Article Text

Abstract
Background Several cancer types harbor alterations in the gene encoding AT-Rich Interactive Domain-containing protein 1A (ARID1A), but there are no approved therapies to address these alterations. Recent studies have shown that ARID1A deficiency compromises mismatch repair proteins. Herein, we analyzed 3403 patients who had tumor tissue next-generation sequencing.
Findings Among nine cancer subtypes with >5% prevalence of ARID1A alterations, microsatellite instability-high as well as high tumor mutational burden was significantly more frequent in ARID1A-altered versus ARID1A wild-type tumors (20% vs 0.9%, p<0.001; and 26% vs 8.4%, p<0.001, respectively). Median progression-free survival (PFS) after checkpoint blockade immunotherapy was significantly longer in the patients with ARID1A-altered tumors (n=46) than in those with ARID1A wild-type tumors (n=329) (11 months vs 4 months, p=0.006). Also, multivariate analysis showed that ARID1A alterations predicted longer PFS after checkpoint blockade (HR (95% CI), 0.61 (0.39 to 0.94), p=0.02) and this result was independent of microsatellite instability or mutational burden; median overall survival time was also longer in ARID1A-altered versus wild-type tumors (31 months vs 20 months), but did not reach statistical significance (p=0.13).
Conclusions Our findings suggest that ARID1A alterations merit further exploration as a novel biomarker correlating with better outcomes after checkpoint blockade immunotherapy.
- ARID1A
- PD-L1
- immune checkpoint inhibitor
- immunotherapy
- tumor mutation burden
- microsatellite instability
- biomarker
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- ARID1A
- PD-L1
- immune checkpoint inhibitor
- immunotherapy
- tumor mutation burden
- microsatellite instability
- biomarker
Introduction
The ARID1A gene encoding AT-Rich Interactive Domain-containing protein 1A is known as a member of the switching/sucrose non-fermentable (SWI/SNF) complex involved in chromatin remodeling.1 Mutations in and loss of the ARID1A gene mostly lead to its inactivation and ARID1A protein loss.2 Certain types of cancer, including clear cell ovarian carcinoma (46%–50%), gastric adenocarcinoma (10%–35%), and cholangiocarcinoma (15%–27%), frequently harbor ARID1A alterations.2–4 To date, clinical and preclinical data indicate that ARID1A alterations may sensitize tumors to drugs targeting the ataxia telangiectasia and Rad3-related (ATR) protein, the enhancer of zeste 2 (EZH2), or the phosphatidylinositol-3-kinase (PI3K) pathway,5–10 but no therapies targeting ARID1A alterations have been approved. Importantly, Shen et al demonstrated that ARID1A alterations interact with the mismatch repair (MMR) protein MSH2 and, hence, compromise MMR.3 Tumors formed by an ARID1A-deficient ovarian cancer cell line in syngeneic mice exhibited higher mutation load, as well as increased numbers of tumor-infiltrating lymphocytes and elevated programmed cell death-ligand 1 (PD-L1) expression. Furthermore, administration of anti-PD-L1 antibody decreased cancer burden and extended survival of mice bearing ARID1A-deficient but not ARID1A wild-type ovarian tumors.3 Interestingly, alterations in the polybromo-1 (PBRM1) gene, which is another member of the SWI/SNF complex, have been reported to correlate with salutary effects in cancer patients receiving checkpoint blockade inhibitors, though the clinical evidence remains controversial.11 12 In gastric cancers, ARID1A alterations are associated with Epstein-Barr virus infection, which is in turn associated with checkpoint blockade response.13 Herein, for the first time to our knowledge, we investigated the clinical correlation between ARID1A alterations and treatment benefit after anti-programmed cell death-1 (PD-1)/PD-L1 immunotherapy in the human pan-cancer setting.
Materials and methods
Study population and next-generation sequence
In a cohort of 3403 eligible patients at the Center for Personalized Cancer Therapy (University of California San Diego Moores Cancer Center), whose tissue DNA was analyzed by next-generation sequencing (NGS) by Foundation Medicine, Inc. (CLIA-licensed and CAP-accredited laboratory. Cambridge, Massachusetts, USA https://www.foundationmedicine.com), we reviewed the clinicopathological and genomic information of patients whose tumors were pathologically diagnosed as one of nine types of cancer that frequently harbored ARID1A alterations (>5% of prevalence in this cohort): non-small cell lung cancer, colorectal adenocarcinoma, breast cancer, melanoma, pancreatic ductal adenocarcinoma, cholangiocarcinoma/hepatocellular carcinoma, gastric/esophageal adenocarcinoma, uterine/ovary endometrial (endometrioid) carcinoma (including clear-cell carcinoma), and urothelial bladder carcinoma. Tissue DNA sequencing at the laboratory was approved by the US Food and Drug Administration in November 2017 and designed to include all genes somatically altered in human solid malignancies that were validated as targets for therapy, either approved or in clinical trials, and/or that were unambiguous drivers of oncogenesis based on available knowledge.14 15 Although the gene panel expanded with time (236–324 genes), the interrogation of the ARID1A gene was considered consistent. Only characterized ARID1A alterations were considered in this study (variants of unknown significant were excluded). In terms of microsatellite instability (MSI) status, 114 intron homo-polymer repeat loci with adequate coverage are analyzed for length variability and compiled into an overall score via principal components analysis.16 17 Measuring genes interrogated on the tissue DNA NGS and extrapolating to the genome as a whole as previously validated determined tumor mutational burden (TMB).18 TMB was classified to three categories: high (≥20 mutations/mb), intermediate (6–19 mutations/mb), and low (<6 mutations/mb).
Statistics
Using the Mann-Whitney U test and Fisher’s exact test, respectively, we compared categorical and continuous data. Progression-free survival (PFS) and overall survival (OS) data were measured from date of the initiation of anti-PD-1/PD-L1 immunotherapy and plotted by the Kaplan-Meier method. Data were censored if patient was progression free or alive (for PFS and OS, respectively) at last follow-up. The curves were compared by using the log-rank test. In multivariate analysis to investigate independent predictive factors for the PFS after anti-PD-1/PD-L1 immunotherapy, we used Cox’s proportional hazard model for estimating HR and its 95% CI (variables with p<0.1 in the univariate analyses were entered into the multivariate analysis). RO performed and verified statistical analysis using SPSS V.24 software.
Results and discussion
Starting with 3403 eligible patients who underwent tissue DNA NGS, we found 1540 patients with nine types of cancer diagnoses that had >5% prevalence of characterized ARID1A alterations in tissue DNA NGS (figure 1A and online supplementary figure 1). Of 161 patients with ≥1 characterized ARID1A alteration in diverse types of cancer, 142 had ARID1A substitution or frameshift alterations, while the remaining 19 had insertions, deletions, allelic loss, rearrangement, or truncation. Endometrial and gastroesophageal cancers were the tumor types in which ARID1A alterations were most frequent—49% and 20% of cases, respectively (figure 1A). The median number of genomic coalterations among tumors with ARID1A alterations was 6 (range, 1–72) (not including ARID1A alterations), which was significantly higher than the median of 4 alterations (range, 0–61) among those cancers with wild-type ARID1A (p<0.001). The rate of MSI-high was significantly higher in tumors with ARID1A alterations than in those with wild-type ARID1A (20% vs 0.9%; p<0.001) and in multiple individual tumor types as well (eg, MSI-high in ARID1A-altered vs wild-type endometrial cancer, 41% vs 0%, p=0.001) (figure 1B). Similarly, TMB-high (≥20 mutations/mb) was more often observed in tumors with ARID1A alterations than in those with wild-type ARID1A (26% vs 8.4%; p<0.001) and in individual tumor types (eg, endometrial cancer, 35% vs 0%, p=0.001) (figure 1C).
Supplemental material
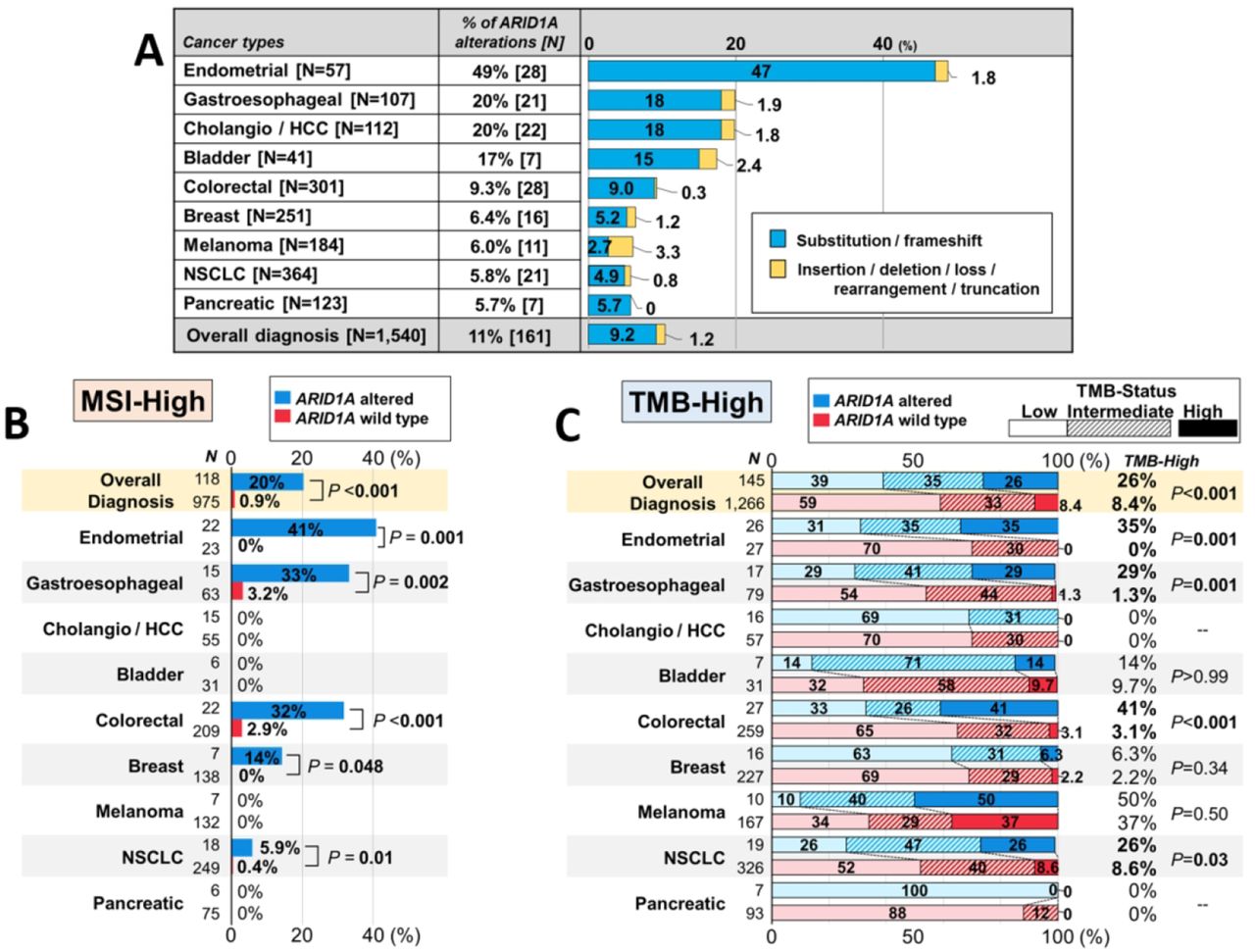
(A) Prevalence of characterized ARID1A alterations in tissue DNA NGS according to cancer types (n=1540). (B) Frequency of MSI-high according to ARID1A status (microsatellite status was available in 1093 patients (71.0%)). (C) Frequency of TMB-high according to ARID1A status (TMB-status was available in 1411 patients (91.6%); p values are for TMB-high rates): TMB-high (≥20 mutations/mb); TMB-intermediate (6–19 mutations/mb); TMB-low (<6 mutations/mb). ARID1A, AT-Rich Interactive Domain-containingprotein 1A; bladder, urothelial bladder carcinoma; breast, breast cancer; cholangio/HCC, cholangiocarcinoma and hepatocellular carcinoma; colorectal, colorectal adenocarcinoma; endometrial, uterine/ovary endometrial (endometrioid) carcinoma; gastroesophageal, gastric/esophageal adenocarcinoma; MSI, microsatellite instability; NGS, next-generation sequencing; NSCLC, non-small cell lung cancer; pancreatic, pancreatic ductal adenocarcinoma; TMB, tumor mutational burden.
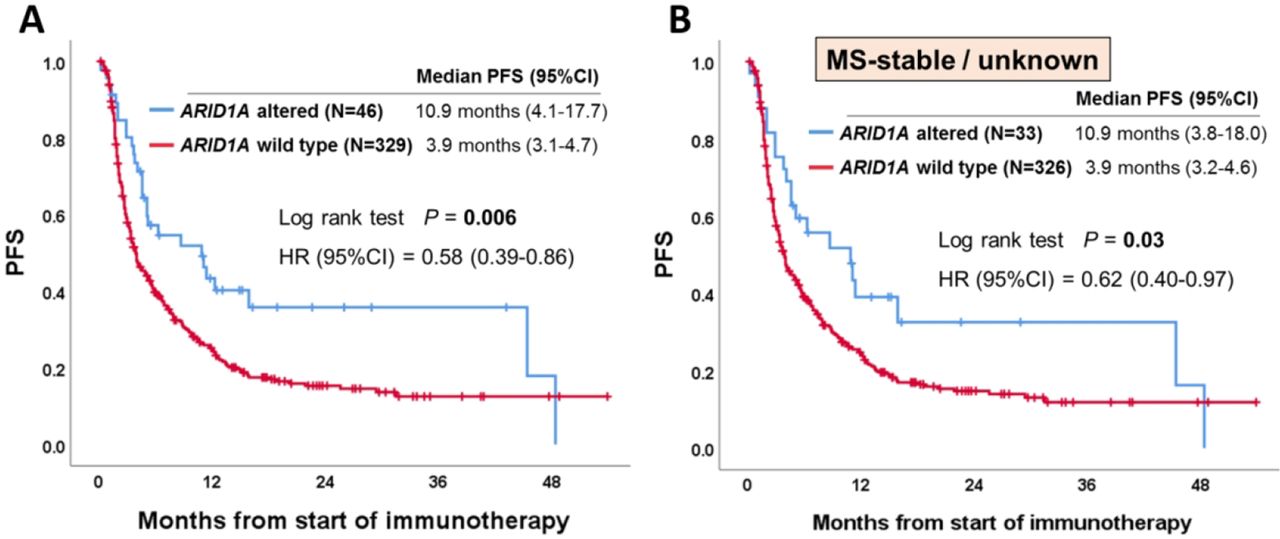
Overall, 375 patients (24%) among the 1540 patients with the nine types of cancer with >5% ARID1A alterations received anti-PD-1/PD-L1 immunotherapy in the advanced/metastatic disease setting (see online supplementary figure 1). MSI-high and TMB-high were seen in 4.3% (n=16) and 17% (n=65) of these 375 patients, respectively. As shown in figure 2A, patients with ARID1A-altered tumors showed a significantly longer PFS than those with the wild-type tumors (10.9 months vs 3.9 months, p=0.006) from the start of anti-PD-1/PD-L1 immunotherapy. When PFS was analyzed according to cancer diagnosis (only tumor types with ≥5 patients with ARID1A alterations), similar sensitivity was observed in individual tumor types (eg, colorectal cancer (5.2 months vs 2.1 months, p=0.005); endometrial cancer (4.6 months vs 3.0 months, p=0.02)) (see online supplementary figure 2). Importantly, even when only patients without MSI-high were included to the analysis, ARID1A-altered tumors showed a significantly longer PFS than those with wild-type tumors: HR (95% CI), 0.62 (0.40 to 0.97); p=0.03 (figure 2B). In the same way, when only patients without TMB-high were included to the analysis, patients with ARID1A-altered tumors (vs ARID1A wild-type) showed a trend towards longer PFS: HR (95% CI), 0.69 (0.43 to 1.08) although not statistically significant (p=0.10) (see online supplementary figure 3) (small numbers of patients precluded analysis of patients with MSI-high or TMB-high who had ARID1A alterations vs not). When examining OS in ARID1A-altered versus the wild-type patients, median OS time was longer in the ARID1A-altered group (30.8 months vs 20 months), but this did not reach statistical significance (p=0.13) (see online supplementary figure 4). In order to better determine if the correlation between ARID1A alterations and longer PFS was independent of specific confounding variables, we performed a multivariate analysis (patient characteristics of ARID1A-altered vs wild-type patients are shown in table 1). Our Cox-regression model demonstrated that ARID1A alterations were selected as an independent predictor of better outcome (PFS) after anti-PD-1/PD-L1 immunotherapy (HR (95% CI), 0.61 (0.40 to 0.94); p=0.03) (table 2).

{kind=link}
{kind=link}
Kaplan-Meier curve of PFS according to ARID1A status. (A) Among patients who received anti-programmed cell death-1 (PD-1)/programmed cell death-ligand 1 (PD-L1) immunotherapy (n=375). (B) Among patients without microsatellite instability-high who received anti-PD-1/PD-L1 immunotherapy (n=359). Similar results were seen even if the MS-unknown (n=60) were excluded (p=0.02). ARID1A, AT-Rich Interactive Domain-containingprotein 1A; MS, microsatellite status; PFS, progression-free survival.
Characteristics of patients who underwent anti-PD-1/PD-L1 immunotherapy (n=375)
Univariate and multivariate analyses for progression-free survival after anti-PD-1/PD-L1 immunotherapy (n=375). Variables with p<0.10 in the univariate analyses were entered into the multivariate analysis
In conclusion, 28% of ARID1A-altered tumors (n=32 of 114 patients whose microsatellite and TMB status were both available) had either MSI-high or TMB-high (or both), and the rate of MSI-high and TMB-high was significantly higher in ARID1A-altered versus wild-type tumors. These findings are consistent with previous reports that ARID1A deficiency is correlated with MMR deficiency.3 19 ARID1A alterations were independently and significantly associated with longer PFS after anti-PD-1/PD-L1 immunotherapy (regardless of microsatellite and TMB status). This study has several limitations such as the small number of patients with each cancer type, which restricted our ability to analyze individual tumor histologies. Nevertheless, the results suggest generalizability across tumor types. Another limitation was that improvement in OS in ARID1A-altered patients (vs wild-type) did not reach statistical significance; larger numbers of patients are needed to validate this endpoint. Therefore, ARID1A alterations may be a genomic marker of checkpoint blockade sensitivity, in addition to other putative markers such as MSI-high and TMB-high.20–22 Our observations indicate that ARID1A alterations warrant further studies with longer follow-up and larger numbers of patients in order to confirm if they can be added to the armamentarium of genomic markers that are exploitable for matching patients to immunotherapy in the pan-cancer setting.23 24
References
Footnotes
Contributors Study conception and design: RO, SK, JKS, and RK; data acquisition: RO, SL, and REJ; statistical analysis: RO and SK; data interpretation: RO, SK, JKS, and RK; drafting the manuscript or revising it critically: all authors; final approval of the manuscript: all authors.
Funding This work was funded in part by the Joan and Irwin Jacobs Fund and NIH P30 CA023100 (RK). We appreciate funding support from Hope for a Cure Foundation, Jon Strong, NIH R01 CA226803, and FDA R01 FD006334 (JKS).
Competing interests SK serves as a consultant fee (Foundation Medicine) and speaker’s fee (Roche). JKS has the following disclosure information: Research funding (Novartis Pharmaceuticals, Amgen Pharmaceuticals, and Foundation Medicine); Consultant fee (Grand Rounds (2015–2019), Loxo Oncology (2017–2018), Deciphera (2019), and Roche (2019)). RK has the following disclosure information: Stock and Other Equity Interests (IDbyDNA, CureMatch, and Soluventis); Consulting or Advisory role (Gaido, LOXO, X-Biotech, Actuate Therapeutics, Roche, NeoMed, Soluventis, and Pfizer); Speaker’s fee (Roche); Research Funding (Incyte, Genentech, Merck Serono, Pfizer, Sequenom, Foundation Medicine, Guardant Health, Grifols, Konica Minolta, DeBiopharm, Boerhringer Ingelheim, and OmniSeq (All institutional)); Board Member (CureMatch).
Patient consent for publication Not required.
Ethics approval This study was approved by the Internal Review Board at UC San Diego Moores Cancer Center. All investigations followed the guidelines of the Profile-Related Evidence Determining Individualized Cancer Therapy study (UCSDPREDICT study: NCT02478931) for data collection and any investigational therapies for which the patient gave consent.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. The data that support the findings of our study are available upon request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.