Article Text

Abstract
Immunosuppressive entities in the tumor microenvironment (TME) remain a major impediment to immunotherapeutic approaches for a majority of patients with cancer. While the immunosuppressive role of transforming growth factor-β (TGF-β) in the TME is well known, clinical studies to date with anti-TGF-β agents have led to limited success. The bifunctional agent bintrafusp alfa (previously designated M7824) has been developed in an attempt to address this issue. Bintrafusp alfa consists of an IgG1 targeting programmed death ligand 1 (PD-L1) moiety fused via peptide linkers to the extracellular domain of two TGF-β receptor II molecules designed to ‘trap’ TGF-β in the TME. This agent is able to bring the TGF-β trap to the TME via its anti-PD-L1 component, thus simultaneously attacking both the immunosuppressive PD-L1 and TGF-β entities. A number of preclinical studies have shown bintrafusp alfa capable of (1) preventing or reverting TGF-β-induced epithelial-mesenchymal transition in human carcinoma cells; this alteration in tumor cell plasticity was shown to render human tumor cells more susceptible to immune-mediated attack as well as to several chemotherapeutic agents; (2) altering the phenotype of natural killer and T cells, thus enhancing their cytolytic ability against tumor cells; (3) mediating enhanced lysis of human tumor cells via the antibody-dependent cell-mediated cytotoxicity mechanism; (4) reducing the suppressive activity of Treg cells; (5) mediating antitumor activity in numerous preclinical models and (6) enhancing antitumor activity in combination with radiation, chemotherapy and several other immunotherapeutic agents. A phase I clinical trial demonstrated a safety profile similar to other programmed cell death protein 1 (PD-1)/PD-L1 checkpoint inhibitors, with objective and durable clinical responses. We summarize here preclinical and emerging clinical data in the use of this bispecific and potentially multifunctional agent.
- TGF-β
- checkpoint inhibition
- immunotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Immuno-oncology (IO) agents have now joined surgery, radiation, chemotherapy and small molecule targeted therapies as an integral component in the management of a range of human cancers. These IO agents include various monoclonal antibodies (MAbs) directed against checkpoint inhibitors such as the programmed cell death protein 1 (PD-1), its ligand 1 (PD-L1) and the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4).1–3 While the use of these checkpoint inhibitor MAbs as monotherapies has led to great strides in the management of diseases such as melanoma, lung, renal and bladder cancer, durable responses have been seen in <20% of treated patients with most solid malignancies.4 One possible reason for this is that checkpoint inhibitors attack only one aspect of a complex system composed of the tumor and its surrounding tumor microenvironment (TME). In addition to the PD-1/PD-L1 axis and the CTLA-4 ligand interactions, there are numerous other immunosuppressive entities in the TME such as regulatory T cells (Tregs), myeloid derived suppressor cells (MDSCs) and cytokines such as interleukin-8 (IL-8) and transforming growth factor-β (TGF-β) that could result in suboptimal antitumor immune responses.
TGF-β is a well-studied pleiotropic cytokine that has a dual role in cancer progression. While it has multiple normal cellular functions, including effects on the cell cycle, motility, angiogenesis and suppression of the initiation of some tumor types, TGF-β can also serve as a driver of tumor progression by suppressing the host antitumor immune response and by inducing tumor cell plasticity, a process rendering epithelial tumors more mesenchymal, stem-like and resistant to immune and other therapies. Excellent review articles have been published on the various functions of TGF-β in both animal models and clinical studies.5–10 Prior and ongoing agents targeting TGF-β in clinical studies involve the use of both small molecule inhibitors of the serine/threonine kinase domain of the TGF-β receptor I (TGF-βRI) and anti-TGF-β neutralizing MAbs. These agents have seen limited success in the clinical setting, indicating the potential need for a tumor-targeted approach to inhibit TGF-β in the TME. Consequently, a novel anti-PD-L1/TGF-β receptor II (RII) fusion protein designated bintrafusp alfa (previously M7824) was designed to simultaneously inhibit PD-1/PD-L1-mediated immunosuppression while decreasing the levels of TGF-β in the TME via a TGF-β ‘trap’ portion. This article reviews preclinical and clinical studies of bintrafusp alfa.
Background on bintrafusp alfa
Avelumab is an IgG1 checkpoint inhibitor that is approved for the treatment of metastatic Merkel cell carcinoma,11 locally advanced or metastatic urothelial carcinoma12 and renal cell carcinoma in combination with axitinib.13 Bintrafusp alfa is a bifunctional agent which also consists of an IgG1 anti-PD-L1 moiety, fused via flexible peptide linker sequences to the extracellular domain of two TGF-βRII molecules (figure 1A). Bintrafusp alfa was developed as part of a Collaborative Research and Development Agreement between the National Cancer Institute (NCI) and EMD Serono. The TGF-βRII moiety of bintrafusp alfa has been shown to bind three TGF-β isoforms (TGF-β1, TGF-β2, TGF-β3) in solution and two isoforms (TGF-β1, TGF-β3) on plate-bound enzyme linked immunosorbent assays.14 Bintrafusp alfa has also been found to bind to murine TGF-β isoforms and murine PD-L1, which has enabled preclinical investigation in syngeneic murine models.15

Bintrafusp alfa (designated in previous publications as M7824) prevents and reverts TGF-β-induced mesenchymal features in lung cancer models in vitro and in vivo. (A) Schema of the structure of M7824, a bifunctional protein consisting of a human IgG1 anti-PD-L1 antibody fused via linkers to two TGF-β receptor II molecules. (B) M7824 blocks and reverts TGF-β1-induced mesenchymal features in A549 cells in vitro. Blockade: A549 cells were treated with TGF-β1 (2 ng/mL)±anti-PD-L1 or M7824 (200 ng/mL) for 72 hours to prevent EMT induction. Reversion: A549 cells were pretreated with TGF-β1 (2 ng/mL) for 72 hours to induce EMT, followed by 72 hours of treatment with TGF-β1±anti-PD-L1 or M7824 (200 ng/mL) to revert established EMT. Indicated EMT markers and loading control GAPDH were visualized by immunoblot. (C) HCC4006 lung cancer cells (4×106 cells) were implanted s.c. in C.B-17 SCID mice (day −6). tumor-bearing mice were treated with i.p. injections of vehicle (HBSS), anti-PD-L1 (400 µg), or M7824 (492 µg; days 0, 2, 4, 6, 8, 10, 14). tumors were harvested, fixed and paraffin embedded (day 15), and stained via IHC for mesenchymal vimentin (shown is a representative tumor from each group (brown=vimentin, haematoxylin counterstain). (D) M7824 blocks and reverts chemoresistance conferred by TGF-β1 in lung PC-9 cells in vitro. PC-9 cells were left untreated (control) or treated with TGF-β1 (2 ng/mL) for 72 hours followed by treatment with indicated doses of chemotherapy for 96 hours. For the blockade experiments, PC-9 cells were simultaneously treated with TGF-β1 (2 ng/mL) and M7824 (200 ng/mL) for the entire duration of the assay; for the reversion experiments, cells were treated with TGF-β1 (2 ng/mL) for 72 hours, followed by TGF-β1+M7824 during the chemotherapy assay to revert a previously induced EMT. Cell viability was assayed by the Cell-Titer-Glo luminescent viability assay. Figures adapted from David et al.18 EMT, epithelial-mesenchymal transition; GAPDH, glyceraldehyde3-phosphate dehydrogenase; HBSS, Hank’s balanced salt solution; IHC, immunohistochemistry; i.p., intraperitoneal; PD-L1, programmed death ligand 1; s.c., subcutaneous; SCID, severe combined immunodeficiency; TGF-βRII, transforming growth factor-β receptor II.
Bintrafusp alfa in preclinical applications
Bintrafusp alfa decreases tumor plasticity and resistance to chemotherapeutics
As a manifestation of the plastic phenotype of cancer cells, epithelial tumors can undergo a phenotypic switch called an epithelial–mesenchymal transition (EMT) in which cancer cells shift from an epithelial state (cuboidal and clustered, apical/basolateral polarity, extensive cell-to-cell adhesion) to an intermediate or mesenchymal phenotype (spindle-like morphology, loss of polarity, minimal cell-to-cell adhesion). These changes promote the acquisition of migratory and invasive capabilities by the tumor cells while enhancing their resistance to several anticancer agents, including chemotherapies, radiation, small targeted therapies and immunotherapy. Both the phenomenon of EMT and EMT-induced resistance to therapeutics can be reversible.16
TGF-β is considered a master regulator of EMT and has been found to promote a phenotypic transition in many cancer models including breast, prostate, colon and melanoma among others.17 In addition, the transcriptional regulators of the process, overall designated as EMT transcription factors, can upregulate TGF-β expression and secretion by cancer cells, thus creating a positive feedback loop that helps tumor cells maintain their mesenchymal-like phenotype while promoting EMT in surrounding cancer cells.16 In a previous study,18 bintrafusp alfa was shown to efficiently suppress TGF-β1-mediated EMT in human non-small cell lung cancer (NSCLC) models. In vitro, bintrafusp alfa prevented the downregulation of the epithelial marker E-cadherin and the up-regulation of the mesenchymal proteins fibronectin and vimentin when administered simultaneously with TGF-β1 to lung cancer cells (figure 1B). In addition, in cells that had already undergone TGF-β1-induced EMT, subsequent treatment with bintrafusp alfa was able to ‘revert’ EMT, thus restoring E-cadherin expression and decreasing expression of mesenchymal proteins to levels similar to control untreated cells (figure 1B).18 In xenograft models of NSCLC in vivo, bintrafusp alfa treatment was also shown to markedly decrease the expression of the mesenchymal protein vimentin in tumor cells (figure 1C). Because these changes were not induced by anti-PD-L1 treatment alone, the anti-EMT effect of bintrafusp alfa was attributed to its TGF-β ‘trapping’ activity.
By suppressing EMT, bintrafusp alfa may potentially improve antitumor efficacy of other therapeutics such as chemotherapy or radiation. The loss of E-cadherin expression is one of the classical signs of EMT and is associated with increased invasion and drug resistance in several cancers.19 20 While the addition of TGF-β1 to lung cancer cell lines in vitro resulted in decreased susceptibility to the cytotoxic effect of docetaxel, paclitaxel and gemcitabine, the addition of bintrafusp alfa simultaneously or following treatment with TGF-β1 was shown to block or revert, respectively, TGF-β1-induced resistance, thus increasing tumor lysis in response to chemotherapy to levels similar to those of control, untreated cells (figure 1D).18 Moreover, as described below, experiments conducted in vivo also demonstrated the ability of bintrafusp alfa to increase anti-tumor efficacy of chemotherapy in a murine model of colon cancer.
Bintrafusp alfa alters natural killer cell phenotype and promotes natural killer-mediated cytotoxicity
The bifunctional blockade of PD-L1 and sequestration of TGF-β offer a novel approach to alter the immune cell compartment in tumors. TGF-β is a master regulator of the immune system; for an in-depth review of this topic, see Batlle and Massagué.21 For the purpose of this review, we will limit our discussion to the natural killer (NK) and T cell compartments because those have been shown to be the most relevant to the antitumor activity of bintrafusp alfa in preclinical models. There is substantial evidence that TGF-β may alter the phenotype of NK cells and suppress their cytotoxic activity.22 In agreement, a recent study of human NK cells in vitro demonstrated the ability of bintrafusp alfa to prevent changes in gene expression induced by TGF-β1 treatment. Moreover, the anti-PD-L1 moiety of bintrafusp alfa has been found in vitro to mediate antibody-dependent cellular cytotoxicity of multiple human tumor cell lines of breast, cervical and urothelial origin, using human NK cells as effectors.23 24 In several murine models in vivo, bintrafusp alfa has also been shown to improve the recruitment of NK cells to the TME.14 15 For example, in a syngeneic breast murine model (EMT6), bintrafusp alfa treatment increased the population of tumor-infiltrating NK cells including those with an activated phenotype (eg, T-bet+; NKG2D+; NKp46+).14 Moreover, treatment with bintrafusp alfa induced antitumor efficacy in multiple tumor models, which was partially suppressed when NK cells were depleted.14 15 Altogether, the bifunctional blockade of PD-1/PD-L1 and TGF-β was shown to increase NK cell activation, cytotoxicity and recruitment to the TME, thus improving host antitumor immune responses.
Bintrafusp alfa alters the T cell compartment and promotes antitumor responses
TGF-β and PD-L1 are both important regulators of the T cell compartment. Binding of PD-L1 to its receptor PD-1 on activated T cells delivers a negative signal that blocks T cell proliferation, survival and effector functions. In addition, TGF-β promotes Treg differentiation and inhibits antitumor responses.25 TGF-β has been shown to cause differentiation to the Treg phenotype by driving FOXP3 expression in vitro.21 26 In melanoma and breast cancer patients, FOXP3 and TGF-β expression are highly correlated, indicating that TGF-β promotes Treg infiltration in the TME.27 Tregs have also been shown to produce TGF-β1 both as a secreted factor and a surface protein26; secreted TGF-β may help Tregs to maintain a steady presence in tissues and to regulate the activity of other immune cells, while TGF-β1 expressed on the membrane of Tregs has been shown to inhibit cytotoxic T cell activity.28 Thus, bintrafusp alfa also offers a method of inhibiting Treg activity by sequestering TGF-β. In agreement, treatment with bintrafusp alfa in vitro was shown to partially alleviate Treg suppression of CD4+ T cell proliferation. In the same study, the activity of each component of the bifunctional bintrafusp alfa was compared by using an anti-PD-L1 antibody versus a mutated version of bintrafusp alfa (designated as M7824mut in previous publications) that traps TGF-β but does not bind PD-L1, demonstrating that the blockade of TGF-β alone was responsible for the restoration of effector CD4+ T cell proliferation in the presence of bintrafusp alfa.24
In addition to its effects on Tregs, bintrafusp alfa added in vitro to CD8+ T cells has been shown to enhance IL-2 production in response to superantigen stimulation14 and to modestly increase antigen-specific T cell-mediated tumor cell lysis.23 In murine models, bintrafusp alfa demonstrated antitumor efficacy and was shown to increase the density of proliferative and cytotoxic CD8+ T cells in the TME.15 In syngeneic murine mammary and colon carcinoma models (EMT6, MC38), bintrafusp alfa treatment was shown to significantly reduce tumor growth compared with control and anti-TGF-β treatment alone with the M7824mut agent (figure 2A,B).15 In order to expand on the mechanism by which bintrafusp alfa controls tumor growth, Knudson et al 15 used depletion studies and demonstrated that the antitumor efficacy of bintrafusp alfa was dependent on NK and CD8+ T cells. Bintrafusp alfa treatment increased effector and effector memory T cell density in the spleen of tumor-bearing mice and induced an active CD8+ T cell phenotype in the TME. A limitation of these studies, however, is the fact that transplanted syngeneic tumor models were used. Given the high expression of TGF-β in the stroma, the use of spontaneous transgenic models with an anatomically intact stroma would be more adequate to evaluate the effects of bintrafusp alfa.
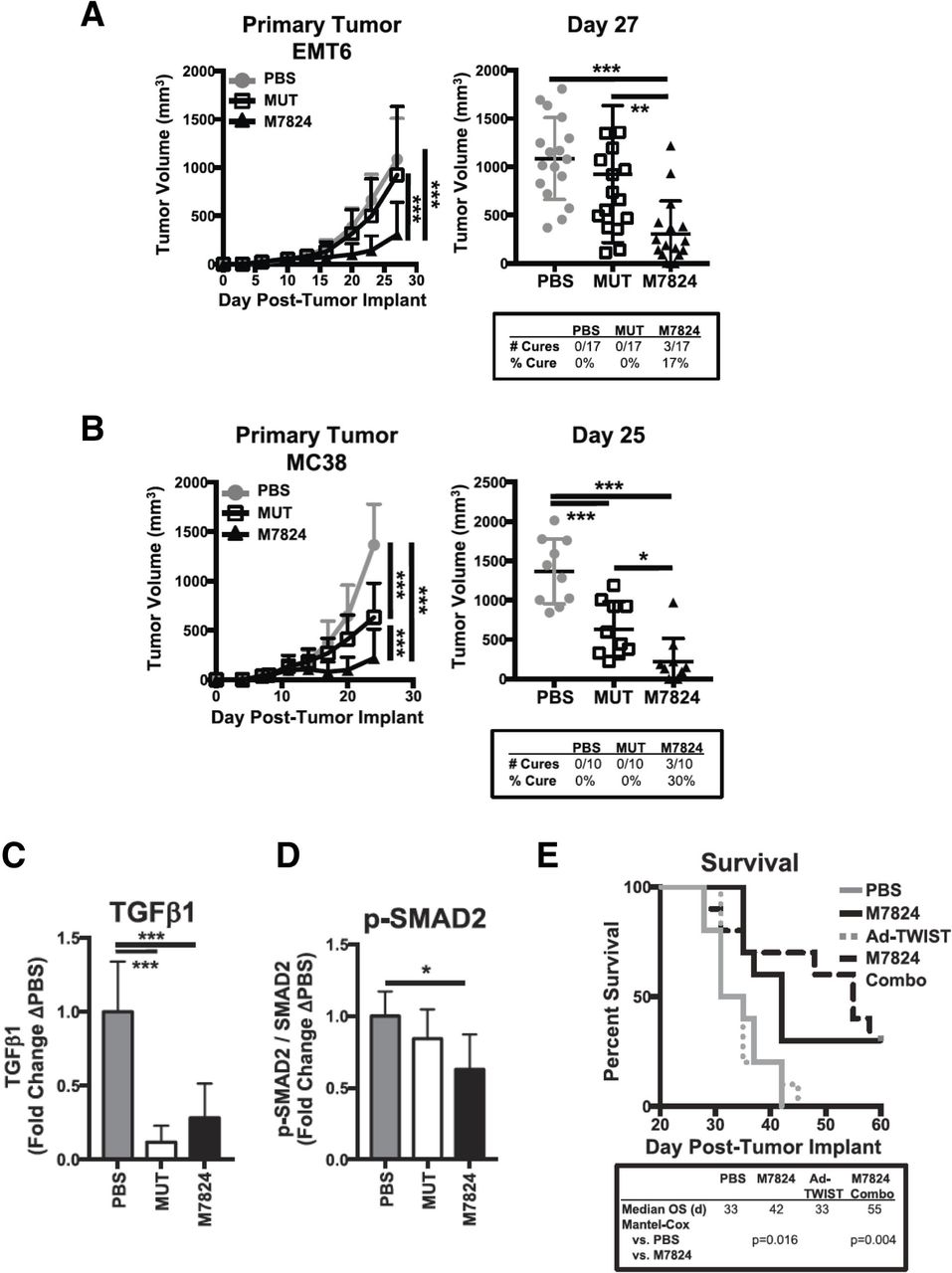
Bintrafusp alfa (designated in previous publications as M7824) reduces TGF-β signaling and growth of murine tumors. (A) Murine carcinoma EMT6 cells (2.5×105 cells) were implanted s.c. in the mammary fat pad of female Balb/c mice, or (B) murine colon cancer MC38 cells (5×105 cells) were implanted s.c. in the right flank of female C57BL/6 mice. Treatment was administered i.p. on days 9, 11 and 13 post-tumor implantation with vehicle (PBS), a modified version of M7824 lacking a functional anti-PD-L1 moiety (MUT, 492 µg), or M7824 (492 µg) (EMT6, n=17 mice per group; MC38, n=10 mice per group). Primary tumor growth curves (left panels) show mean±SD; tumor weights on indicated days for individual mice (right panel) with mean±SD and compared by one-way ANOVA with Tukey’s multiple comparisons test. Tables below show the number and per cent of cured mice per group. (C) M7824 reduces plasma levels of TGF-β1 in EMT6-bearing mice. EMT6 tumors were implanted as in (A); mice were treated i.p. with PBS, MUT, or M7824 (492 µg; days 10, 12, 14). Platelet-poor plasma was collected 24 hours after last treatment. Total TGF-β1 levels in the plasma were normalized to the mean of the PBS control group. (D) M7824 reduces phosphorylated SMAD2 in the TME of EMT6-bearing mice. EMT6 tumors were implanted as in (A); mice were treated i.p. with vehicle, MUT, or M7824 (492 µg; days 17, 19, 21). Six hours after the last treatment, tumors were sectioned and snap-frozen and subsequently analyzed for total and phosphorylated SMAD2 by capillary Western blot. Phosphorylated SMAD2 relative to total SMAD2 levels were then normalized to the control group. Data in (C, D) combine two independent experiments (n=3–6 mice per experiment); graphs show mean±SD and analysis by one-way ANOVA with Tukey’s multiple comparisons test. (E) Combination of M7824 and Ad-TWIST vaccine improves survival in EMT6-bearing mice. EMT6 tumors were implanted as in (A); mice were treated i.p. with either PBS, M7824 (492 µg; days 10, 12, 14), Ad-TWIST (1×1010 virus particles, days 16, 23, 30), or M7824+Ad-TWIST. Survival curves show per cent survival and table below shows median OS in days. Data are representative of 2–3 independent experiments, n=10 mice. Statistical significance: *P<0.05, **P<0.005, ***P<0.001. Figures adapted from Knudson et al.15 ANOVA, analysis of variance; EMT, epithelial-mesenchymal transition; i.p., intraperitoneal; OS, overall survival; MUT, M7824 lacking a functional anti-PD-L1 moiety; PBS, phosphate buffered saline; PD-L1, programmed death ligand 1; p-SMAD2, phosphorylated SMAD2; s.c., subcutaneous; TGF-β, transforming growth factor-β.
Plasma levels of TGF-β1 were reduced in mice treated with bintrafusp alfa or M7824mut (figure 2C) indicating the ability of these agents to sequester murine TGF-β1 in vivo.15 In addition, bintrafusp alfa significantly reduced intratumoral TGF-β signaling, as shown by decreased levels of phosphorylated SMAD2 in breast tumors (figure 2D 15), thus demonstrating the ability of bintrafusp alfa to accumulate at the site of the tumor via binding to PD-L1. It is important to note that while these experiments did not include an anti-PD-L1 monotherapy group, separate research in the EMT6 or MC38 models from Lan et al 14 extensively compared single anti-PD-L1 or single M7824mut treatment against bintrafusp alfa. In those studies, bintrafusp alfa displayed statistically significantly better tumor control than either anti-PD-L1 or M7824mut alone, and was more efficacious than each individual therapy to increase the density of CD8+ T cells in the TME with an activated, proliferative and effector memory phenotype.14
Several recent studies demonstrated the importance of TGF-β as a mechanism of resistance to checkpoint inhibitor therapies. One such study, for example, evaluated tumors from patients with metastatic urothelial cancer who were treated with atezolizumab and identified a signature of TGF-β signaling positively associated with lack of response.29 The same study evaluated a combination of anti-PD-L1 and anti-TGF-β in the EMT6 murine breast cancer model, demonstrating that the combination of both blocking antibodies could afford better antitumor effect than that observed with each single treatment alone. In another study,30 blockade of TGF-β signaling was shown to improve the antitumor activity of anti-PD-L1 treatment in a murine model of colon cancer characterized by low mutation burden, T cell exclusion and a TGF-β activated stroma, thus supporting the role of stromal TGF-β as a mechanism of resistance to PD-1/PD-L1 blockade. In light of these studies and the results with bintrafusp alfa, one important question is whether using a dual, bifunctional molecule such as bintrafusp alfa could afford a better outcome as compared with the combination of single anti-PD-L1 and anti-TGF-β agents. Using the MC38 model, Lan et al have shown a modest advantage of using bintrafusp alfa over a combination of anti-PD-L1 or anti-TGF-β alone.14 Studies performed with a similar bifunctional agent that blocks PD-L1 and sequesters TGF-β via a TGF-βRII (designated as Y-trap) have also recently shown that the fusion protein is more efficient than the combination of anti-PD-L1 and IgG-TGF-βRII agents in achieving antitumor activity against human cancer xenografts in humanized mice.27
Bintrafusp alfa in combination therapies
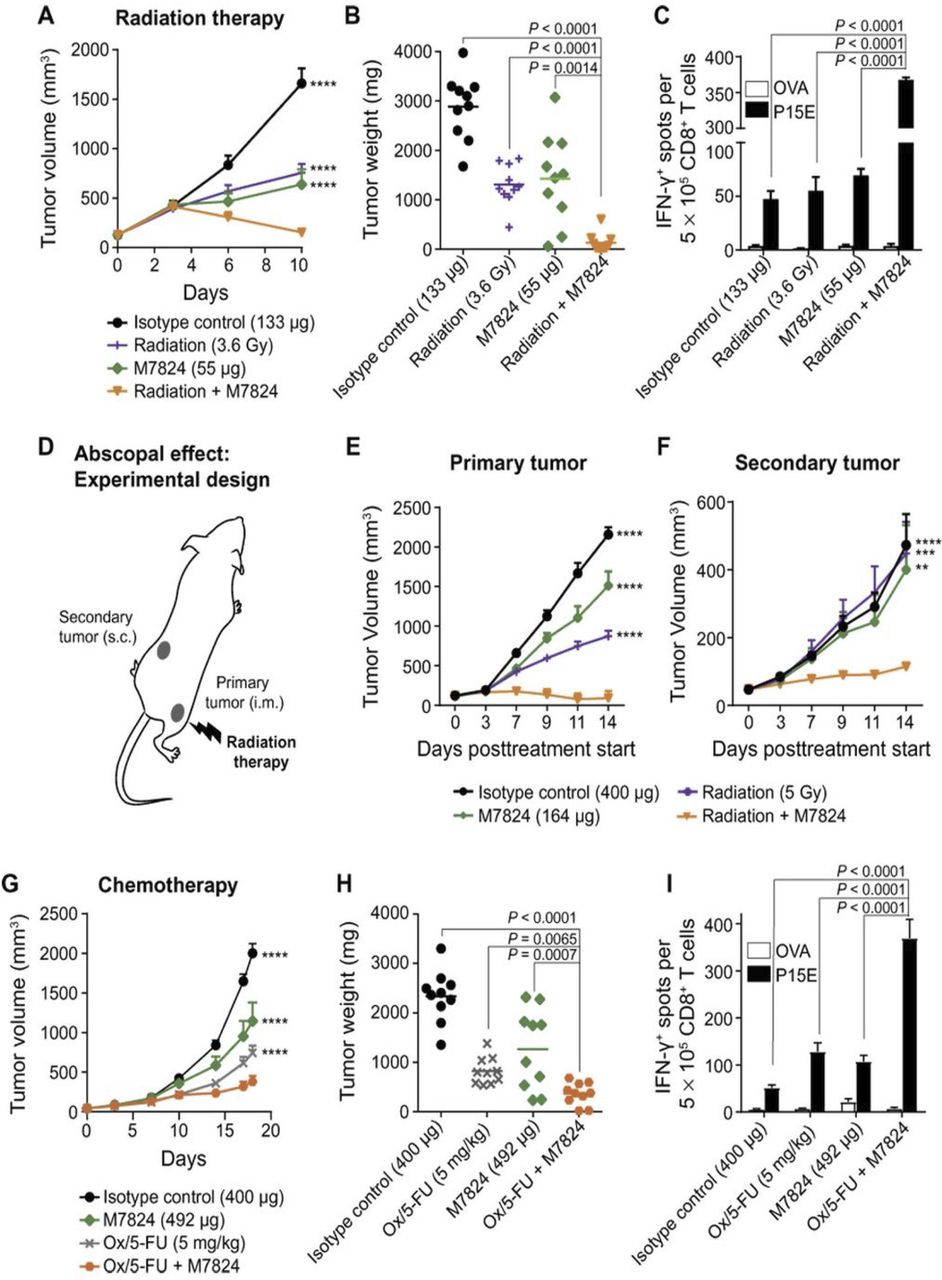
By sequestering TGF-β, bintrafusp alfa has the ability to modulate tumor cell plasticity, proliferation and susceptibility to other agents. One potential combination that has shown promise in preclinical studies is the use of bintrafusp alfa with tumor-associated antigen (TAA)-targeting vaccines. In a syngeneic breast model, bintrafusp alfa and the TAA-targeting vaccine Ad-TWIST combination significantly increased overall survival above that induced by each monotherapy (figure 2E), demonstrating that bintrafusp alfa may be used in combination with other IO agents. Another potential scenario for combination therapies involving bintrafusp alfa is in the context of standard of care treatment. Both chemotherapy and radiation have been shown to induce the upregulation of TGF-β and PD-L1 expression in various carcinoma models.31–34 Thus, bintrafusp alfa may be particularly effective in combination with these therapies. In preclinical studies,14 bintrafusp alfa was shown to improve antitumor responses in combination with radiation in the syngeneic colon cancer model MC38 in vivo (figure 3A–C). In this model, the combination therapy also demonstrated significant systemic immune activation, shown by an increase of activated CD8+ T cells in the spleen above either treatment alone. Concurrent bintrafusp alfa and radiation treatment were also shown to confer antitumor efficacy at the primary tumor site that received radiation and at a secondary location (immune-mediated abscopal effect, figure 3D–F). These data suggested that bintrafusp alfa may be used in combination with local radiation to generate a systemic antitumor immune response that may target metastasis.

Bintrafusp alfa (designated in previous publications as M7824) in combination with radiation and chemotherapy. (A–C) B cell-deficient mice (μMt−) were inoculated i.m. with 0.5×106 MC38 cells (day −8) and treated with isotype control (133 µg i.v.; day 2), radiation (3.6 g/day; days 0–3), M7824 (55 µg i.v.; day 2), or radiation+M7824 (n=10 mice per group). (A) tumor growth curves. (B) tumor weight for individual mice on day 14. (C) ELISpot of IFN-γ–producing, p15E-responsive CD8+ T cells: CD8+ T cells were isolated from spleens (day 14; n=5) followed by culture with irradiated antigen-presenting cells derived from naïve solenocytes pulsed with KPSWFTTL (p15E) peptide or irrelevant peptide (OVA). (D–F) C57BL/6 mice were inoculated i.m. in the right thigh with 0.5×106 MC38 cells (primary tumor) and s.c. in the left flank with 1×106 MC38 cells (secondary tumor). Seven days following tumor inoculation, mice were treated with isotype control (400 µg; days 0, 2, 4), radiation (5 g/day; days 0–3), M7824 (164 µg; day 0) or M7824+radiation. (D) Radiation was only applied to the primary tumor site; tumor measurements were taken at the primary (E) and secondary (F) tumor sites. (G–I) µMt- mice were inoculated s.c. with 1×106 MC38 cells (day −7). Mice were treated with isotype (400 µg; days 3, 6, 9, 12, 17), M7824 (164 µg; days 3, 6, 9, 12, 17), oxaliplatin/5-fluorouracil (Ox/5-FU; 5 mg/kg i.p. and 60 mg/kg i.v.; day 0), or M7824 +Ox/5-FU (n=10). tumor volume (G) and tumor weights on day 18 (H). (I) ELISpot of IFN-γ–producing, p15E-responsive CD8+ T cells (day 18; n=5) as described in (C). Statistical significance: **P<0.01, ***P<0.001, ***P<0.0001. (B, H) tumor weights are presented for individual mice with mean indicated by lines and compared by unpaired t-test. (C, I) IFN-γ ELISpot data show means±SEM of three replicates analyzed by two-way ANOVA. From Lan et al.14 Reprinted with permission from the American Association for the Advancement of Science (AAAS). ANOVA, analysis of variance; IFN-γ, interferon-γ; i.m., intramuscular; i.p., intraperitoneal; i.v., intravenous; OVA, ovalbumin; s.c., subcutaneous.
In addition to radiation, combination of bintrafusp alfa with chemotherapy (oxaliplatin/5-fluorouracil) has also been shown in preclinical models to be effective at improving antitumor efficacy, where concurrent treatments synergistically induced CD8+ T cell activation and effective immune-mediated antitumor effect (figure 3G–I).14 In addition to enhancing CD8+ T cell activation, it could also be hypothesized that at least a fraction of the antitumor efficacy observed with combinations of chemotherapy and bintrafusp alfa may be due to the ability of bintrafusp alfa to directly impact the phenotype of the tumor cells. As previously discussed, tumor cells that are highly plastic or mesenchymal are more resistant to cytotoxic lysis, including cell death induced by chemotherapy. Since TGF-β induces EMT, bintrafusp alfa may increase tumor cell susceptibility to chemotherapeutics by depleting TGF-β in the TME and reducing the phenomenon of EMT. This hypothesis is supported by preclinical evidence as shown in figure 1 that bintrafusp alfa effectively decreases mesenchymal features of tumor cells both in vitro and in vivo and increases tumor susceptibility to chemotherapy in vitro.
Clinical applications of bintrafusp alfa
TGF-β-targeting therapies in the clinic
Numerous inhibitors targeting the TGF-β pathway have progressed to clinical trials including small molecule inhibitors, antibodies and receptor-based TGF-β traps.35 Among the small molecule inhibitors that have been evaluated in preclinical and clinical studies is galunisertib, a small molecule inhibitor targeting the TGF-βRI (ALK5) kinase. This agent has been tested in several cancer types with mixed results. In phase II clinical trials, galunisertib demonstrated some antitumor efficacy in patients with unresectable pancreatic cancer and in a subset of patients with advanced hepatocellular carcinoma;36 37 however, no meaningful antitumor activity was observed in a phase II trial of recurrent glioblastomas.38 Galunisertib has exhibited an acceptable safety profile in phase I testing and in phase II studies.39
Several antibodies against TGF-β have also progressed to clinical trials. Of these, fresolimumab has been most extensively studied. Fresolimumab (GC1008) is a fully human anti-TGF-β MAb that sequesters all isoforms of TGF-β. It has completed a phase I clinical trial (NCT00356460) with acceptable safety signals in renal cell carcinoma, melanoma and glioma.40 Fresolimumab-related adverse events (AEs) that were reported included gingival bleeding, headaches, epistaxis and skin disorders (actinic keratosis, hyperkeratosis and reversible cutaneous keratoacanthomas).40 Of the 29 enrolled patients, one partial response (PR) was observed in a patient with advanced malignant disease, and six patients had stable disease (SD). Fresolimumab is currently being investigated in ongoing phase I/II trials in advanced and metastatic solid tumors and mesothelioma.
While anti-TGF-β therapeutics offer a path to treat cancers, as monotherapies these agents have had limited success in clinical application thus far. The limited number of patients who responded indicates the need for further treatment combinations; the bifunctionality of bintrafusp alfa thus offers a novel approach.
Bintrafusp alfa in clinical trials
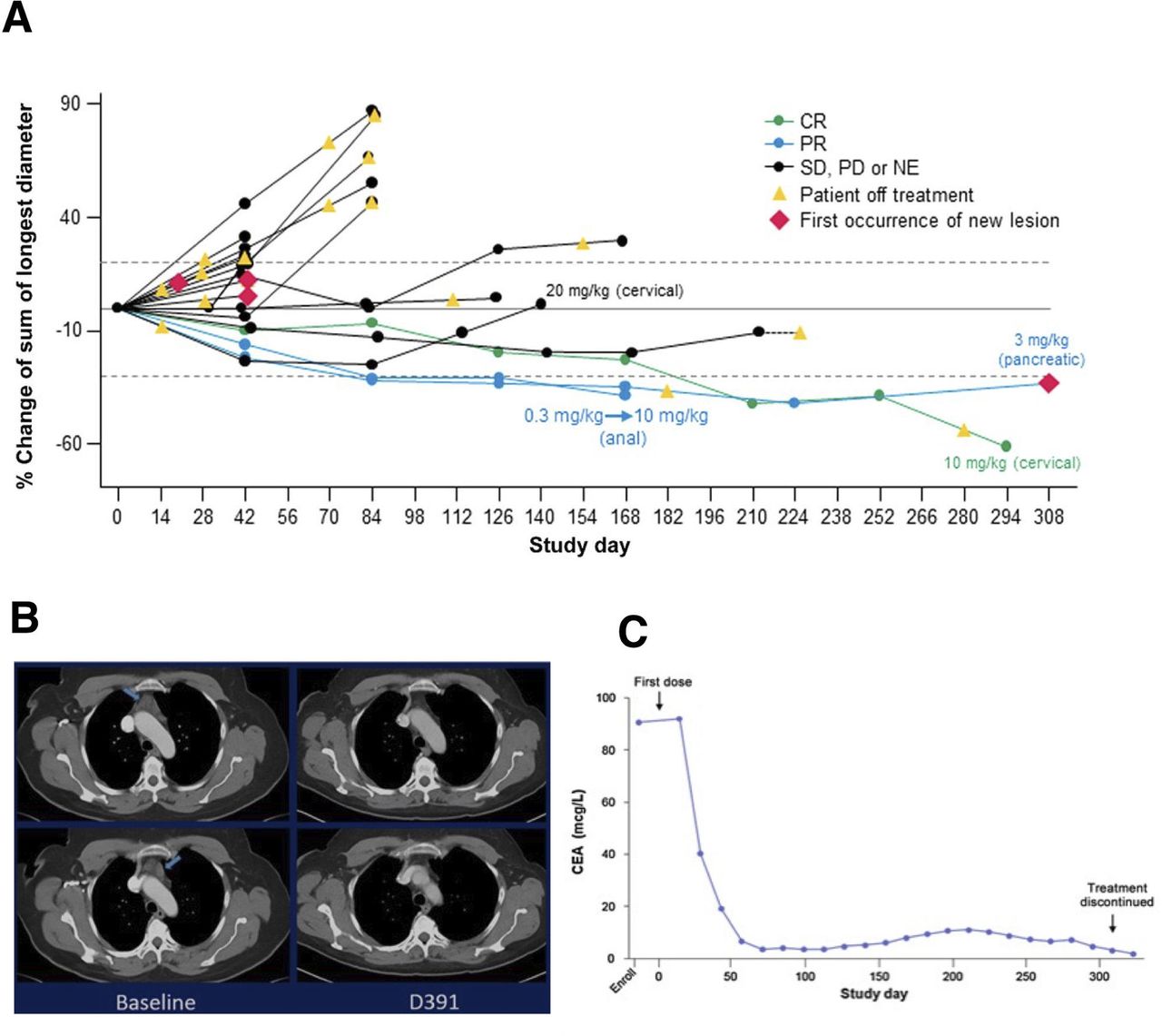
Bintrafusp alfa is currently being studied in clinical trials at multiple institutions. The dose-escalation portion of a phase I, open-label clinical trial of bintrafusp alfa (MSB0011359C) has been completed, showing promising antitumor efficacy in heavily pretreated patients with metastatic or locally advanced solid tumors without prior checkpoint inhibitor treatment.41 A total of 19 patients were enrolled in that trial at the Center for Cancer Research at the NCI (Bethesda, MD) with primary tumor types including pancreatic, cervix uteri, colorectal, anal and adenoid cystic carcinoma, among others. Bintrafusp alfa showed a safety profile similar to other anti-PD-1/PD-L1 monotherapies with the addition of skin-related AEs characteristic of TGF-β blockade. Evidence of clinical activity was seen with bintrafusp alfa across all evaluated doses (figure 4A); of the 19 patients enrolled, one case (cervical cancer) demonstrated a durable complete response (figure 4B,C), two patients (pancreatic cancer, anal cancer) had a durable PR, and two cases (pancreatic cancer, carcinoid) experienced prolonged SD. Analyses of plasma TGF-β1, -β2 and -β3 levels demonstrated that bintrafusp alfa reached sufficient levels to completely sequester all three isoforms of TGF-β in circulation for the entire dosing period at doses >1 mg/kg.41 This study also reported results of the analysis of exploratory biomarkers conducted in peripheral blood collected at various time points. Though the study included a small heterogenous group of patients, results from a flow cytometry-based analysis of 131 immune cell subsets in peripheral blood mononuclear cells prior to therapy and following administrations of bintrafusp alfa demonstrated a trend in those patients with evidence of clinical benefit toward an increase in peripheral B cells and CD4+ T cells, and decreases in MDSCs expressing CD16 at the time of best overall clinical response versus pretherapy levels. However, the limited number of patients analyzed in the phase I dose-escalation study precluded any conclusions about the potential clinical utility of bintrafusp alfa over other anti-PD-1/PD-L1 therapies. In addition, further studies with larger number of patients of defined tumor types will need to be conducted to determine potential biomarkers of response to bintrafusp alfa.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Phase I trial of bintrafusp alfa shows antitumor efficacy in advanced solid tumors. (A) Spider plot showing changes in sum of the longest tumor diameter from baseline according to Response Evaluation Criteria in Solid tumors (V.1.1) for each patient who received bintrafusp alfa. Threshold for PR indicated by the dotted line at −30%. Threshold for PD indicated by the dotted line at 20%. (B) A metastatic cervical cancer patient treated with bintrafusp alfa shows durable complete CR. (C) Carcinoembryonic antigen (CEA) curve for patient with confirmed, ongoing, durable CR. CR, complete response; NE, not evaluable; PD, progressive disease; PR, partial response; SD, stable disease. Figures adapted from Strauss et al.41
Currently, numerous clinical studies are ongoing involving the use of bintrafusp alfa in patients with a variety of cancer types including breast, NSCLC, prostate, pancreas, biliary and HPV-associated cancers. Several of these trials are investigating combinations of bintrafusp alfa with other immunotherapeutics or other therapeutic modalities such as radiation or chemotherapy. An important question moving forward is whether certain patients could particularly benefit from receiving treatment with bintrafusp alfa versus the use of other anti-PD-1/PD-L1 agents. As one of the features associated with TGF-β signaling in tumors is the occurrence of phenotypic plasticity, the evaluation of biomarkers of EMT could potentially be used for selection of patients who may be more responsive to bintrafusp alfa. Gene signatures of EMT have been defined for specific tumor types, including gastric, colorectal and bladder tumors in association with worse overall survival42–44 and more recently, a signature enriched in genes involved in EMT was found to be associated with resistance to anti-PD-1 therapy in metastatic melanoma patients.45 The use of such signatures could be prospectively used in future clinical studies to investigate potential associations between the occurrence of EMT in tumors and treatment outcome with bintrafusp alfa; if validated, the approach could also be used for selection of patients to be included in bintrafusp alfa clinical studies.
Conclusions
The bifunctional blockade of the PD-1/PD-L1 and TGF-β pathways offers a unique approach to engaging the immune system and promoting antitumor efficacy. In preclinical studies, bintrafusp alfa has also shown promising synergy in combinations with other therapeutics, and clinical investigation of combinations between bintrafusp alfa and radiation, chemotherapy, and/or TAA-targeting vaccines are ongoing. Thus far, bintrafusp alfa has shown the ability to promote antitumor immune responses in preclinical studies, particularly by increasing cytotoxic NK and T cell recruitment to the TME. In clinical studies, bintrafusp alfa has shown a promising antitumor efficacy in patients with heavily pretreated advanced solid tumors with a manageable safety profile similar to anti-PD-1/PD-L1 monotherapies.
Acknowledgments
The authors thank Debra Weingarten for her editorial assistance in the preparation of this manuscript.
References
Footnotes
Twitter @gulleyj1
Contributors HL, SRG, CJ, RND, JSt, JLG, CP and JSc all contributed to the conception, writing and revisions, and approved the final version. CP and JSc contributed equally to this work.
Funding This study was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; internally peer reviewed.