Article Text

Abstract
Background The programmed cell death-1/programmed cell death ligand-1 (PD-1/PD-L1) axis plays a central role in suppressing antitumor immunity; axis dysregulation can be used by cancer cells to evade the immune system. Tislelizumab, an investigational monoclonal antibody with high affinity and binding specificity for PD-1, was engineered to minimize binding to FcγR on macrophages to limit antibody-dependent phagocytosis, a potential mechanism of resistance to anti-PD-1 therapy. The aim of this phase IA/IB study was to investigate the safety/tolerability, antitumor effects and optimal dose and schedule of tislelizumab in patients with advanced solid tumors.
Methods Patients (aged ≥18 years) enrolled in phase IA received intravenous tislelizumab 0.5, 2, 5 or 10 mg/kg every 2 weeks; 2 or 5 mg/kg administered every 2 weeks or every 3 weeks; or 200 mg every 3 weeks; patients in phase IB received 5 mg/kg every 3 weeks. Primary objectives were to assess tislelizumab’s safety/tolerability profile by adverse event (AE) monitoring and antitumor activity using RECIST V.1.1. PD-L1 expression was assessed retrospectively with the VENTANA PD-L1 (SP263) Assay.
Results Between May 2015 and October 2017, 451 patients (n=116, IA; n=335, IB) were enrolled. Fatigue (28%), nausea (25%) and decreased appetite (20%) were the most commonly reported AEs. Most AEs were grade 1–2 severity; anemia (4.9%) was the most common grade 3–4 AE. Treatment-related AEs led to discontinuation in 5.3% of patients. Grade 5 AEs were reported in 14 patients; 2 were considered related to tislelizumab. Pneumonitis (2%) and colitis (1%) were the most common serious tislelizumab-related AEs. As of May 2019, 18% of patients achieved a confirmed objective response in phase IA and 12% in phase IB; median follow-up duration was 13.6 and 7.6 months, respectively. Pharmacokinetics, safety and antitumor activity obtained from both phase IA and IB determined the tislelizumab recommended dose; ultimately, tislelizumab 200 mg intravenous every 3 weeks was the dose and schedule recommended to be taken into subsequent clinical trials.
Conclusions Tislelizumab monotherapy demonstrated an acceptable safety/tolerability profile. Durable responses were observed in heavily pretreated patients with advanced solid tumors, supporting the evaluation of tislelizumab 200 mg every 3 weeks, as monotherapy and in combination therapy, for the treatment of solid tumors and hematological malignancies.
Trial registration number NCT02407990.
- tumors
- oncology
- programmed cell death 1 receptor
- immunotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
The programmed cell death-1/programmed cell death ligand-1 (PD-1/PD-L1) axis plays a central role in suppressing antitumor immunity; dysregulation of the PD-1/PD-L1 axis can be used by cancer cells to evade the immune system.1 2 PD-L1 is an immune checkpoint protein that is often overexpressed on the surface of tumor and immune cells in the tumor microenvironment.3 4 PD-1, the cell receptor for PD-L1, is mainly expressed in activated T cells.5 An increase in PD-1 expression in the tumor microenvironment has been reported in many cancer types.6–8 Increased expression of PD-1 and PD-L1 is often associated with poor survival but may be predictive of anti-PD-1/PD-L1 antitumor activity.9–11
Tislelizumab is an investigational humanized IgG4 monoclonal antibody with high affinity and binding specificity for PD-1.12 Tislelizumab was engineered to minimize binding to FcγR on macrophages in order to limit antibody-dependent phagocytosis, a potential mechanism of resistance to anti-PD-1 therapy.1 Preclinical data suggest tislelizumab does not bind to FcγRI, whereas other anti-PD-1 antibodies bind to FcγRI in a manner consistent with human IgG4 antibody affinity.12 Furthermore, in cell-based assays, tislelizumab enhanced the functional activity of human T cells and pre-activated primary peripheral blood mononuclear cells.12
This first-in-human (FIH), dose-escalation/dose-expansion study assessed the safety/tolerability, pharmacology and clinical activity of tislelizumab in patients with advanced solid tumors. The primary objective was to evaluate the safety and tolerability of tislelizumab (phase IA), as well as the antitumor response (phase IB). Secondary end points included determining the maximum tolerated dose (MTD) and the optimal dose and treatment regimen. Confirmed objective response rate (ORR) to tislelizumab by PD-L1 status was an exploratory end point.
Methods
Study design and treatment administration
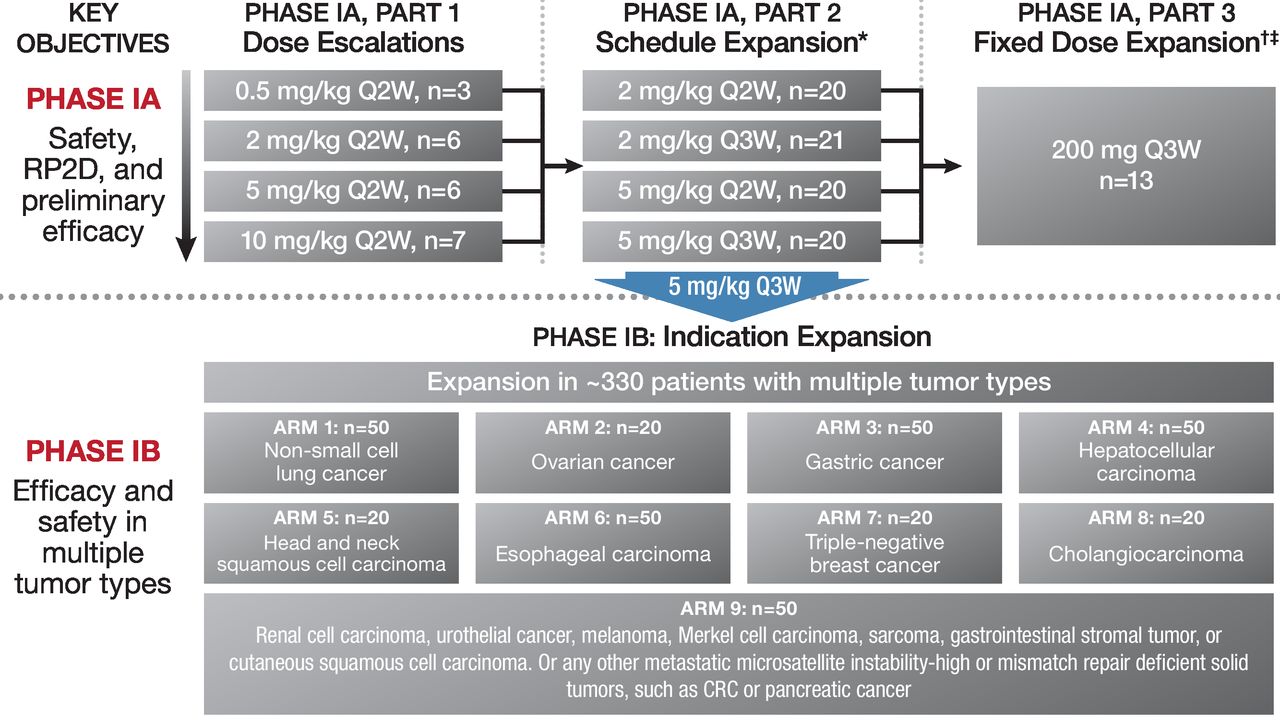
The design of this phase IA/IB study is detailed in figure 1. Phase IA comprised three parts. Part 1 was a dose-escalation/dose-finding phase that followed a modified 3+3 design. Four weight-based dose levels (0.5, 2, 5 and 10 mg/kg every 2 weeks) were assessed for dose-limiting toxicity (DLT). If a DLT was observed in the first three patients completing one cycle of treatment at a given dose level, three additional patients were enrolled. A DLT was defined as an adverse event (AE) occurring in the first 28-day cycle that met the predefined criteria based on grading per NCI-CTCAE V.4.03. DLT criteria included any grade ≥4 non-hematological toxicity or grade 4 laboratory anomaly irrespective of duration; any grade ≥3 immune-related AE (irAE), grade 3 tumor flare (ie, local pain, irritation or rash localized at sites of known/suspected tumor) for >7 days; grade 3 AEs irrespective of duration, except for laboratory abnormalities, diarrhea, nausea and vomiting; asymptomatic biochemical abnormalities that improve to grade ≤2 within 3 days of institution of supportive care and grade 2 ophthalmological toxicities. Hematological toxicities meeting DLT criteria include febrile neutropenia (defined as absolute neutrophil count <1000/mm3 with a single temperature of 38.3°C or a sustained temperature of 38°C for >1 hour); grade 4 thrombocytopenia, grade 4 anemia or grade 4 neutropenia lasting >7 days; grade 3 thrombocytopenia with bleeding and grade 3 neutropenic infection. No additional patients were required if a DLT was observed in one of six patients. Dose escalation continued until two out of six patients in each dose cohort experienced a DLT. If an MTD could not be established after evaluation of all planned doses, then the dose for subsequent studies would be determined based on safety and pharmacokinetic (PK) profile from aggregated data across the study. Part 2 of phase IA was a schedule expansion that evaluated tislelizumab at 2 and 5 mg/kg every 2 weeks and every 3 weeks. Part 3 of phase IA (fixed-dose expansion) evaluated tislelizumab 200 mg administered every 3 weeks. The phase IB component of this study was an expansion phase of tislelizumab at 5 mg/kg every 3 weeks in nine disease-specific cohorts. The initial administration of tislelizumab was administered via a 60 min infusion, if tolerated, the infusion was shortened to 30 min for all subsequent administrations. Dose modifications/delays were allowed; criteria for dose modifications are presented on page 2 of the online supplementary appendix.
Supplemental material

Study design. CRC, colorectal cancer; Q3W, every 3 weeks.
Approximately 120 patients were planned to be enrolled in phase IA. Twenty-four patients were needed for part 1, and ~20 patients were planned for the schedule-expansion and fixed-dose cohorts. Approximately 330 patients were planned to be enrolled in phase IB in the nine disease-specific cohorts.
Study population
Patients (aged ≥18 years) with previously treated histologically/cytologically confirmed advanced solid tumors with measurable disease (as defined by Response Evaluation Criteria in Solid Tumors (RECIST) V.1.1 criteria), an Eastern Cooperative Oncology Group performance status ≤1 and adequate organ function, were eligible for enrollment. Patients with previously treated brain metastases that was/were asymptomatic and radiographically stable and not requiring steroid medications for 4 weeks prior to enrollment were also eligible. Patients who received prior anti-PD-1/PD-L1 treatments; had a history of hypersensitivity reactions to monoclonal antibodies; a history of, or active, autoimmune disease; a history of interstitial lung disease or non-infectious pneumonitis, except for radiation-induced cases; a prior malignancy within the previous 2 years except for locally curable cancers that have apparently been cured (eg, basal or squamous cell skin cancer, superficial bladder cancer or carcinoma in situ of the cervix or breast cancer); patients who had prior liver transplant, allogeneic organ transplantation or bone marrow transplant and patients with a requirement for systemic treatment with corticosteroids (<10 mg daily prednisone equivalents) or other immunosuppressive medications within 2 weeks of study drug administration were excluded. PD-L1 expression was assessed retrospectively.
Study objectives and assessments
The primary objectives of this study were to assess the safety and tolerability of tislelizumab (phase IA) as well as antitumor response (phase IB). Secondary objectives included determining the MTD and the optimal dose and treatment regimen, characterization of the tislelizumab PK profile and assessment of host immunogenicity to tislelizumab; assessment of confirmed ORR by PD-L1 status was exploratory.
Safety/tolerability was assessed across the entire study and included continuous monitoring of AEs, as well as vital signs, physical examinations, electrocardiograms and laboratory investigations at specific study visits. AEs were categorized according to severity (NCI-CTCAE V.4.03), seriousness and relationship to the study treatment. Antitumor activity was assessed by CT imaging at screening, every 8 (every 2 weeks) or 9 (every 3 weeks) weeks for the first 12 months, and every 12 weeks thereafter. Tumor response was evaluated by RECIST V.1.1 criteria13 for all tumor types. Formalin-fixed, paraffin-embedded tumor tissue from biopsies and resections collected before tislelizumab treatment were used for PD-L1 analysis by immunohistochemistry with the VENTANA PD-L1 (SP263) Assay (Ventana Medical Systems, Tucson, Arizona, USA). Definition of PD-L1 positivity was different for individual tumor types based on previously reported cut-offs. The rationale of cut-off selection for each indication was based on published PD-L1 algorithms. PD-L1 expression was scored as: 1) the percentage of tumor cells (TC) with any membrane staining; the percentage of tumor-associated immune cells (IC) with staining at any intensity or 3) the percentage of tumor area occupied by tumor-associated IC (TC/IC) with staining at any intensity (see online supplementary appendix, p3).
Standard PK parameters (eg, area under the concentration-time curve (AUC), maximal plasma concentration (Cmax) and time to maximal plasma concentration) were estimated. As an indication of target engagement, PD-1 receptor occupancy assay was centrally assessed on samples taken from select patients enrolled in arm 2 in phase IB. Immunogenic responses to tislelizumab were assessed for antidrug antibody (ADA) formation. Blood samples for PK, receptor occupancy and immunogenicity assessment were collected as described below.
In part 1, cycles 1 and 4, predose (within 1 hour before administration) and postdose (0.5, 1.5, 6, 24, 72 (or 96), 168 and 336 hours after the end of infusion) blood samples were collected on days 1, 2, 4 (or 5), 8 and 15 to assess the PK profile of tislelizumab. For additional cycles, blood samples were collected on day 1 of each cycle at the following time points: predose (within 1 hour before administration) and postdose (end infusion to 30 min). In part 2, predose (within 60 min before the start of infusion) and postdose (within 30 min after the end of infusion) samples were collected at cycle 1 day 1 and day 15 (only for every 2 weeks), day 1 of cycle 2, every subsequent cycle in the first 12 months and approximately every 6 months thereafter. Additional PK samples were collected at cycle 1 day 4 (or 5), day 8, day 15 (only for every 3 weeks) and at the mandatory safety follow-up visit. In part 3, predose (trough, within 24 hours before the start of infusion) and postdose (within 30 min after the end of infusion) samples were collected on day 1 of cycle 1, cycle 2, cycle 3, cycle 5 and every two cycles in first 6 months, every four cycles in the next 6 months and approximately every 6 months thereafter. Additional PK samples were collected at day 2, day 4 (or 5), day 8 and day 15 of cycle 1 and cycle 5; a predose sample was also collected on cycle 5 day 22 (cycle 6 day 1). Further PK samples were collected at the mandatory safety follow-up visit.
For subjects in phase IA part 1, blood samples for immunogenic analysis were collected before starting the day 1 infusion every cycle in the first 6 months, every two cycles in the next 6 months, approximately every 6 months thereafter and at the mandatory safety follow-up visit. For subjects in phase IA part 2, blood samples were collected within 24 hours before starting the day 1 infusion every cycle in the first 6 months, every two cycles (every 2 weeks) or three cycles (every 3 weeks) in the next 6 months, approximately every 6 months thereafter and at the mandatory safety follow-up visit. For subjects in phase IA part 3, blood samples were collected before starting the day 1 infusion every two cycles in the first 6 months, every four cycles in the next 6 months, approximately every 6 months thereafter and at the mandatory safety follow-up visit. For subjects in phase IB, blood samples were collected before starting the day 1 infusion of cycle 1 and every two cycles in the first 6 months, every four cycles in the next 6 months, approximately every 6 months thereafter and at the mandatory safety follow-up visit. The immunogenicity results were summarized using descriptive statistics by the number and percentage of patients who developed detectable ADA. The incidence of positive ADA and neutralizing ADA is reported for patients in the ADA analysis set.
For select patients in arm 2 in phase IB, blood samples for receptor occupancy assessment were taken on cycle 1 day 1 (predose) and day 2 (approximately 24 hours after the first dose) as well as predose on day 1 of cycles 2 and 3.
Statistical analyses
SAS V.9.4 was used for statistical analyses. Descriptive statistics were used to summarize all study data. The Kaplan-Meier method estimated median time and 95% CI for progression-free survival (PFS) and overall survival (OS). The safety/tolerability profile and antitumor activity of tislelizumab were assessed in the safety analysis set (SAF; patients who received at least one dose of tislelizumab); DLTs were determined from the DLT set (patients who experienced a DLT during cycle 1 and received at least 90% of the planned doses during the DLT observation period (cycle 1)) and the PK profile was established from the PK analysis set (patients who had received at least the first dose of tislelizumab and provided PK samples as per protocol following first dosing).
Results
Population disposition, demographics and disease baseline characteristics
Between May 2015 and October 2017, 27 sites in Australia, Korea, New Zealand and Taiwan enrolled 451 patients (phase IA, n=116; phase IB, n=335) (see online supplementary figure S1). In phase IA part 1, 22 patients received one of four escalating doses of tislelizumab every 2 weeks (0.5 mg/kg (n=3), 2 mg/kg (n=6), 5 mg/kg (n=6) and 10 mg/kg (n=7)). In part 2, 81 patients were treated with tislelizumab (2 mg/kg every 2 weeks (n=20), 5 mg/kg every 2 weeks (n=20), 2 mg/kg every 3 weeks (n=21) and 5 mg/kg every 3 weeks (n=20)). In part 3, 13 patients were treated with fixed-dose tislelizumab (200 mg every 3 weeks). All 335 patients enrolled in phase IB received tislelizumab 5 mg/kg every 3 weeks.
Median age of all enrolled patients was 61.0 years (range: 18–81) with more male than female patients (table 1). This was a heavily pretreated population with >95% of patients having received ≥1 prior anticancer drug treatments, including platinum compounds (66.1%), pyrimidine analogs (50.6%), taxanes (38.8%), anthracyclines (25.9%) and protein kinase inhibitors (17.5%).
Patient demographics and baseline disease characteristics
The median treatment duration of tislelizumab was 2.8 months (range: 0.13–43.7); 65 patients (14.4%) received tislelizumab for ≥12 months. The median duration of study follow-up was 8.6 months (range: 0.1–40.8) in all patients: 13.6 months (range: 0.7, 40.8) in phase IA and 7.6 months (0.1–35.9) in phase IB. Of the 451 enrolled patients, 123 (27.3%) experienced ≥1 AE leading to dose modification (ie, treatment interruption or dose delay). Overall, infusion-related reactions (n=12; 2.7%) was the most common AE leading to dose modification (see online supplementary table S1). Treatment with tislelizumab was discontinued in 24 patients (5.3%) due to tislelizumab-related AEs, most commonly due to pneumonitis (n=8; 1.8%). One DLT was observed during dose escalation (grade 3 colitis in the 5 mg/kg every 2 weeks cohort); the maximum administered dose was 10 mg/kg every 2 weeks.
Pharmacokinetic profile of tislelizumab
The single-dose and multiple-dose PK profiles of tislelizumab were characterized from 109 patients from phase IA (table 2). Mean steady-state PK profiles for the every 2 weeks and every 3 weeks dosing regimens are presented in online supplementary figure S2. Drug exposure (Cmax and AUC0–14) increased in a dose-proportional manner from 0.5 to 10 mg/kg tislelizumab; no correlation was reported between clearance and baseline body weight, supporting fixed dosing of tislelizumab. At the 5 mg/kg dose, PD-1 receptor occupancy was >90%.
Summary of PK parameters of single-agent tislelizumab following single-administration and multiple-administration to patients with advanced tumors (PK analysis set)
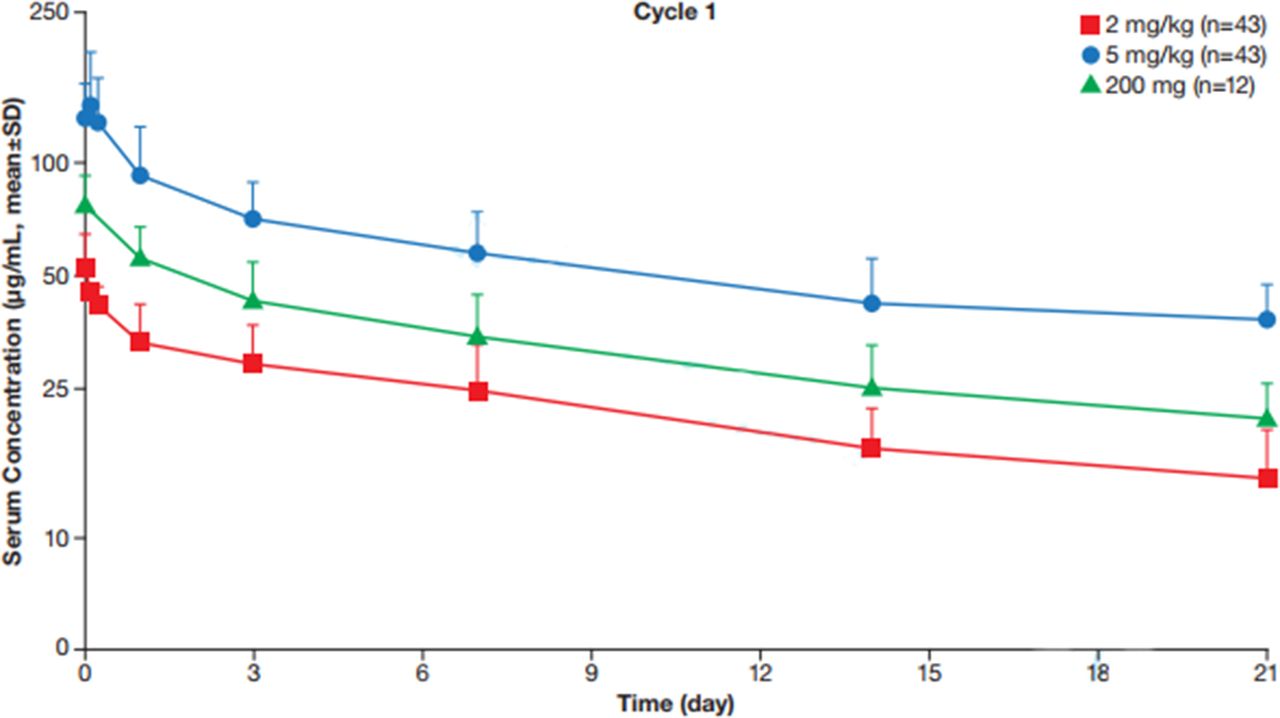
Data from the 200 mg every 3 weeks cohort demonstrated that tislelizumab concentrations after the first dose fell within 2 and 5 mg/kg dose range (figure 2). Tislelizumab demonstrated a 1.69-fold accumulation ratio, which was consistent with the estimated terminal half-life following tislelizumab 200 mg every 3 weeks (t1/2=16.8±5.5 days). Steady-state trough concentrations from patients with various tumor types in phase IB generally fell within a similar range of concentrations (see online supplementary figure S3), suggesting the tislelizumab PK profile is independent of tumor type.

{kind=link}
{kind=link}
Concentration of tislelizumab following a single 200 mg dose vs single 2 and 5 mg dose.
Tislelizumab safety/tolerability profile
Of the 451 enrolled patients, 436 (96.7%) experienced ≥1 AE. Safety/tolerability profiles were generally consistent across study phases as well as dose levels and schedules (see online supplementary tables S2–3). Commonly reported grade 1–2 AEs included fatigue, nausea and decreased appetite (table 3). Fatigue was the only AE considered related to treatment in ≥10% of the total population (13.1%), and was the most commonly reported tislelizumab-related AE (TRAE) across doses and schedules (see online supplementary tables S4 and S5). Potential irAEs occurred in 35.0% of patients; irAEs occurring in ≥5% of patients were rash (all terms; 14.6%), diarrhea (6.9%) and hypothyroidism (6.0%). The emergence of ADAs occurred in 18.7% of patients across both phases, with one patient (0.3%) testing positive for neutralizing antibodies.
Treatment-emergent adverse events* (SAF; n=451)
AEs of grade 3–4 severity, regardless of attribution, were reported in 198 patients (43.9%); these AEs were consistently reported across both phases. Grade 3–4 AEs occurring in ≥2% of patients included anemia (n=22; 4.9%), pneumonia (n=21; 4.7%) and hypokalemia (n=9; 2.0%). Thirty-nine patients (8.6%) experienced a grade 3–4 TRAE; increased alanine aminotransferase (n=6; 1.3%), pneumonitis (n=5; 1.1%), increased aspartate aminotransferase (n=4; 0.9%), colitis (n=3; 0.7%), diarrhea (n=3; 0.7%) and fatigue (n=3; 0.7%) were TRAEs of grade 3–4 occurring in >2 patients. Serious AEs considered related to tislelizumab were reported in 35 (7.8%) patients. Pneumonitis (n=8; 1.8%) and colitis (n=5; 1.1%) were the only serious AEs related to tislelizumab reported in ≥5 patients. Overall, 14 patients (3.1%) experienced a fatal AE. Two patients experienced fatal TRAEs (pneumonitis, n=1; fulminant hepatitis, n=1); both AEs were considered potentially immune-related and both cases were complicated by underlying disease. The pneumonitis event occurred in a patient with NSCLC with compromised pulmonary capacity at baseline; the hepatitis event occurred in a patient with hepatocellular carcinoma (HCC) with rapidly progressing disease.
Antitumor activity of tislelizumab
Confirmed responses were observed across phases (table 4) and dose/dosing schedules (see online supplementary table S6). Overall, the confirmed ORR was 13.3% (95% CI: 10.31 to 16.79) (table 4). While responses were primarily driven by patients who achieved partial response (PR; n=54; 12.0%), six patients achieved complete response (CR; 1.3%) in esophageal cancer, clear cell carcinoma of the endometrium, Merkel cell carcinoma, cutaneous squamous cell carcinoma, renal cell carcinoma and urothelial carcinoma (n=1 each). The overall disease control (CR+PR+stable disease (SD)) and clinical benefit (CR+PR+SD ≥24 weeks) rates were 44.6% and 25.9%, respectively. The median duration of response (DoR) was 16.0 months (95% CI: 11.1 to 25.6). Median DoR was 14.6 months (95% CI: 8.3 to not estimable (NE)) in phase IA and 16 months (95% CI: 9.2 to NE) in phase IB.
Clinical response as assessed by investigator by phase and overall (SAF)
When responses per tumor type were evaluated by PD-L1 status using previously published cut-offs, there was a trend of higher ORRs in PD-L1-positive (PD-L1+) patients with HCC, as well as gastric, esophageal, ovarian, non-small cell lung, colorectal, head and neck squamous cell and urothelial cancers (table 5). Notably, all patients in the HCC cohort who responded to tislelizumab were PD-L1+ (ORR=23.1%). In patients with renal carcinoma, ORR was identical between PD-L1+ and PD-L1− groups (33.3% vs 33.3%). No patients from the triple-negative breast cancer or cholangiocarcinoma cohorts responded to treatment, and thus the impact of PD-L1 status could not be assessed.
Response by PD-L1 expression status in tumor types with ≥10 patients
Estimated survival associated with tislelizumab treatment
As of the data cut-off of May 20, 2019, 58.3% of patients with an objective response had progressed or died. Median OS across the study was 10.3 months (95% CI: 8.5 to 11.6); median OS was 13.6 months (95% CI: 9.9 to 17.5) and 9.3 months (95% CI: 7.7 to 11.1) in phases IA and IB, respectively (see online supplementary figure S4A). Overall survival rates at 6 and 12 months were 67.3% (95% CI: 62.7 to 71.6) and 43.1% (95% CI: 38.3 to 47.8), respectively. Across the study, median PFS was 2.1 months (95% CI: 2.1 to 2.7), with a median PFS of 3.5 months (95% CI: 2.1 to 3.8) in phase IA and 2.1 months (95% CI: 2.1 to 2.2) in phase IB. The proportion of patients with PFS at 6 and 12 months was 29.5% (95% CI: 25.2 to 33.8) and 15.6% (95% CI: 12.3 to 19.2), respectively (see online supplementary figure S4B).
Discussion
This phase IA/IB study was conducted to characterize the safety/tolerability, PK and preliminary antitumor activity of tislelizumab monotherapy. Across the 451 patients enrolled from Australia, Korea, New Zealand and Taiwan, tislelizumab was generally well tolerated with a safety/tolerability profile consistent with other anti-PD-1 antibodies.10 14–19 The majority of tislelizumab-related AEs observed in this study were grade 1–2 in severity; 9% of patients had grade ≥3 AEs considered possibly related to tislelizumab. irAEs were reported in 35% of patients; rash was the only reported irAE with an incidence of >10%. Severe and fatal treatment-related AEs were uncommon. Clinical responses were observed across multiple solid tumors. These responses were durable, with a median duration of 16 months.
A key study objective was to determine the appropriate dosing for future clinical studies. Ultimately, a fixed 200 mg dose of tislelizumab every 3 weeks was selected for further evaluation based on comparable safety, PK and efficacy profiles between the 200 mg fixed-dose and the 2 and 5 mg/kg weight-based doses. One DLT was observed (grade 3 colitis) in the 5 mg/kg every 2 weeks cohort. No clear dose-related safety/tolerability concerns were observed between patients receiving 2 and 5 mg/kg every 2 weeks and every 3 weeks, and no unexpected tislelizumab-related AEs occurred in the 200 mg fixed-dose cohort compared with body weight-based cohorts, and the practicalities provided by fixed dosing. Additionally, tislelizumab clearance was found to be independent of body weight, ethnicity and sex; observed exposure of a 200 mg dose fell within the bounds of serum exposure observed after 2 and 5 mg/kg doses. Finally, ORRs in patients treated with tislelizumab 2 and 5 mg/kg every 2 weeks ranged between 10% and 15%, compared with a range of 15%–38% for patients treated at 2 and 5 mg/kg every 3 weeks. Of the evaluable patients treated at 200 mg every 3 weeks (n=13), three (23%) had achieved partial response, consistent with results seen with the weight-based cohorts.
As these data are from a non-randomized, open-label study, the data have some inherent limitations. Response and survival of tislelizumab were assessed in a heterogeneous group of patients with a variety of advanced solid tumors with different levels and patterns of PD-L1 expression. Patients with PD-L1− tumors achieved objective responses, suggesting that PD-L1 expression, as a single biomarker for response, may not be sufficient to identify those who are more likely to respond to checkpoint inhibition. It is important to note that key differences in PD-L1 antibody clones and various immunohistochemical platforms have raised questions about comparability. In terms of assessment of PD-L1 expression, specific tumor-type cutoffs vary depending on the antibody clone used. As different anti-PD-L1 antibody clones can have different levels of sensitivity and specificity, and can recognize different epitopes, using the same cut-off for different clones may cause results to be skewed, leading to an inappropriate treatment selection. Finally, the clinical impact of tislelizumab’s lack of binding to FcγRI compared with other anti-PD1 antibodies was difficult to determine in this phase IA/IB trial. Additional studies are needed to enhance our understanding of the tumor microenvironment and the effects of anti-PD-1 antibodies and further evaluation of tislelizumab in specific tumor types will provide a more accurate assessment of its antitumor activity.
In this study, single-agent tislelizumab demonstrated a favorable safety/tolerability profile and induced durable clinical responses in ≥10% of patients, which is promising in this heavily pretreated population, including a balanced population of both Caucasian and Asian patients. Furthermore, these responses were observed in patients with either PD-L1+ or PD-L1− tumors; additional studies are needed to further characterize the predictive value of PD-L1, and optimize a diagnostic/treatment algorithm for different tumor types. Based on the strength of these data, tislelizumab is currently being evaluated as monotherapy and in combination therapy for the treatment of solid tumors and hematological malignancies, including in registrational studies in esophageal, gastric, HCC, lung and urothelial cancers.
Acknowledgments
The authors would like to thank the investigative center study staff, the study patients and their families. The authors would like to thank Regina Switzer, PhD and Elizabeth Hermans, PhD (OPEN Health Medical Communications, Chicago, Illinois, USA) for providing writing and editorial assistance.
References
Footnotes
Contributors Concept and design: JD, BM, MM. Provision of study materials or patients: all authors. Collection and assembly of data: all authors. Data analysis and interpretation: JD, YZ, LL, VP. Manuscript writing and critical evaluation: all authors. Final approval of manuscript: all authors. Accountable for all aspects of the work: all authors.
Funding The study protocol was developed by BeiGene, Ltd. in collaboration with the study investigators. BeiGene, Ltd. was also involved in data collection, analysis and interpretation of results. Statistical analyses were performed by statisticians at BeiGene, Ltd.
Competing interests JD has served in a consulting or advisory role for Amgen, BeiGene, Bionomics, Eisai, Eli Lilly and Novartis, and their institution has received research funding from Bionomics, GlaxoSmithKline, Novartis and Roche. SD has received personal fees from Roche; C-CL has served in a consulting or advisory role for Novartis and has received travel, accommodations and expenses from BeiGene; BK has received grants and personal fees from AstraZeneca and MSD Oncology, grants from Ono Pharmaceutical and personal fees from Genexin; MJ's institution has received research funding from BeiGene, Merck Sharp & Dohme, Pfizer and Bristol-Myers Squibb; BM has served in a consultancy or advisory role for Novartis; LH has received grants from Astellas; MF has received honoraria from AstraZeneca, Merck Sharp & Dohme, Lilly, Takeda and Novartis, serves in a consulting or advisory role for AstraZeneca, Merck Sharp & Dohme and has received research funding from BeiGene and AstraZeneca. AH is employed by, has a leadership role in and has stock or other ownership in, Tasman Healthcare/Tasman Oncology Research. SS has received grants from Merck, Britsol-Meyers Squibb, Amgen, Endocyte, AstraZeneca and Roche; PB has received personal fees from Roche; LL, C-YW, JW, VP and YZ are employees of BeiGene; MM serves in a consulting or advisory role for AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Merck Sharp & Dohme, Novartis and Roche, and has received travel, accommodations and expenses from AstraZeneca, Bristol-Myers Squibb, Merck Sharp & Dohme and Roche.
Patient consent for publication Not required.
Ethics approval This study was conducted according to the ethical principles of the Declaration of Helsinki, Good Clinical Practice guidelines, the principles of informed consent and the requirements of the public registration of clinical trials. Written informed consent was obtained from each patient prior to screening. The protocol was approved by the institutional ethics committee and was monitored by a safety monitoring committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. On request, and subject to certain criteria, conditions and exceptions, BeiGene will provide access to individual de-identified participant data from BeiGene-sponsored global interventional clinical studies conducted for medicines (1) for indications that have been approved or (2) in programs that have been terminated. BeiGene will also consider requests for the protocol, data dictionary and statistical analysis plan. Data requests may be submitted to medicalinformation@beigene.com.