Article Text

Abstract
Background Preclinical evaluation of drugs targeting the human immune system has posed challenges for oncology researchers. Since the commercial introduction of humanized mice, antitumor efficacy and pharmacodynamic studies can now be performed with human cancer cells within mice bearing components of a human immune system. However, development and characterization of these models is necessary to understand which model may be best suited for different agents.
Methods We characterized A375, A549, Caki-1, H1299, H1975, HCC827, HCT116, KU-19–19, MDA-MB-231, and RKO human cancer cell xenografts in CD34+ humanized non-obese diabetic-scid gamma mice for tumor growth rate, immune cell profiling, programmed death ligand 1 (PD-L1) expression and response to anti-PD-L1 therapy. Immune cell profiling was performed using flow cytometry and immunohistochemistry. Antitumor response of humanized xenograft models to PD-L1 therapy was performed using atezolizumab.
Results We found that CD4+ and CD8+ T-cell composition in both the spleen and tumor varied among models, with A375, Caki-1, MDA-MB-231, and HCC827 containing higher intratumoral frequencies of CD4+ and CD8+ T cells of CD45+ cells compared with other models. We demonstrate that levels of immune cell infiltrate within each model are strongly influenced by the tumor and not the stem cell donor. Many of the tumor models showed an abundance of myeloid cells, B cells and dendritic cells. RKO and MDA-MB-231 tumors contained the highest expression of PD-L1+ tumor cells. The antitumor response of the models to atezolizumab was positively associated with the level of CD4+ and CD8+ tumor-infiltrating lymphocytes (TILs).
Conclusions These data demonstrate that there are tumor-intrinsic factors that influence the immune cell repertoire within tumors and spleen, and that TIL frequencies are a key factor in determining response to anti-PD-L1 in tumor xenografts in humanized mice. These data may also aid in the selection of tumor models to test antitumor activity of novel immuno-oncology or tumor-directed agents.
- immunology
- oncology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Immunotherapy is a promising therapeutic intervention for cancer treatment. Activation of the immune system via checkpoint blockade, through the use of antibodies against programmed death receptor 1 (PD-1) and programmed death ligand 1 (PD-L1), and cytotoxic T-lymphocyte-associated protein 4, has been shown to produce antitumor responses in patients with a range of cancer types.1 2 However, many patients do not respond to checkpoint blockade, and additional therapies are needed to treat these patients. The use of in vivo preclinical models is critical for establishing activity of drug candidates prior to entering the clinic. For many therapeutic targets, the use of syngeneic models can be appropriate, assuming that the therapeutic agent functionally modulates mouse isoforms of the target. However, many times, the therapeutic agent under evaluation only recognizes human isoforms or the expression level of targets of interest are different between human and mouse immune cell subsets. Therefore, there is a need for well-characterized humanized tumor models to inform rational model selection.
Significant advancement in the development of humanized mouse models has been made.3–5 One type of humanized mouse is generated through the use of CD34+ stem cells from cord blood donors. In this model, lymphocytes and dendritic cells have been shown to be at least partially functional; however, in general, lesser functionality has been observed for natural killer (NK), B and myeloid cells in CD34+ humanized mice.3 6 Despite this, the use of antibodies against PD-1 has shown efficacy in human xenograft and patient-derived xenograft tumors in these mice, demonstrating that these cells can generate antitumor responses.7–10 Whereas checkpoint modulators have shown antitumor activity in humanized mice, there are limited data reporting characterization on human immune cell subsets within these tumors. To this end, we characterized several commonly used xenograft models across a range of tumor types in CD34+ humanized non-obese diabetic-scid gamma (NSG) mice. We show that these models represented both ‘hot’ and ‘cold’ tumors with high and low levels of intratumoral infiltrates, respectively. We also demonstrate that tumor-intrinsic factors play a predominant role in determining immune cell residency in tumors and spleen.
Materials and methods
Cell lines and materials
Cell lines were obtained from the American Type Culture Collection. Growth media supplemented with 10% fetal bovine serum to maintain cells was used for the following: Dulbecco's Modified Eagle's medium (A375, MDA-MB-231), RPMI (H1299, H1975, HCC827, HCT116, and KU-19–19), F-12K (A549), McCoy’s 5A (Caki-1), and EMEM (RKO). Human IgG1 antibody (#BE0297; BioXCell, West Lebanon, New Hampshire, USA) and atezolizumab (#50242-0917-01; Myoderm, Norristown, Pennsylvania, USA) were formulated at 5 mg/kg in sterile injectable saline. Cell line human leuokocyte antigen (HLA) typing data were obtained from the TRON Cell Line Portal.11
Immunohistochemistry (IHC)
IHC was performed by Indivumed (Hamburg, Germany). See online supplementary file 1 for additional methods.
Supplemental material
In vivo studies
Animals were housed in a barrier facility fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. For the assessment of tumor growth and take rate, the following cell numbers were used: 3×106 cells (H1975 and MDA-MB-231), 5×106 cells (A375, H1299, and KU-19–19), and 1×107 cells (A549, Caki-1, HCC827, HCT116, and RKO). All cell inoculations included Matrigel (#354248; Corning, New York, USA) in a 1:1 vol ratio in phosphate-buffered saline. Cells were implanted subcutaneously into the right-hind flank of female human CD34+ reconstituted NSG mice 20–24 weeks old (The Jackson Laboratory, Bar Harbor, Maine, USA). When tumor volumes averaged ~150 mm3, mice were randomized by both tumor volume and donor (2–5 donors per group) into groups of 10 and were treated with either human IgG1 antibody or atezolizumab. The control arm for the HCC827 study was 5% Dimethylacetamide (DMAC) in 0.5% (w/v) methylcellulose (# MC430; Sigma, St. Louis, Missouri, USA), which was dosed once daily. Antibodies were dosed every 5 days intraperitoneally at 5 mg/kg. Tumor growth inhibition (TGI) was calculated using the formula (1−(VT/VC))×100, where VT is the tumor volume of the treatment group on the last day of treatment and VC is the tumor volume of the control group on the last day of treatment. CD45+ engraftment and HLA typing data were provided by the Jackson Laboratory (Sacramento, California, USA). Statistical analysis was performed using two-way analysis of variance.
Immunophenotyping
Tumors and spleens were harvested from mice when tumor volumes averaged 150–525 mm3, with the exception from one mouse in MDA-MB-231 study 1, which had a tumor volume of ~900 mm3. Tumor and spleens were dissociated according to the manufacturer's protocol (#130-096-730; Miltenyi Biotec, Bergisch Gladbach, Germany). Red blood cells were lysed in Pharm Lyse (#555899; BD, Franklin Lakes, New Jersey, USA). Samples were transferred into 96-well plates for staining. Samples were incubated for 5 min with Fc block (#564220, #553142; BD) at room temperature. The following antibodies were added to the samples for 30 min at 4°C: CD25 (#555434, BD), human CD45 (#563879, BD), mouse CD45 (#559864, BD), CD279 (#563245, BD), CD274 (#557924, BD), CD3 (#654307, BD), CD8 (#557834, BD), CD4 (#557852, BD), NKp46 (#557991, BD), HLA-DR (#555811, BD), CD11b (#50-165-703; BioLegend, San Diego, California, USA), CD15 (#563872, BD), and CD14 (#5601; ThermoFisher, Waltham, Massachusetts, USA). Cells were washed and resuspended in stain buffer (#554657, BD) for acquisition. Data were acquired on the CytoFLEX (Beckman Coulter, Brea, California, USA) and analyzed with FlowJo software (FlowJo, LLC, Ashland, Oregon, USA). Percentages reflected live CD45+ single cells. Cell populations were gated as follows: regulatory T cells: CD4+CD25+, NK cells: CD3-Nkp46+, myeloid cells: CD11b+, B cells/dendritic cells: CD3−CD14−HLA DR+, monocytes/macrophages: CD11b+CD33+CD14+ CD15−HLA DR+, monocytic myeloid-derived suppressor cells (M-MDSCs): CD11b+CD33+CD14+CD15−HLA DR−, and granulocytic myeloid-derived suppressor cells (G-MDSCs): CD11b+CD33+CD14−CD15+. Cells that were CD45+ but not positive for CD3, CD4, CD8, NKp46, CD11b or HLA-DR are labeled as ‘other’. Statistical significance was determined via Student’s t-test.
Results
Our initial study evaluated the tumor growth and take rate of 10 xenograft models in CD34+ humanized mice. Cell lines commonly used as tumor models in immunodeficient mice were implanted in humanized mice and monitored for tumor growth (figure 1A). All models (H1975, H1299, A375, A549, HCT116, RKO, KU-19–19, Caki-1, HCC827 and MDA-MB-231) grew and had a 100% take rate. Compared with the rest of the models examined, however, HCC827 did not grow to a large size. Tumor growth data in immunodeficient mice were obtained from 8 of the 10 models. Compared with growth in humanized mice, five out of eight tumor models in immunodeficient mice had similar growth kinetics, and the other three models (H1299, A549 and MDA-MB-231) grew about twice as slow in immunodeficient mice (data not shown), suggesting that, as Wang et al have shown, humanization is not an impediment to tumor growth.7 Immunophenotyping was performed to determine the frequencies of lymphocyte, myeloid and NK cell populations within the tumor. The models were found to contain a wide range of tumor-infiltrating CD45+ cells, ranging from <2% to >20% of viable cells within the tumor, with MDA-MB-231, HCC827, H1299, and Caki-1 containing the highest frequencies of CD45+ cells (figure 1B). No correlation was observed between the extent of HLA matching between the tumor and stem cell donor and the frequency of CD45+ cells within the tumors (online supplementary additional file 2, table S1). In addition, no correlation was observed between the hCD45+ initial engraftment percentage in the periphery and the percentage of human CD45+ cells within tumors (online supplementary table S1). Within the CD45+ cell subset, MDA-MB-231, Caki-1, HCC827, and A375 had the highest frequencies of CD4+ and CD8+ T cells (figure 1C). Surprisingly, all tumors had a high level of B-cell and dendritic cell infiltrates (figure 1C). The percentage of regulatory T cells as a percentage of human CD45+ cells ranged between 0.39% and 5.0%, while the percentage of NK cells ranged between 1% and 5% among the models. The frequencies of myeloid-derived suppressor cell (MDSCs) (M-MDSCs and G-MDSCs) were extremely low, with some models containing no MDSCs to some containing up to 1%–2% (online supplementary figure S1). In addition, very few monocytes/macrophages were found in tumors, with the exception of the MDA-MB-231 model, which was found to contain the highest frequency of macrophages among the models (online supplementary figure S1). In the spleen, it was observed that the relative levels of each of the immune cell populations within each model were similar to those found in the tumors (figure 1D).
Supplemental material

Tumor growth and immunophenotypical characterization of xenograft models in humanized mice. (A) Mean tumor volume of xenografts inoculated in CD34+ humanized mice. n=10 mice per group. Each group contained mice engrafted with two to four donors. X-axis indicates the day of measurement after tumor inoculation. Error bars reflect the SEM. (B) Tumors engrafted in CD34+ humanized mice (n=3) were dissociated and samples were stained for human CD45 according to the Materials and methods section. Percentages are of total live cells. Error bars reflect the SD. (C) Tumors and (D) spleens from mice (n=3) were dissociated and samples were stained according to the Materials and methods section. Percentages reflect percent of CD45+ cells. For MDA-MB-231 tumors, n=12 mice were averaged. NK, natural killer; Treg, regulatory T cell.
To determine whether the variation in tumor-infiltrating lymphocyte (TIL) populations was influenced by the tumor or the stem cell donor, frequencies of CD3+, CD4+, and CD8+ T cells within the CD45+ cell population were determined with respect to donor. Figure 2A shows the frequency of CD3+, CD4+, and CD8+ T cells in three different experiments in the MDA-MB-231 model. These data show that the frequencies of these populations were based on the tumor, as the percentages of each cell population were similar within each study despite mice having different stem cell donors. A similar finding was also observed with respect to the %CD3 (figure 2B), %CD4 (figure 2C), and %CD8 (figure 2D) among the other nine tumor models. We also assessed whether there was a relationship between the number of mutations per tumor and the percent of tumor-infiltrating CD8+ T cells. This analysis revealed that there was no such relationship observed in these models (online supplementary figure S2). The relative levels of CD4+ and CD8+ T cells were also assessed via IHC. MDA-MB-231, Caki-1, and HCC827 were found to have the highest infiltration of CD4+ and CD8+ T cells (figure 3A,B), and were largely in agreement with the TIL frequencies as measured by flow cytometry (figure 1C).

Frequencies of TILs are based on tumor and not donor. (A) Frequency of CD3+, CD4+ and CD8+ TILs in three different experiments in the MDA-MB-231 model. (B) %CD3+, (C) %CD4+ and (D) %CD8+ TILs from n=3 mice in the A375, A549, Caki-1, H1299, H1975, HCC827, HCT116, KU-19–19 and RKO models. MDA-MB-231 data are from three separate studies with n=4 per study. Percentages are among the CD45+ population. Tumors engrafted in CD34+ humanized mice were dissociated and samples were stained according to the Materials and methods section. The donor is represented by color as well as shape on the plots. TIL, tumor-infiltrating lymphocyte.

Immunohistochemistry of intratumoral CD4+ and CD8+ T cells. (A) Tumors (n=3 per model) were fixed in 10% formalin followed by transfer to 100% ethanol. Three tissue sections per tumor were stained with antihuman CD4 (clone SP35) and antihuman CD8 antibody (clone SP16) (Indivumed, Hamburg, Germany). Representative images are shown. Scale bar=200 µm. (B) Digital quantification of CD4+ and CD8+ T cells was performed according to the Materials and methods section. The number of positive cells per square millimeter is shown.
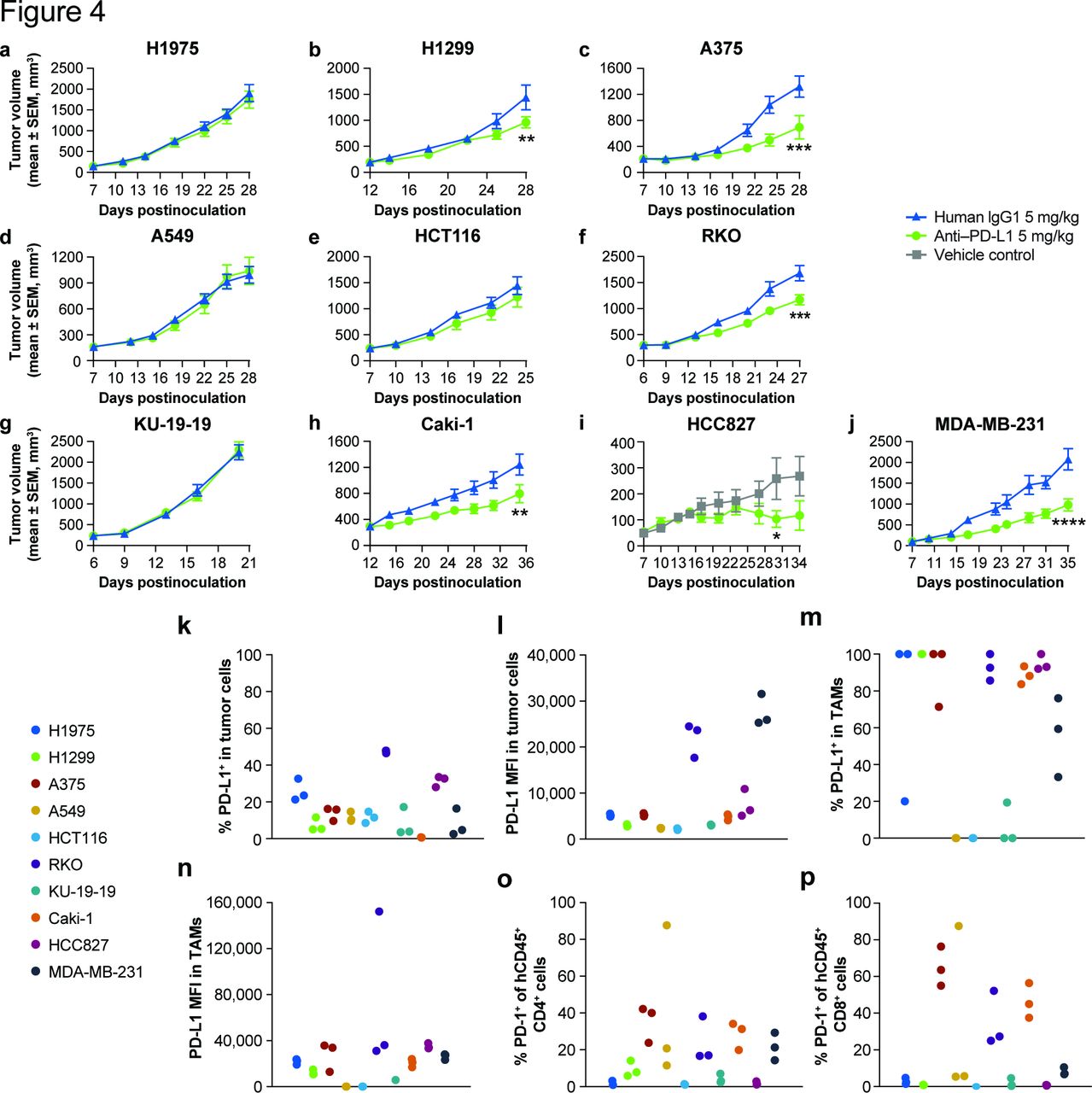
To explore the sensitivity of each tumor model to checkpoint blockade, efficacy studies were performed with the anti-human PD-L1 antibody, atezolizumab (figure 4). Statistically significant TGI was observed in H1299 (figure 4B), A375 (figure 4C), RKO (figure 4F), Caki-1 (figure 4H), HCC827 (figure 4I), and MDA-MB-231 (figure 4J) tumor models, and no growth inhibition was observed in the H1975 (figure 4A), A549 (figure 4D), HCT116 (figure 4E), or KU-19–19 (figure 4G) tumor models. The TGI values are reported in table 1.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Antitumor efficacy of atezolizumab in humanized tumor models and characterization of PD-L1 and PD-1 expression. Antitumor efficacy of atezolizumab in (A) H1975, (B) H1299, (C) A375, (D) A549, (E) HCT116, (F) RKO, (G) KU-19–19, (H) Caki-1, (I) HCC827 and (J) MDA-MB-231. Mice were randomized by both tumor volume and donor into groups of 10 (two to four donors per group). (K) Frequency of PD-L1 on tumor cells; (L) mean fluorescence intensity (MFI) of PD-L1 on tumor cells; (M) frequency of PD-L1 on tumor-associated macrophages (TAMs); (N) MFI of PD-L1 on tumor-associated macrophages; (O) frequency of PD-1 within CD4+ cells; (P) frequency of PD-1 on CD8+ cells. Tumors engrafted in CD34+ humanized mice were dissociated and samples were stained according to the Materials and methods section. Tumor cells were gated from live, human CD45− and mouse CD45− cells. Statistical analysis was performed using two-way analysis of variance. *P<0.05, **P<0.01, ***P<0.001, ****P=0.0001. PD-1, programmed death receptor 1; PD-L1, programmed death ligand 1.
TGI of humanized models to atezolizumab
Antitumor responses were not well correlated with HLA matching of tumor and stem cell donors. In the A375 model, both responders and non-responders included tumor/stem cell donors that were only partially HLA matched (online supplementary figure S3a,b). In the MDA-MB-231 model, however, antitumor responses were observed with complete tumor/stem cell donor mismatches (online supplementary figure S3c,d). Therefore, although an antigen-mediated T-cell response cannot be ruled out in certain models, our data demonstrate that HLA matching is not required for antitumor responses.
As PD-L1 expression, at least in some tumor types, has been linked to response to checkpoint blockade,12 13 PD-L1 expression on both tumor cells as well as macrophages was determined from untreated tumor-bearing animals. All tumor models expressed PD-L1, with H1975, RKO, and HCC827 tumors containing the highest frequency of tumor cells with PD-L1 (figure 4K), with RKO and MDA-MB-231 containing the highest level of PD-L1 expression based on mean fluorescence intensity (MFI) (figure 4L). Most Ttumor-associated macrophages expressed PD-L1 among the tumor models, except A549 and HCT116 (figure 4M,N), which did not have appreciable levels of TAMs (online supplementary file 2). We also evaluated the level of PD-1 expression on intratumoral CD4+ and CD8+ T cells. In CD4+ T cells, PD-1 expression was variable, and most CD4+ T cells expressed PD-1; however, low levels were observed in the H1975, HCT116, KU-19–19, and HCC827 models (figure 4O). In contrast, expression of PD-1 on CD8+ T cells was high in a select number of models, including A375, RKO, and Caki-1 models (figure 4P). Treatment of tumor-bearing mice with atezolizumab increased the percentage of CD4 and CD8+ T cells expressing PD-1 in the MDA-MB-231 model, demonstrating T-cell activation (online supplementary figure S4a). Atezolizumab treatment also increased the percentage of CD3+, CD4+, and CD8+ T cells within tumors (online supplementary figure S4b). These data demonstrate that a subset of diverse xenograft models examined responded to anti–PD-L1 and that models represented both ‘hot’ and ‘cold’ tumors based on the level of TILs.
Discussion
Humanized models represent a powerful tool to assess preclinical activity of immuno-oncology (IO) agents. In this report, we characterized several commonly used xenograft models spanning a range of tumor types for immunophenotypic profile, expression of PD-L1 and PD-1 on tumor and immune cell subsets, and response to atezolizumab. We observed that a subset of models responded to atezolizumab and that the overall level of TILs was associated with response. The relative frequencies of CD4+ and CD8+ T cells within the CD45+ population were a factor in determining response to atezolizumab. In one example, A375 had a low overall frequency of intratumoral CD45+ cells; however, most of these cells were CD3+ T cells. The robust response of this model to atezolizumab is likely therefore due to the high frequencies of CD4+ and CD8+ TILs, despite the low overall frequency of CD45+ cells. The low frequency of CD45+ cells in this model also explains why relatively little IHC staining of CD4+ and CD8+ cells were observed in this model, whereas high levels of CD4+ and CD8+ within the CD45+ cell population by immunophenotyping were observed. Our preclinical finding that the efficacy of atezolizumab therapy was correlated to TIL levels is consistent with reports that associate TIL frequency (eg, tumors with an inflamed phenotype) with patient response to PD-1/PD-L1 antibody therapy.13–15
It was observed that that the RKO model responded to atezolizumab, despite relatively lower levels of intratumoral CD4+ and CD8+ T cells. One reason may be due to the presence of high levels of PD-L1 expression in this model. Although all tumor models expressed PD-L1, high tumor PD-L1 expression was not distinctly associated for response to anti-PD-L1 as high PD-L1 expression was only observed in the RKO and MDA-MB-231 models. Also, our data demonstrated that HLA matching between tumor and stem-cell donor is not required for an antitumor response, although an allogeneic mechanism may not be the exclusive mechanism of antitumor response to PD-L1 blockade in all models. This finding is similar to the recent report by Wang et al, which did not observe a pattern between HLA matching and response to PD-1 therapy.7 Further studies to elucidate the precise mechanisms for anti-PD-L1 activity in humanized mice are needed.
Interestingly, our data demonstrated a moderately high frequency of myeloid cells within many of the tumor models, which is consistent with a recent report.8 In addition, we found a high level of B cells and dendritic cells, particularly in the spleen. The splenic B cells are likely to be B-cell progenitors; however, as it is known that B-cell development is partially impaired in CD34+ humanized mice.16 The levels of intratumoral macrophages and MDSCs were very low in the models, which is not surprising since CD34+ NSG mice are not efficient at supporting myeloid cell engraftment. Other humanized tumor models that better support myeloid cell engraftment may be a better experimental setting to test preclinical agents directed towards drug targets residing on these cell populations.17 18
Unexpectedly, we discovered that the immune cell profile was dependent on the tumor model, not the host or donor. When analyzing levels of TILs, similar levels were observed within each tumor model despite mice being engrafted with different stem cell donors. Therefore, there are tumor-intrinsic factors in these models that determine the immune cell repertoire in the tumor microenvironment. It is known that oncogenic signaling, as well as loss of tumor suppressor genes, influences T-cell exclusion and drives immunosuppressive phenotypes.19 20 For example, when the tyrosine kinase AXL was ablated using CRISPR in radio-resistant transgenic mouse mammary tumors, greater infiltrating CD8+ T cells were observed, which increased further after radiation.21 In addition, activation of the β-catenin pathway, along with the presence of mutated FGFR3, was among the most frequent alterations found in non-T-cell-inflamed bladder cancer.22 These data suggest that targeting oncogenic pathways that drive T-cell recruitment or support immunosuppressive mechanisms may be of therapeutic benefit. Therefore, evaluation of not only novel IO agents but also tumor-directed agents in these humanized models may induce stronger antitumor responses in combination with checkpoint blockade. We would suggest that humanized models be strongly considered for all preclinical antitumor studies since these will capture both tumor-directed and immunomodulatory drug activity. In summary, this characterization may provide guidance for rational model selection to evaluate novel drug candidates.
Conclusions
In this study, we demonstrated successful growth of 10 xenograft models in humanized mice, and generated data aimed to increase understanding of immune cell composition in the tumor microenvironment in these mice. The immune cell composition varied between the models, yet several models stood out as phenotypically representative of inflamed tumor models (MDA-MB-231, Caki-1, and HCC-827). Our finding that response to anti–PD-L1 is correlated with the level of TILs is consistent with patient clinical responses. Finally, our data strongly indicate that the immune cell repertoire is driven by the tumor, and not the stem cell donor, which provides support for rational combinations of agents that target oncogenic signaling pathways with IO agents.
Acknowledgments
We thank Sunkyu Kim for critical reading of the manuscript, Philip Czerniak and Laboratory Animal Resources at Incyte for operational support, and Peggy Scherle for intellectual contributions when initiating these studies.
References
Footnotes
Contributors JR-D and HKK contributed to the conception and design of the study. CS, KL and CM collected the data. JR-D and CS analyzed the data. JR-D drafted the manuscript. All authors revised the manuscript and approved the final version of the manuscript for submission.
Funding All studies were funded by Incyte.
Competing interests All authors are full-time employees and shareholders of Incyte.
Patient consent for publication Not required.
Ethics approval All animal studies were conducted with prior approval from the Incyte Animal Care and Use Committee and in a facility accredited by the American Association for Accreditation of Laboratory Animal Care. All procedures were conducted in accordance with the US Public Service Policy on Human Care and Use of Laboratory Animals and with Incyte Animal Care and Use Committee Guidelines.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information. All datasets supporting the conclusions of this article are included in this published article and its additional files.