127+ T-cell populations from baseline to on-treatment compared with mCRPC (table 1 and figure 1).

Modulation of different T-cell populations for mCRPC and melanoma patients with treatment. T-cell counts for specific populations were assessed by flow cytometry pretreatment after one cycle of treatmemt. (A) The count of CD4+ T cells. (B) The count of CD8+ T cells. (C) The count of CD25+ T cells. (D) The count of CD69+ T cells. (E) The count of CD127+ T cells. (F) The count of PD1+ T cells. (G) The count of CD25+CD69+CD127+PD1− T cells. (H) The count of CD25−CD69+CD127+PD1+ T cells. (I) The count of CD25−CD69+CD127+PD1− T cells. (J) The count of CD25−CD69−CD127+PD1+ populations. Each figure has two panels representing mCRPC (left) and metastatic melanoma (right). Each panel plots the population count at baseline and on-treatment with connected lines for each patient. The figures with * indicate that there is a significant difference in the ratio of on-treatment count and baseline count of each respective T-cell population between mCRPC and melanoma patients (p<0.05). mCRPC, metastatic castration resistant prostate cancer.
Treatment induces greater change in the T-cell repertoire in melanoma patients than in mCRPC patients
To assess the changes in TCR repertoire induced by treatment, we assessed cryopreserved PBMC by TCR sequencing at baseline and on-treatment. The TCR repertoire of melanoma patients undergoes greater change compared with mCRPC patients (figure 2A). While the proportion of clones present in both pretreatment and on-treatment were similar in metastatic melanoma and mCRPC patients, metastatic melanoma patients have a significantly higher proportion of clones that increased more than fourfold after the treatment compared with mCRPC patients (18.9% vs 11.0%, p=0.017) (figure 2B). For clones present at both pretreatment and on-treatment, the median Morisita’s distance tended to be lower in melanoma patients compared with mCRPC patients (0.59 vs 0.72 p=0.077), which indicates that there is more change within the T-cell repertoire of melanoma patients compared with mCRPC patients (figure 2C). In addition, the ratio of TCR convergent frequency comparing convergence from baseline to post was greater in melanoma patients, suggesting that the TCR repertoire converges more in melanoma patients with treatment (1.22 vs 0.60, p=0.012) (figure 2D).
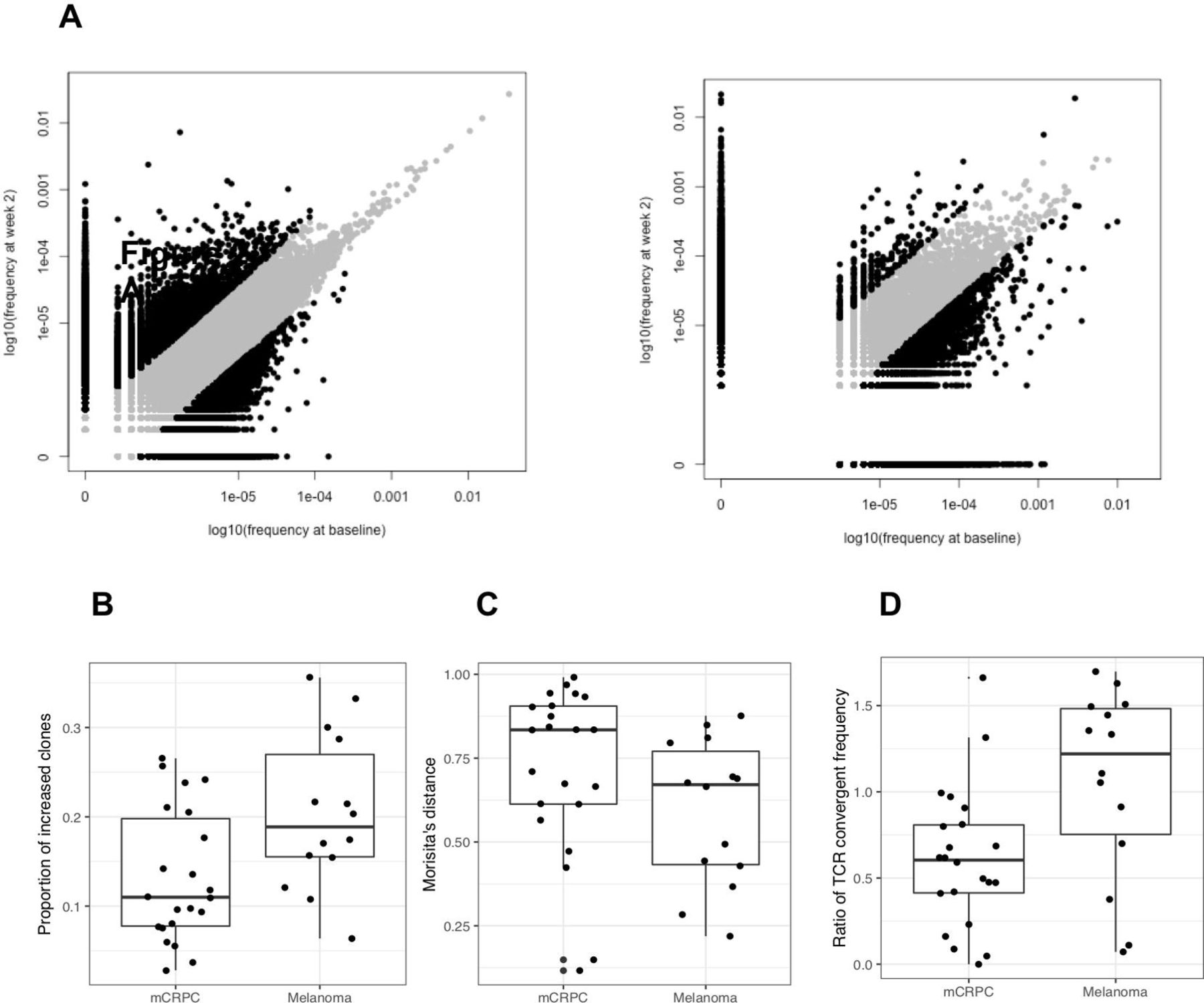
T-cell repertoire in melanoma patients undergo greater change with treatment compared with mCRPC patients. Changes in T-cell repertoire were assessed using NGS-based TCR sequencing. (A) The frequency of unique TCR for mCRPC and melanoma at baseline and with treatment are shown. Left and right scatter plots are from representative mCRPC and metastatic melanoma patients, respectively. Gray dots represent the clonotypes whose absolute log2FC less than 2, where FC is defined as the ratio of on-treatment frequency versus baseline frequency. (B) Comparison of the proportion of T-cell clones that increased in frequency by more than fourfold after the treatment are shown for metastatic melanoma and mCRPC patients (18.9% vs 11.0%, respectively, p=0.017). (C) Morisita’s distance of clones present at both pretreatment and on-treatment is shown for metastatic melanoma and mCRPC patients (0.59 vs 0.72, respectively, p=0.077). (D) Ratio of TCR convergent frequency for metastatic melanoma and mCRPC patients (1.22 vs 0.60, p=0.012). mCRPC, metastatic castration resistant prostate cancer; NGS, next-generation sequencing; TCR, T-cell receptors.
Treatment results in distinct VJ gene usage in melanoma and mCRPC patients
With the TCR sequence, we could classify clonotypes in 877 VJ genes. We could then assess whether VJ gene usage within the T-cell repertoire changes with treatment. Random forest method shows the changes in gene usage of 12 VJ genes from baseline to on-treatment that were significantly changed in the individual melanoma and mCRPC patients (figure 3). With unsupervised hierarchical clustering, the 12 VJ usages largely segregated based on disease. This included a greater decrease in the TCRV20.1 gene usage from baseline to on-treatment in mCRPC patients compared with metastatic melanoma patients. On the other hand, there was a greater decrease in the TCRJ2.6 gene usage from baseline to on-treatment in metastatic melanoma patients compared with mCRPC patients whom showed a greater increase.
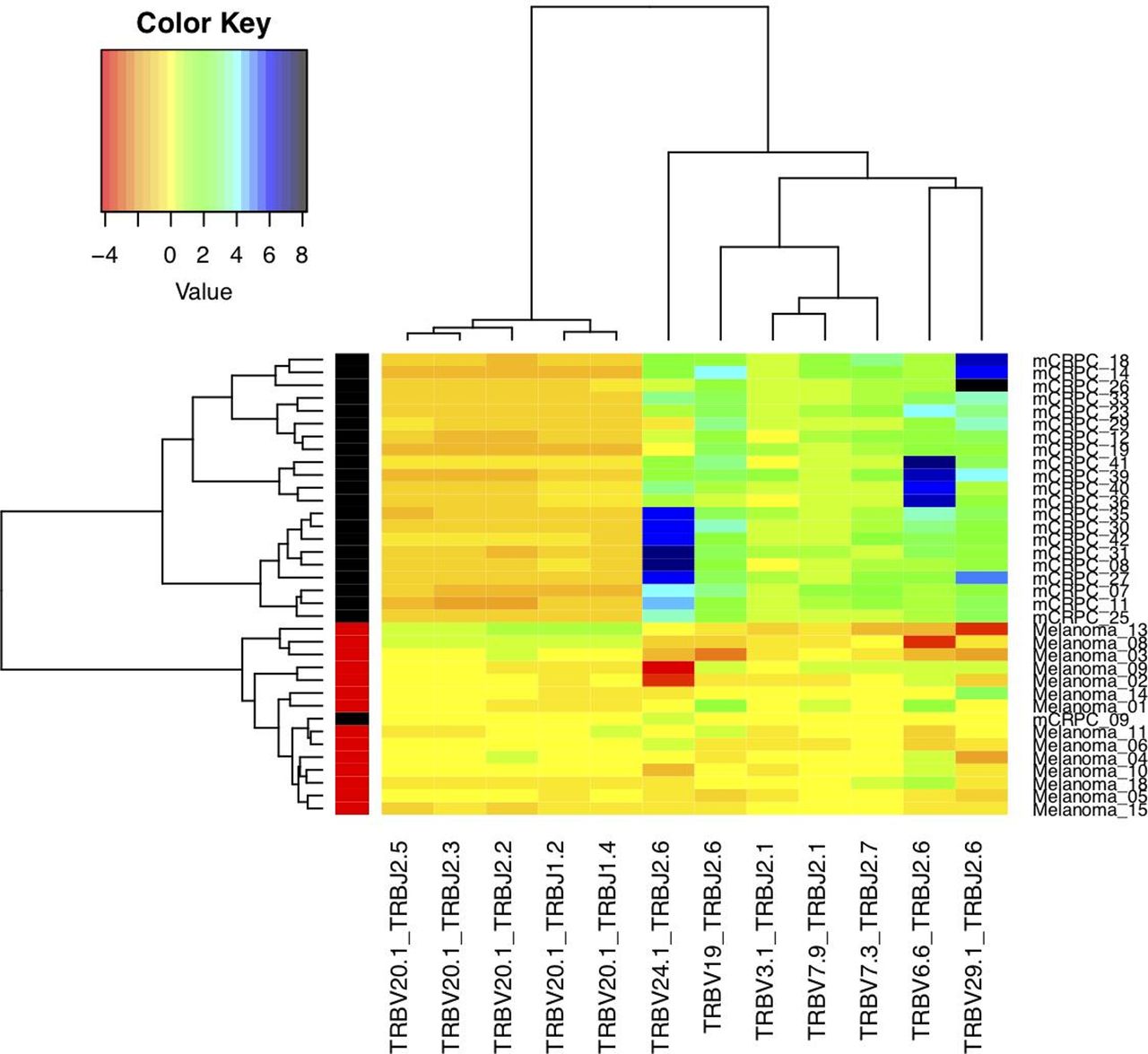
Changes in TCR VJ gene usage induced with treatment in mCRPC and melanoma patients. VJ gene usage was assessed within pretreatment and on-treatment T cells. Each column represents an individual single VJ gene combination, and each row represents an individual patient with red bar and black bar in left side of the heat map representing melanoma and mCRPC patients, respectively. The color code represents the log2(FC), where FC is the ratio of normalized gene usage at on-treatment versus baseline, with red to dark blue representing decreased to increased gene usage from baseline to on-treatment, respectively. mCRPC, metastatic castration resistant prostate cancer; TCR, T-cell receptors.
Treatment results in greater change in the high frequency T-cell clonotypes in melanoma patients than in mCRPC patients
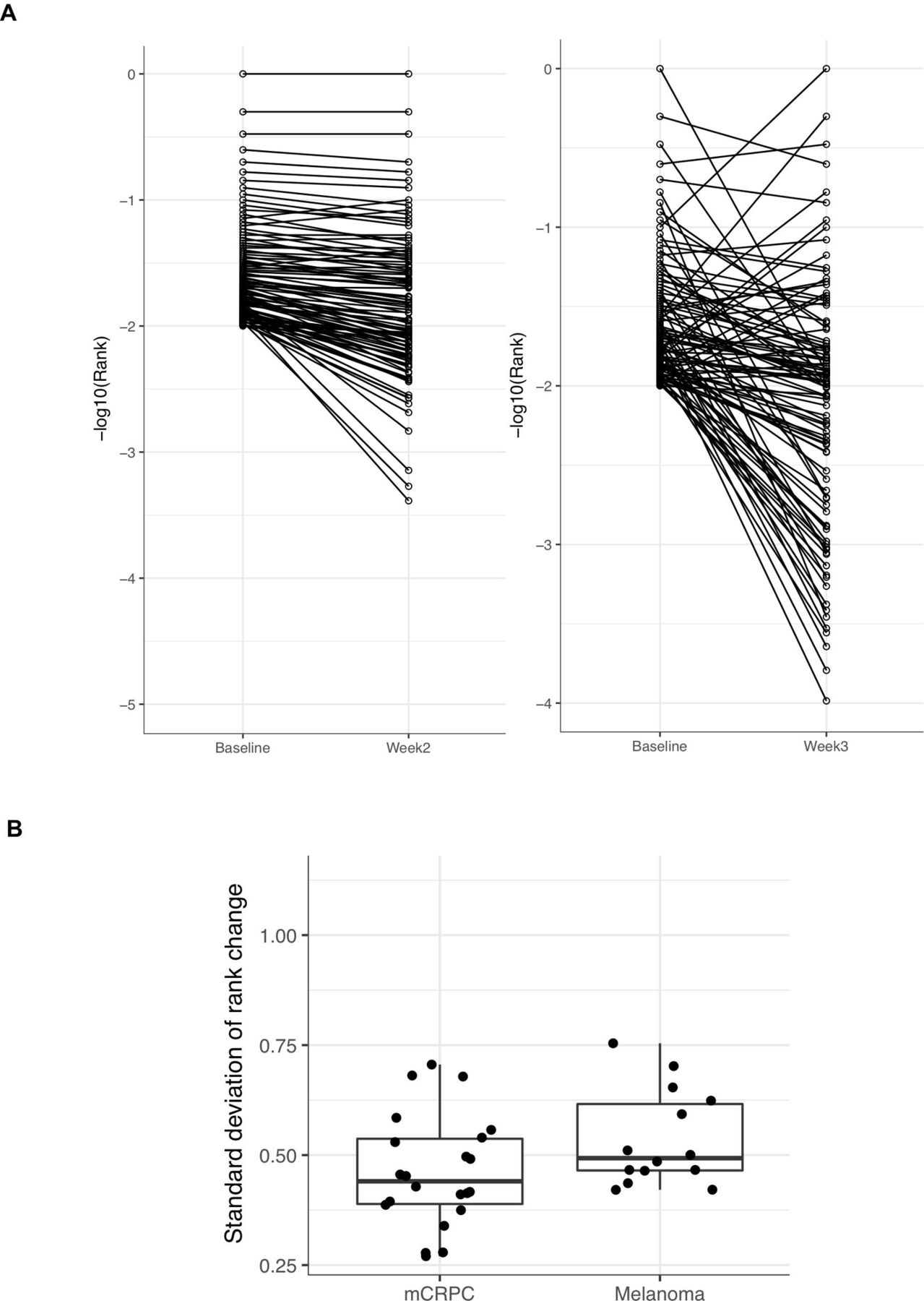
When we focus on the top 100 most frequent clones at baseline for each study, we found that there was considerable change in frequencies of these most abundant clones post-treatment (representative patients shown in figure 4A). The SD of the rank change from baseline to on-treatment was significantly greater in metastatic melanoma patients compared with mCRPC patients (1.05 vs 0.82, p=0.037) (figure 4B). This further shows that despite receiving the same treatment, metastatic melanoma patients undergo greater rearrangement of ranks within high frequency T-cell clonotypes than mCRPC patients.

Greater reshuffling of high frequency T-cell clones in melanoma patients with treatment. (A) The change in rank of matched T-cell clones pretreatment and on-treatment for the top 100 T-cell clones at baseline is shown. (B) The SD of rank change in the top 100 clones in mCRPC and metastatic melanoma are shown. The left and right boxplots represent the SD of rank changes in the top 100 clones at baseline for mCRPC and metastatic melanoma patients, respectively. The SD of rank change is greater in melanoma patients than in mCRPC patients (p=0.045).
Correlation of T-cell frequencies with T-cell repertoire changes
There was a significant positive correlation between the CD8+ T-cell counts and clonality at baseline for both melanoma patients (r=0.81, p=0.0004) and mCRPC patients (r=0.70, p=0.003), indicating that at baseline, the greater the CD8+ T-cell counts, the more clonal the overall TCR repertoire was (figure 5A and B, table 2). Melanoma patients had a stronger and more significant correlation compared with mCRPC patients. There was also a significant negative correlation between the CD8+ T-cell counts at baseline and relative clonality (r=−0.81, p<0.001) for melanoma patients, suggesting that a greater CD8+population at baseline predicted a more diverse TCR repertoire after the treatment. A similar correlation was also observed in mCRPC patients though much weaker and less significant (r=−0.59, p=0.02) (figure 5C and D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Associations between T-cell counts and T-cell repertoire. (A) CD8+ T-cell counts plotted against clonality at baseline for mCRPC patients where each point represents an individual patient and the line represents the linear correlation (r=0.71, p=0.03). (B) CD8+ counts plotted against clonality at baseline for metastatic melanoma patients (r=0.81, p<0.001). (C) CD8+ counts plotted against relative clonality for mCRPC patients (r=−0.59, p=0.022). (D) CD8+ counts plotted against relative clonality for metastatic melanoma patients (r=−0.82, p<0.001). mCRPC, metastatic castration resistant prostate cancer.
Associations between T-cell counts and T-cell repertoire
Finally, the ratio of CD8+ T-cell counts from baseline to on-treatment was positively correlated with the proportion of TCR clones that decreased from baseline to on-treatment (r=0.65, p=0.011) and relative clonality (r=0.55, p=0.041), indicating that the greater the increase is in absolute count of CD8+ population, the more focused the TCR repertoire becomes in metastatic melanoma patients. In contrast, the ratio of CD4+ T cells from baseline to on-treatment was not significantly correlated with the proportion of clones that decreased in frequency (r=0.25, p=0.383) or in relative clonality (r=0.24, p=0.42).
Discussion
CTLA-4 blockade has been shown to result in long-lasting regression in some cancers. The addition of GM-CSF in combination with CTLA-4 blockade has been demonstrated to potentially be more efficacious than anti-CTLA-4 blockade monotherapy in metastatic melanoma.5 Additionally, it has been shown to enhance antitumor immunity in prostate cancer.15 Here, we show that its effect on circulating T cells differs based on the cancer type. Melanoma patients had a greater proportion of clonotypes increasing in frequency and a more convergent repertoire with treatment compared with mCRPC patients. This could reflect the high neoantigen burden seen in melanoma that would presumably help drive the expansion of antigen-specific T cells. Additionally, GM-CSF is a signal of proliferation, maturation, and migration for antigen-presenting cells such as dendritic cells, a potent activator of T cells. The combination of GM-CSF with CTLA-4 blockade might have further augment the observed increases in the T-cell repertoire.
The differential changes in VJ gene combinations from baseline to on-treatment are unexpected. TCR recombination and family usage should be relatively stochastic events. Nevertheless, we could distinguish between mCRPC and melanoma patients based on these changes. This could reflect a distinct antigen milieu, or perhaps other factors including prior treatments that the patients might have received (eg, hormonal therapy with prostate cancer). Among the 12 VJ gene combinations, V20.1 and J2 were the most highly used, which also happen to be the predominant V and J gene segments found in pancreatic cancer and in hepatitis B virus (HBV(-associated hepatocellular carcinoma.26 27 While these gene segments may be associated with clones specific for common infections such as cytomegalovirus (CMV), given that they are differentially affected by treatment, these clones may actually play a larger role in targeting tumor.
While conventional CD4+ T cells are known to have greater repertoire diversity compared with CD8+ T cells, the absolute CD4+ T- cell count at baseline was not correlated with relative clonality. However, for both cancer types, a greater CD8+ T-cell count at baseline was associated with a more focused T-cell repertoire. In addition, the negative correlation between CD8 at baseline and relative clonality suggests that the CD8+ T cells drive the change in the diversity of the T-cell repertoire after treatment. Interestingly, the greater the change in CD8+ T-cell counts, the more focused the TCR repertoire in metastatic melanoma patients, an association not for mCRPC patients. This result further supports the notion that ipilimumab may affect different T-cell subset populations depending on the disease type. Performing TCR sequencing on sorted T-cell populations would help further characterize the effect on specific T-cell populations. In addition, analysis of specific sequences and motifs within the CDR3 region might also reveal tumor specificity.28
One major limitation of this study is that the on-treatment samples were taken at different times, 2 weeks for mCRPC population and 3 weeks for the melanoma population. This 1 week difference may be critical for the T-cell repertoire to undergo the changes seen in the melanoma population. Additionally, later time points were not analyzed in this study due to insufficient samples. Further studies need to be performed at later time points to determine long-term effects on the T-cell repertoire particularly as tumor responses are commonly evaluated at 12, 16, or 24 weeks post-treatment.1
The significant changes that the T-cell repertoire undergo in metastatic melanoma patients compared with mCRPC patients in on-treatment timeframe may parallel the relative effectiveness of CTLA4 blockade in these two diseases: CTLA-4 results in a survival benefit in metastatic melanoma but not in mCRPC patients where we see less of an impact on circulating T cells. As more trials with the same immunotherapies are performed in different diseases, assessment of the T-cell repertoire may further reveal how the immune system responds to different combination therapies in different disease contexts.
References
Footnotes
Contributors JC, LZ, and LF designed and conducted the study including data collection, data analysis. JC prepared the manuscript draft with important intellectual input from LZ, LF, SK, AP, and DYO. All authors approved the final manuscript. JC, LZ, and LF had complete access to the study data. LZ, AP, TH, and GF provided statistical support in analyzing the data.
Funding SK is supported by the Peter Michael Foundation. DYO is supported by NIH 4T32 CA177555, 1K08 AI139375, the Harry F. Bisel, MD Endowed Young Investigator Award from the Conquer Cancer Foundation of the American Society of Clinical Oncology, the Bladder Cancer Advocacy Network Palm Beach New Discoveries Young Investigator Award, and the Prostate Cancer Foundation Young Investigator Award. LF is supported by NIH R01CA223484, U01CA233100, and the Prostate Cancer Foundation. LZ receives the support of the UCSF Academic Senate Committee on Research. TH receives the support of San Francisco State University Development of Research and Creativity Grant and a Presidential Award.
Competing interests LF has received research funding from Oncosec, Abbvie, Bavarian Nordic, BMS, Janssen, Merck and Roche/Genentech.
Patient consent for publication Not required.
Ethics approval This study was approved by the UCSF internal review board (IRB approval #10-02217 CC#02558 ZZA Phase I Study of Repetitive Dosing of anti-CTLA-4 Antibody (MDX-010) in Combination with GM-CSF and IRB approval #15-16385 CC#03015 GM-CSF and Ipilimumab as Second-line Therapy in Metastatic Melanoma, a Phase II Study). All human subjects gave written informed consent to participate in the protocol.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.