Article Text

Abstract
The rapid rise to fame of immuno-oncology (IO) drugs has generated unprecedented interest in the industry, patients and doctors, and has had a major impact in the treatment of most cancers. An interesting aspect in the clinical development of many IO agents is the increasing reliance on nonconventional trial design, including the so-called ‘master protocols’ that incorporate various adaptive features and often heavily rely on biomarkers to select patient populations most likely to benefit. These novel designs promise to maximize the clinical benefit that can be reaped from clinical research, but are not without costs. Their acceptance as solid evidence basis for use outside of the research context requires profound cultural changes by multiple stakeholders, including regulatory bodies, decision-makers, statisticians, researchers, doctors and, most importantly, patients. Here we review characteristics of recent and ongoing trials testing IO drugs with unconventional design, and we highlight trends and critical aspects.
- clinical trials as topic
- immunotherapy
- biomarkers
- tumor
- biostatistics
- drug therapy
- combination
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
The last decade has witnessed a rapid surge in the number of new immune-modulating drugs, which we termed next generation immune modulators (NGIMs), that have entered clinical development in immuno-oncology (IO).1 These drugs encompass a large and variegated group of novel chemical structures including not only monoclonal antibodies, but also conjugated antibodies, antibody-like molecules, small molecules, interleukins and even microbial-mimetic compounds to activate innate immune system, for example via toll-like receptors (TLRs).1 In this issue of the journal, many of these novel targets are reviewed in depth, including T-cell immunoglobulin and mucin-domain containing-3 (TIM-3), Neuropilin-1, T cell immunoreceptor with Ig and ITIM domains, CD39, NKG2A, lymphocyte-activation gene 3 (LAG3) and MerTK. Preliminary results for many of these drugs are promising, as in the case of agents targeting LAG3, TIM-3, natural killers and others.2–4 NGIMs are usually employed in combination with classic checkpoint inhibitors, such as monoclonal antibodies directed against cytotoxic T-lymphocyte antigen-4 (CTLA-4) or programmed of death-1 (PD-1) and its ligand (PD-L1), as they are often devoid of meaningful clinical activity when used alone.
Regulatory approval and commercialization of these agents require generally the demonstration of superior efficacy as measured through defined endpoints and with a statistical methodology that is deemed adequate for the clinical context. Traditionally, this was achieved by progressing through a three-phase route culminating with a randomized clinical trial that demonstrates some advantage (superior efficacy, inferior toxicity or cost) over what is considered standard of care at the time. However, some experts consider this canonical model of clinical experimentation less suited for developing novel immune modulators and especially their combinations.5–7 Reasons are manifold but we argue that they can be grouped in to three main categories: a perceived inadequacy of classical toxicity and efficacy endpoints, the increasing reliance on biomarkers, and the rapidity with which the treatment indications and disease classification are changing.
Here we will first review in further detail these three factors and how they influence trial design specifically in IO. We will then introduce the concept of master protocol and its defining features, highlighting how these novel designs can be better suited for NGIM development. We will finally provide examples of currently ongoing master protocols and briefly discuss pro et contra of existing trials.
Problems with classical endpoints
Toxicity
Immune-related adverse events (IrAEs) can develop with significant latency and have been systematically underestimated in trials, becoming evident only with late follow-up or pharmacovigilance studies, particularly for neurological, cardiologic and nephrological events.8–11 Thus, the classical reliance on specific and predictable events (dose-limiting toxicity, DLT) within the first cycle is commonly inadequate to define the correct dose to be tested for efficacy. Still, it would be unfeasible to wait for such long time to determine the correct dose to be tested for efficacy. Interest is rising in immediately integrating safety and early antitumor activity (clinical responses, pharmacodynamic and pharmacokinetic parameters, markers of immune activation) in composite endpoints such as the optimal biologically active dose,12 but this requires better understanding of drug activity in the preclinical phase, which will be facilitated by more sophisticated models of tumor–environment interaction such as organoids and humanized immunocompetent mice.13 Overall, as early DLTs and dose-dependent toxicities rarely occur with immunotherapy, the decision of the dose for expansion based merely on IrAEs is largely considered inappropriate.
Response criteria
IO agents typically can produce different patterns of response compared with standard systemic therapies,14 which cannot be properly evaluated by means of the conventional response evaluation criteria in solid tumors (RECIST V.1.1) criteria.15 The immune-RECIST (iRECIST) criteria have been developed in order to capture the unique pattern of response of the IO agents16 and take into account atypical patterns of responses such as pseudoprogressions and hyperprogressions.17 The distinction between ‘real’ progression and pseudo-progressions is an active field of research and renders even more important the identification of adequate and noninvasive biomarkers, especially those that are measured dynamically over time.18–21 However, it must be emphasized that pseudo-progression events are rare events, described in less than 5%–10% of patients receiving immune checkpoint inhibitors (ICIs).22 Notably, in clinical setting and trials, more than 50% of the patients receive treatment beyond progression, possibly underlining an overemphasis of the event and related expected benefit.23 Overall, around 4% of patients with melanoma who were treated with ICI seem to derive a benefit in terms of disease response beyond progression, suggesting that a selected subgroup truly have an advantage. The coexistence of different systems for response evaluation poses additional problems for interim decisions (particularly relevant for master protocols, as discussed below). While these criteria are currently adopted in the majority of clinical trials investigating IO agents, their harmonization and standardization also for regulatory purposes and drug approval represent an absolute priority. The recent American Society of Clinical Oncology - Society for Immunotherapy of Cancer (ASCO-SITC) statement on trial reporting in immuno-oncology suggests to report responses according to standard RECIST criteria in parallel with the IO-specific response criteria like iRECIST in order to provide comparisons with prior clinical trials, ensuring consistency in the estimation of the benefit.6
Efficacy endpoints
Although some IO agents received approval on the basis of response rate in the early phases of clinical development,24 25 short-term efficacy endpoints like response rate and progression-free survival (PFS) are increasingly called into question. For instance, a meta-analysis on PD-1 inhibitors failed to demonstrate a correlation between PFS and Overall Survival (OS), suggesting an imperfect role of PFS as decisional parameter ad interim.26 Moreover, an exploratory analysis from another work suggested a potential surrogacy of 6 month PFS rate for OS, while overall response rate (ORR) did not seem a robust surrogate endpoint.27 28 Additionally, reporting HRs might not properly represent the treatment benefit due to nonproportional hazards of survival and long-term survival with IO agents.29 30 Other statistical models, including restricted mean survival time analyses and Cox regression models with arm–time interaction coefficients, have been proposed as more appropriate for evaluating survival outcomes in IO trials.30 More recently, a Food and Drug (FDA) analysis of 10 randomized controlled enrolling patients with melanoma treated with ICI, assessed the relation between the depth of response and survival.31 At 24 months, 92% of the patients reaching a maximal tumor shrinkage of 75% or more were alive, suggesting a role of profound responders in the identification of patients deriving a greatest benefit. Finally, patient-reported outcomes are likely to gain increasing importance given the peculiar trade-off between toxicity and efficacy of IO agents, but still suffer from poor standardization as a recent review of IO studies reported.32
Biomarkers and the trade-off between sample size and specificity
Experience with targeted agents has shown that early implementation of biomarker-based stratification can enhance the clinical benefit and the likelihood of successful drug approval.33–35 Translational studies with first-generation ICIs against PD-(L)1/CTLA-4 have revealed that also in the IO context, biological parameters can strongly influence efficacy or predict toxicity.36 These factors include mutational signatures and specific mutations obtainable from DNA sequencing of the tumor sample or circulating free DNA,37–42 transcriptional signatures,43–45 circulating factors (neutrophil to lymphocytes ratio, LDH and others46 47), cytokines,48 intratumoral expression of immune markers (PD-L1, infiltrating lymphocytes, myeloid cell infiltration and others49 50), early changes in peripheral immune cells21 or more sophisticated immune parameters like T-cell receptor repertoire changes,51 52 radiomic patterns,18–20 and the gut microbiota.53–55
Enrichment for biomarker-positive subgroups is a logical approach to achieve large effect size in clinical trials. The overall viability of the chosen biomarker can inform trial design in key ways. If a biomarker is easily implemented in the workflow and/or has strong evidence for a good negative or positive predictive values and/or has sizeable prevalence, it may be advisable to implement it early and solidly in the design, possibly in the inclusion criteria, to immediately define the treatment space for the investigational drug and limit costs. If these conditions do not hold, and especially if biomarker negativity would not fully justify exclusion from treatment, it may be advisable to only include it as an exploratory endpoint or use for stratification. The decision to restrict clinical development to biomarker-selected populations may be key to delimiting the clinical space in which that drug will operate, as very well exemplified by the different results (and consequent FDA approvals) of nivolumab (tested on patients unselected for PD-L1 expression) and pembrolizumab (tested on patients expressing PD-L1 on ≥1% cancer cells) in first-line non-small cell lung cancer (NSCLC).56 57 Three main factors affect the clinical viability of a biomarker: performance (positive/negative predictive value and sensitivity), feasibility (technical, economical, ethical) and prevalence.
Performance
For many biomarkers currently tested in IO trials, predictive value is still speculative. More complex is the discussion over biomarkers for NGIMs. Some of these drugs seem to perform better when their molecular target is highly expressed (eg, LAG3) but given their indirect mechanism of action on the tumor, it is not surprising that local target expression correlates sub-optimally with efficacy. Probably, a combinatorial approach with probabilistic algorithms integrating multiple data dimensions will be required,58 but this of course will require a trade-off with feasibility, as multiple data will require multiple laboratory tests. Still poorly defined are early markers of immune toxicity, an issue of great importance given the peculiar toxicity profile of these drugs48
Feasibility
Biomarker feasibility can be affected by several factors. Technical complexity varies significantly from simple but poorly predictive tests to complex transcriptional signatures or dynamically assessed. Importantly, biomarkers require biological specimens, which can not always be collected, especially in specific cases like dynamic biomarkers requiring serial sampling. Sampling issues may be at least partially overcome by the growing capabilities of liquid biopsies, which have for instance shown to be an adequate surrogate of conventional biopsies for some biomarkers.39 59
Prevalence
Targeted therapies offer numerous examples of drugs with exceptional efficacy in exceptionally rare populations (eg, NTRK-mutated or RET-mutated). Given the relatively low rate of long-term responders in IO-treated patients,60 61 it is not unlikely that responses could be predicted by single or combined biomarkers, each with extremely low prevalence. This may be of particular importance in multi-arm trials where unequal biomarker co-occurrence can lead to unbalanced arms.62
Clinical research in a rapidly changing landscape
The advent of targeted and immune therapies has caused rapid and dramatic changes in the criteria for disease classification and treatment. The gradual shift towards a histology-agnostic, biomarker-driven disease classification, best exemplified by the approval of anti-PD1 agents for mismatch repair-deficient cancers irrespective of the tissue of origin,63 has created novel clinical entities in which preliminary data to formulate testable hypotheses (eg, historical controls) are not or are poorly available. This can have significant implications on the design of clinical trials, when the pre-test assumptions are established. Also, the increasing adoption of IO in the early settings64–66 creates a growing cohort of patients with a history of treatment with ICI. How prior failure to ICIs can influence subsequent response to NGIMs is an open question. On one hand, subsequent success rate may be predicted to be lower if the patient has already progressed to an initial IO line. On the other hand, prior failure may itself be due to the induced upregulation of additional checkpoints that may be specifically targeted in the second line. Besides, IO sequencing may be relevant for toxicity. Unfortunately, preclinical models are mostly inadequate to study these issues, as IO sequencing is difficult to model.
Master protocols as an overarching framework for testing multiple hypotheses
The factors described above critically undermine the canonical model in specific ways. In phase 1, where the aim of the investigation is to establish safety and a recommended phase 2 dose, the opportunity to increase dosage is classically evaluated based on DLTs identified within the first cycle. If however toxicities are manifested with a delayed kinetics and with little dose-dependency, the discriminatory ability of classical ‘3+3’ designs is severely impaired and may lead to significant underestimation of toxicity rates and difficulty in identifying the optimal dose.60 In phase 2, when preliminary efficacy should be evaluated, three factors are of major importance: (1) the lack of a direct relationship between short-term (response rate and PFS) and long-term (overall survival) endpoints; (2) the increasing difficulty in identifying an appropriate historical control to gage efficacy, given the rapidly changing treatment landscape; and (3) the need for biomarkers, which complicates screening procedures and further restricts inclusion criteria, making it more difficult to subsequently extend the results to wider patient populations. Finally, randomized phase 2 and 3 trials become hard to carry out because (1) biomarker stratification restricts the fraction of suitable patients, increasing dramatically the number of patients needed to screen; and (2) the choice of an adequate control arm becomes difficult and sometimes ethically questionable, as the standard of care may be rapidly changing and/or radically different in terms of expected outcome and quality of life.
Master protocols have arisen as a way to overcome these issues and provide statistical and organizational tools to test multiple hypotheses efficiently. Master protocols encompass trials designed to evaluate single drugs across multiple populations (‘basket trials’), multiple drugs on a single population (‘umbrella trials’), or complex multi-arm, multi-stage designs that evaluate multiple treatments simultaneously, also referred to as ‘platform trials’.67 In a certain sense, basket and umbrella trials can be considered special cases of master protocols/platform trials where number of tested drugs or populations is equal to 1. Populations can be defined by classical anatomical/histological parameters or by more sophisticated biomarkers. Examples of trials with multiple biomarker-defined populations include the I-SPY2 trial in the neoadjuvant setting for breast cancer, which includes gene expression signatures among the parameters used to stratify and adapt randomization,68 or the Molecular Analysis for Therapy Choice of the National Cancer Institute (NCI-MATCH trial),69 that uses a histology agnostic approach based on actionable mutations.
The use of a single infrastructure to evaluate multiple drugs and populations has the potential for efficient and accelerated drug development, reducing the exposure of patients to less effective or unacceptably toxic interventions.70 In trials with multiple biomarker-defined arms, simultaneously testing for all biomarkers within the same framework minimizes the number of patients needed to screen and thus maximizes the chances of each patient to receive treatment within the trial.62 Furthermore, if the same drug or combination is used across multiple biomarker-determined groups with staggered enrolment, toxicity events can still be cumulated across arms, obtaining a more faithful description of the safety profile. On the other hand, the statistical design becomes extremely complex and relies critically on interim analyses when efficacy/futility within and across treatment arms is determined; this intermediate evidence is used to decide whether to continue, stop early or add new treatment options. Statistical strategies to modify in medias res the design of a clinical trial are collectively called ‘adaptive designs’. For the FDA, the defining property of an adaptive design is the possibility to ‘adjust to information that was not available when the trial began’. In some cases, new evidence from other trials or from the same trial can be used to adjust the design of the platform, in opposition to the traditional trial design, where all the parameters are prespecified.71 The type of information that can be used to guide trial conduct can in principle be of heterogeneous nature and include efficacy, toxicity or a composite of the two, in the overall population or in biomarker-defined groups. Specific designs have been devised to continuously reassess efficacy and/or toxicity and adapt recruitment accordingly; these continuous reassessment designs may be better suited to adequately capture efficacy and toxicity of IO agents, with their peculiar, delayed kinetics.12 72–78 Master protocols do not strictly require adaptive designs, but in general a certain degree of design flexibility is desirable to accommodate the heterogeneity associated with multiple patient populations. A peculiar feature of platform trials is the possibility for patients to be each counted multiple times within the same trial, since they are assessed for eligibility once and can potentially remain in treatment within multiple arms of the same trial. However, paradoxically this may limit the possibility for some therapies to be proven effective, especially if the multiple agents tested have common mechanisms of action and, consequently, common mechanisms of resistance, a plausible scenario for IO combinations.
Regulatory bodies are increasingly aware of the shifting paradigms in clinical research but also of the need to provide clear frameworks for the road to drug approval. Thus, FDA launched initiatives to help streamline alternative regulatory pathways, like the breakthrough designation pathway. These novel regulatory pathways have been exploited frequently in recent years, representing 43% of the novel FDA approvals between 2012 and 2017.79 However, these expedite tracks are mostly still based on conventional trial design, with either short-term endpoints or nonrandomized but sufficiently large trials. Recognizing the lack of regulatory approvals based on complex innovative trial designs (CIDs), the FDA recently launched the CID pilot project.80 CIDs are defined loosely to include multiple novel design techniques incorporating adaptive and Bayesian statistics, innovative use of external or historical control data, formal incorporation of prior knowledge into the study design (in this context, biomarkers and/or treatment history) and adaptive designs allowing prespecified modifications to the designs as evidence accumulates.80
Some forms of adaptive trials (eg, Simon's two-stage or its derivations81) have entered widespread application, but are not yet considered sufficient evidence for regulatory approval. Many authors have raised concerns over the actual utility or even ethical soundness of adaptive designs.82 83 Most obviously, unplanned variations in sample size and inclusion criteria affect the possibility to interpret, validate and generalize results. Furthermore, although part of the rationale for adaptive designs is to reduce the number of patients on trial, the lack of fixed sample sizes may actually result in larger, rather than smaller sample sizes.82 83
Mapping of recent or ongoing master protocols in IO
To map the current trends in trial designs for NGIMs, we reviewed clinical trials using a loose definition of an IO-centered master protocol combining different drugs. We included phase 1 and 2 interventional clinical trials in our analysis in which:
Three or more drugs were tested.
Two or more combinations of different agents were considered, including at least one IO agent.
t least two different immunotherapy drugs were included, of which at least one was ICI.
We excluded trials that provided for one or more arms in which chemotherapy was administered alone, with the exception of studies considering more than two arms. This choice was made to exclude all trials that compared chemotherapy to a unique combination of the same chemotherapy plus an IO agent. Trials including adoptive therapies and vaccines were not considered. We excluded also trials that enrolled only hematological cancers.
This results in 119 trials being considered for review(figure 1). Most are phase 1 (n=51) or phase 1/2 (n=41) and are nonrandomized. In online supplementary table 1, we categorized these trials based on setting (mostly advanced and metastatic), treatment history (prior IO allowed, required or not allowed); the use of biomarkers (prospectively included), and the timing of their assessment (baseline vs pretreatment or posttreatment), and scope (for inclusion, stratification or exploratory); the presence of adaptive features; if the combination strategy included IO drugs only or also other modalities like chemotherapy, targeted agents, or radiation therapy. Specific methodology is included in the online supplementary file 1.
Supplemental material
Supplemental material

PRISMA diagram describing criteria chosen for literature review. PRISMA, Preferred Reporting Items for Systematic Reviews and Meta-Analyses
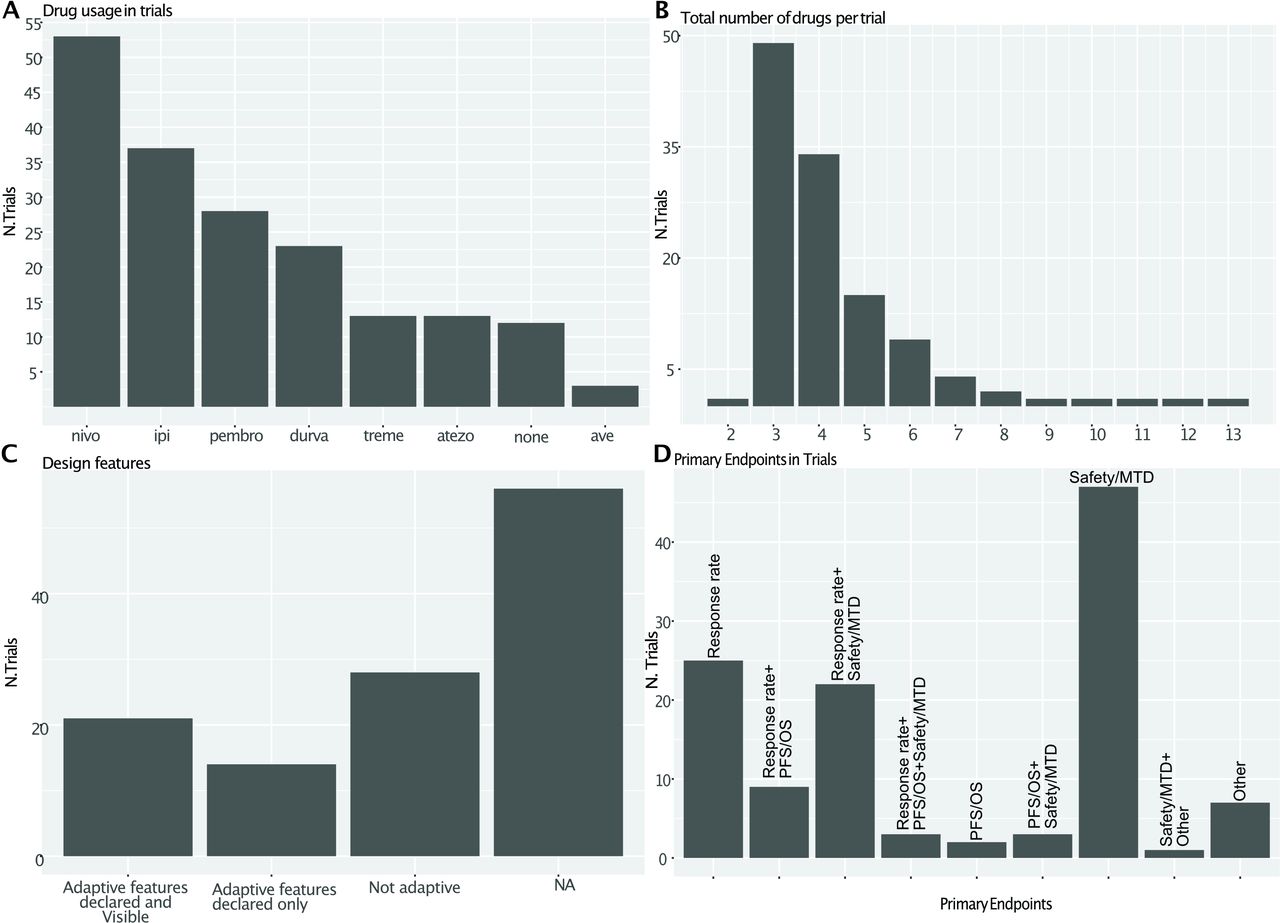
The analysis shows a dramatic rise in the total number of trials and patients enrolled in IO-centered master protocols (figure 2A), the increasing reliance on biomarkers (figure 2B) and the allowance or even requirement for prior IO from 2016 (figure 2C). Most likely these trends were pushed by the approval of anti-PD1 agents nivolumab and pembrolizumab in melanoma and NSCLC, occurred in late 2014/early 2015. These agents represent the most common backbone for combination therapies, together with ipilimumab and durvalumab (figure 3A).
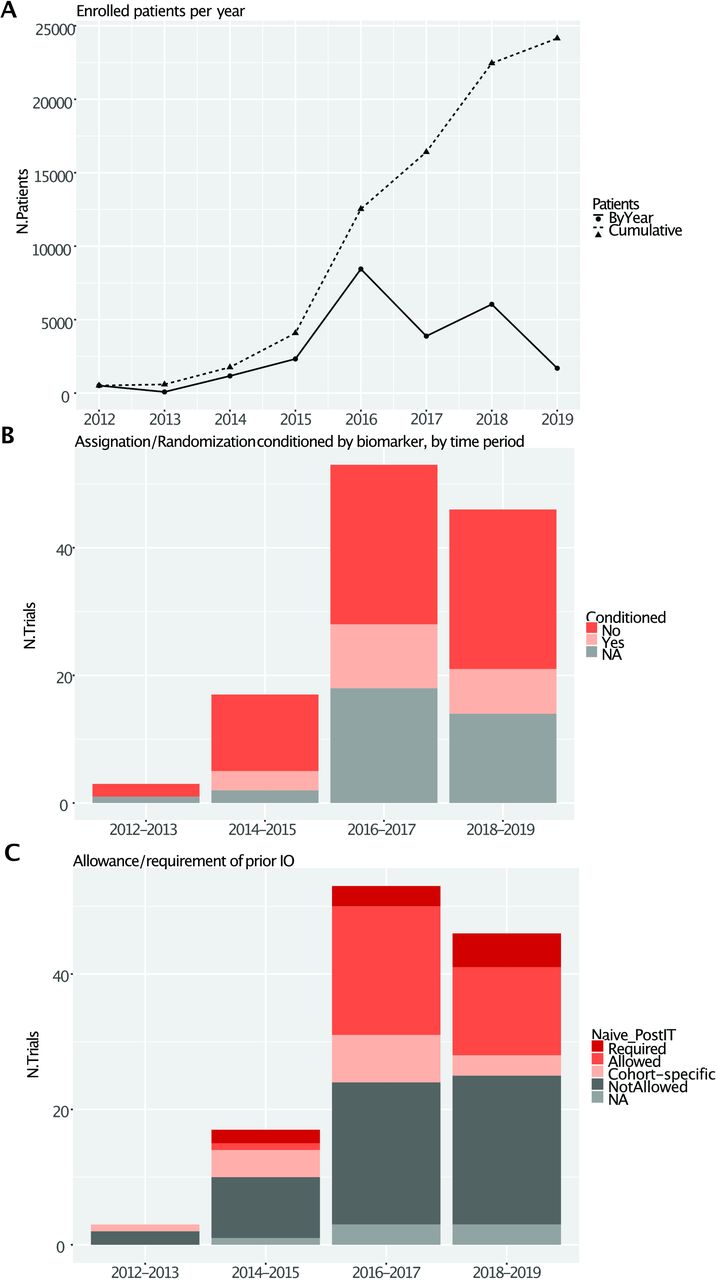
General characteristics of papers included in the review. (A) Yearly and cumulative planned patient enrolment over time. (B) Trial classification by biomarker use by biennia (C) Allowance/requirement of prior IO by biennia.

{kind=link}
{kind=link}
{kind=link}
Specific characteristics of papers included in the review. (A) Drug inclusion. (B) Distribution of total number of drugs per trial. (C) Inclusion of adaptove features in the design. (D) Primary endpoints: is it a crossover when the patient moves in another arm?. MTD = Maximum Tolerated Dose
The number of agents being tested in each trial is most commonly three, but some trials test even more than 10 drugs at the same time (figure 3B).
Statistical design of the trials
Adaptive features in the design of most trials are difficult to evaluate as complete statistical design is not routinely reported in public databases, a practice that will probably change in light of the more positive attitude of regulatory bodies toward adaptive designs, demanding more transparency in the conduct of clinical trials and disclosure of data (figure 3C). In our study, 47% (56/119) of the studies did not publicly disclose sufficient elements of their statistical design, 23% (28/119) use nonadaptive designs, 12% (14/119) included some adaptive features that were not disclosed, and only 18% (21/119) reported verifiable adaptive features (mostly Simon's two-stage design). Unsurprisingly, long-term outcomes like overall survival very rarely constitute the primary endpoint (figure 3D), perhaps related to the prominence of earlier phase of trials.
Specific examples
The most paradigmatic examples of ongoing IO master protocols are those centered around single pharmaceutical companies' and their compounds, mainly based on anti-PD-L1 drugs: BMS' FRACTION (nivolumab)84 Roche's MORPHEUS (atezolizumab)85 and Merck/Pfizer's Javelin Medley (avelumab).86
FRACTION
FRACTION is the only platform whose design is published and described in detail. FRACTION incorporates all elements discussed above: multiple NGIMs (including anti-CTLA4, LAG3, IDO1, cytokines and others) are tested in combination with a nivolumab backbone, in multiple patient populations defined by diseases (lung, gastric and renal cell cancer), biomarkers and prior treatment history. Several adaptive features are incorporated, including the possibility for internal crossover and premature arm termination or expansion. Interim decisions are not solely based on short-term efficacy but also include safety and pharmacodynamics. Biomarkers are investigated both as inclusion criteria (PD-L1) and as exploratory endpoints (gene expression signatures, mutational burden, immune infiltration, oncogenic mutations, germline makeup and peripheral markers). An interesting feature of FRACTION is its ‘modular’ architecture: alongside a ‘backbone’ master protocol defining general rationale and common inclusion criteria, study conduct for each specific disease is regulated by subprotocols, an agile structure that allows the introduction of modifications by amending the subprotocol without perturbing the master protocol.
MORPHEUS
MORPHEUS is a platform with disease-oriented multiple arms of study. Multiple combinations of atezolizumab and other IO and non-IO agents or chemotherapy are assessed for NSCLC, triple-negative breast cancer, colorectal, gastric and pancreatic cancer and transitional-cell malignancies of the urinary tract. The published primary endpoint of the arms to assess the activity of the combination regimens is ORR, across the studies. Secondary endpoints explore longer-term outcomes, including PFS and OS.85 87
Javelin Medley
Javelin Medley was designed as an open-label, phase 1b/2 study evaluating the combination of avelumab with multiple combinations of IO and other agents. The goal of this study was to determine safety, tolerability, and clinical activity of these combination therapies, based on endpoints of safety (DLT) and activity (ORR).86 Avelumab is combined with multiple agents across several immunecheckpoints anti-4 IBB (utomilumab), M-CSF (PD 0360324), agonist of OX-40 (PF-04518600) and TLR9 (CMP-001), in doublets or triplets.
Advantages and disadvantages of master protocols in IO
Advantages of complex, multilayered designs like FRACTION's are many. The amount of information that can be squeezed out from a single study increases dramatically, including longitudinal information after crossover that is not typically collected in traditional trials and that may inform subsequent strategies for treatment sequencing. The administrative work is minimized compared with what would be needed if each hypothesis was tested independently. Perhaps most importantly, likelihood of exposing each patient to the best therapy is increased, both because clearly ineffective or toxic arms are promptly terminated and because crossover allows to receive multiple treatments that also take into account initial responses.
However, some important prices are to be paid. Statistical strength is sacrificed, because of small arm size, frequent crossover, and poor reproducibility as decision criteria are variegated; thus, results are always highly exploratory, and the risk of an inconclusive or misleading trial is high,83 88 as experienced in the field of targeted therapy where such novel designs have been explored first. For instance, I-SPY 2 predicted 88% probability of success in a phase 3 trial for neoadjuvant carboplatin and veliparib, but this was not confirmed in the randomized controlled phase 3 clinical trial BrighTNess.89
Conclusions
As we have shown above, reliance on nonconventional, complex trial designs, especially at the intermediate stages of drug development, is increasing noticeably. Such designs have been called into question from a methodological and even ethical standpoint.83 88 An argument can be made, however, that judging the success of a trial solely on the basis of a primary survival endpoint can be reductive, especially for disease eligible to multiple lines of therapies as platinum-sensitive ovarian cancer or endocrine-positive breast cancer, in a landscape with ever-multiplying treatment options and a pressing need of criteria for adequate patient selection. Finally, increasing patient awareness raises pressure from the final users to gain access to therapies. Time will tell if the recent opening to nonconventional designs by FDA will smoothen or ignite the well-known ‘access vs evidence’ regulator's dilemma.82
References
Footnotes
Twitter @matsadoc
Contributors LM and GC conceived the study. LM, SM, AM, DT, GT, PP and GC collected and analyzed data. LM and GT performed statistical analysis. All authors contributed to writing. All authors read and approved the final manuscript.
Funding Research in LM's lab is funded by the EU ERAPerMed JTC2018 - PEVOSQ-data project.
Competing interests GC received honoraria for speaker, consultancy or advisory role from Roche, Pfizer, Novartis, Seattle Genetics, Lilly, Ellipses Pharma, Foundation Medicine, and Samsung.
Patient consent for publication Not required.
Provenance and peer review Commissioned; externally peer reviewed.