Article Text

Abstract
Background Immunological checkpoint blockade is effective in treating various malignancies. Identifying predictive biomarkers to assist patient selection for immunotherapy has become a priority in both clinical and research settings.
Methods Mutations in patients who responded to immunotherapy were identified through next-generation sequencing. Relationships among protein kinase, DNA-activated, catalytic polypeptide (PRKDC) mutations, mutation load and microsatellite instability (MSI) were analyzed using datasets from The Cancer Genome Atlas. These relationships were validated by conducting an in vitro study and by using tissue samples from 34 patients with gastric cancer. The CT26 animal model was used to evaluate the role of PRKDC as a predictive biomarker and the efficacy of the DNA-PK inhibitor.
Results From the published literature, we found that among patients whose tumors harbored PRKDC mutations, 75%, 53.8%, and 50% of those with lung cancer, melanoma, and renal cell carcinoma, respectively, responded to immunotherapy. Most of these mutations were truncating and located in functional domains or in a destabilizing PRKDC protein structure. Additional analysis showed that a PRKDC mutation was significantly associated with a high mutation load in cervical cancer, colon adenocarcinoma, head and neck squamous cell carcinoma, lung adenocarcinoma, gastric adenocarcinoma and endometrial cancer. Patients with gastric cancer or colon cancer harboring PRKDC mutations were also highly associated with MSI-high status. Finally, we found that knockout PRKDC or DNA-PK inhibitor (PRKDC encodes the catalytic subunit of DNA-dependent protein kinase) enhanced the efficacy of the anti-programmed cell death protein one pathway monoclonal antibody in the CT26 animal model.
Conclusions PRKDC is not only a predictive biomarker but also a drug target for immune checkpoint inhibitors.
- biomarkers, tumor
- gastrointestinal neoplasms
- genome instability
- immunotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Immunological checkpoint blockade using antibodies that target cytotoxic T-lymphocyte-associated protein 4 (CTLA4) or the programmed cell death protein one pathway (PD-1/PD-L1) may be effective in treating various malignancies.1–7 However, treatment response rates for these immunological checkpoint blockades are relatively low, regardless of the cancer type. Thus, patient selection through the identification of predictive biomarkers has become a priority for administering immunotherapy in both clinical and research settings. Currently, several potential biomarkers have been identified, including mutation load,8 microsatellite instability (MSI) status,9 neoepitope load,8 10 PD-L1 expression,11 CD8+ T-cell density,12 13 and interferon-γ gene signature.13 Among these, MSI status has been identified as an effective indicator of patients who might benefit from immunotherapy. However, microsatellite instability—high (MSI-H) status is observed relatively infrequently (4%) in metastatic gastrointestinal cancers,14 leading to limited use of MSI as a predictive biomarker in clinical practice.
Protein kinase, DNA-activated, catalytic polypeptide (PRKDC) encodes a 465 kDa catalytic subunit of DNA-dependent protein kinase (DNA-PKcs)15 16 that plays a pivotal role in the maintenance of genomic stability17 and is a critical component of DNA double-strand break repair and recombination.18 DNA repair genes may serve as potential biomarkers of malignancies or therapeutic targets.19 The ‘mutator phenotype’ theory states that early mutations in critical DNA repair genes lead to genomic instability, subsequent hypermutability, and a high mutation rate in cancer.20 This theory is supported by reports on mismatch repair (MMR), where MMR-deficient colorectal cancers harbored 10–100 times as many somatic mutations compared with MMR-proficient colorectal cancers.21 Loss of PRKDC expression is associated with impaired DNA repair.18 However, whether the role of other DNA repair genes, including PRKDC, in cancers is similar to that of alterations in MMR genes remains unclear. Therefore, in this study, we explored the role of PRKDC mutations in the era of immunotherapy.
Methods
Whole-exome and targeted sequencing
For whole-exome sequencing, genomic DNA was isolated from formalin-fixed paraffin-embedded (FFPE) tissue and peripheral blood samples by using a QIAamp DNA FFPE tissue kit. DNA was quantified using the Quant-iT dsDNA assay (Advanced Analytical Technologies) and quantitative real-time PCR. A library was constructed using the Ion AmpliSeq Exome RDY primer pool. Whole-exome sequencing was performed on the Ion Proton sequencer, with an average read depth of 200×.
For targeted sequencing, the extracted DNA was amplified using four pools of primer pairs (Ion AmpliSeq Comprehensive Cancer Panel, Life Technologies) targeting the coding exons of analyzed genes. Amplicons were ligated with barcoded adaptors by using the Ion AmpliSeq library kit (Life Technologies). The barcoded libraries were subsequently conjugated with sequencing beads through emulsion PCR and enriched using Ion Chef system (Life Technologies) according to the Ion PI IC 200 protocol (Life Technologies). Targeted sequencing was performed on the Ion Proton, with an average read depth of 1000×.
Resulting reads were mapped to the hg19 reference genome by using the Ion Torrent Suite V.4.4. Variants were identified using a Torrent Variant Caller Plug-in V.4.4 and were annotated with Variant Effect Predictor V. 78. Common variants (minor allele frequency (MAF) ≥1%) in the single nucleotide polymorphism (dbSNP) database (build 138) or 1000 Genome project (phase I), but not in the Catalog of Somatic Mutations in Cancer (COSMIC) database, were filtered out. Variants were further filtered to remove those with low frequencies (<5%), single-nucleotide polymorphisms, germline mutations, and synonymous mutations. Only somatic nonsynonymous variants were retained and analyzed.
Data collection and analysis from the published literature and public domains
Mutation and response data from patients treated by immunotherapy were obtained from the published literature.8 22–26 The Cancer Genome Atlas (TCGA) data, including DNA mutation, MSI status, and mRNA sequences, were downloaded from Broad GDAC Firehose/Firebrowse (http://firebrowse.org/). Variant annotation from TCGA data was obtained using cBioPortal.27 The mutation lollipop diagram was drawn using cBioPortal Mutation Mapper. The functional impact of PRKDC variants was predicted using Grantham,28 PolyPhen,29 and SIFT30 with default parameters. The mutation load for a patient is defined as the total number of non-synonymous mutations. For expression analysis and estimated cell proportion analysis, patients were grouped as those harboring PRKDC mutations, those not harboring PRKDC mutations but with MSI-H, and those harboring PRKDC mutations but with microsatellite stable (MSS) or microsatellite instability—low (MSI-L). mRNA expression was based on RSEM-normalized RNA-seq data and then log transformed. Cell proportions that may contribute to mRNA expression were estimated using CIBERSORT31; only data with statistical significance were considered (p≤0.05).
Patient demographics
Patients with gastric cancer who underwent curative resection between May 1988 and October 2003 were enrolled in this study. Any patient with a pathological diagnosis other than that of adenocarcinoma was excluded. A total of 34 patients with gastric cancer were enrolled.
Microsatellite analysis
DNA was extracted through PCR for D5S345, D2S123, BAT25, BAT26, and D17S250 and detected using an ABI 3730 automated sequencer (Applied Biosystems, Foster City, California, USA), as described previously.32
Microsatellite analysis by MSIseq tool
The MSI status (MSI-H or MSS) for each sample in the TCGA MC3 dataset was predicted by the MSIseq tool ADDIN EN.CITE.33 In order to verify the prediction accuracy of MSIseq, we first tested it on the five cancer types with MSI status in the clinical data from TCGA, that is, colon adenocarcinoma (COAD), rectum adenocarcinoma (READ), stomach adenocarcinoma (STAD), uterine corpus endometrial carcinoma (UCEC), and uterine carcinosarcoma (UCS). The results indicated that MSIseq can assess the MSI status with high accuracy (98.5%, 97.3%, 98.6%, 94.9%, and 96.5% in COAD, READ, STAD, UCEC, and UCS, respectively). Therefore, we reasoned that samples lacking MSI status specified in their clinical data can also be predicted of their MSI-H or MSS status using MSIseq with high confidence. Furthermore, since the mutation status of the PRKDC can be classified as mutated or wild type (WT), we could then analyze the association between PRKDC mutation status and MSI status by Fisher’s exact test for each cancer type.
Functional consequences of PRKDC mutations
To calculate the change in the free energy of mutations, the FoldX V.3.0 algorithm was applied.34 As a starting point, the protein data bank (PDB) structure 5Y3R of the DNA-dependent protein kinase catalytic subunit (PRKDC) was used. FoldX focuses on the prediction of free energy changes and calculates the free energy of the WT and the mutant (MT); the difference between the two is calculated as follows: (ΔΔG(change)=ΔG(MT)−ΔG(WT)). ΔΔG(change)>0 indicates that the mutation is destabilizing, whereas ΔΔG(change)<0 indicates that the mutation is stabilizing.
Cell line
Human gastric cancer cell line (SNU-1, obtained from the Korean Cell Line Bank, Seoul, Korea) and human colon cancer cell line (HT29, obtained from ATCC, Rockefeller, Massachusetts, USA) were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium. All mediums were supplemented with 10% fetal bovine serum (GIBCO, Grand Island, New York, USA), penicillin (100 unit/mL) and streptomycin (100 µg/mL). Cells were incubated in a humidified atmosphere at 37°C with 95% air and 5% CO2.
Western blot analysis
Cells were lysed at 4°C in RIPA buffer supplemented with protease and phosphatase inhibitors. Equal loads of 30 µg proteins were electrophoretically separated using sodium dodecyl sulfate-polyacrylamide gels and then transferred onto the PVDF membrane (Millipore, Bedford, Massachusetts, USA). After blocking with 5% non-fat milk, the membrane was made to react with specific antibodies (PRKDC, GTX9673, 1:5000; tubulin, 1:5000, Cell Sigma, USA; Histone H2A.X, cell signaling #7631, 1:1000; phospho-Histone H2A.X (Ser139), cell signaling #2577, 1:1000; actin, Sigma-Aldrich A2228, 1:1000) overnight at 4°C and then incubated with horseradish peroxidase-conjugated secondary antibody for 1 hour. Blots were visualized using the ECL-Plus detection kit (PerkinElmer Life Sciences, Boston, Massachusetts, USA).
Virus Production and Infection shPRKDC clones were amplified from the pGIPZ gene bank (Thermo Company, from Dr Michael Hsiao’s library). Plasmids were transfected into 293 T cells with pCMV△8.91 and pMD.G. Target cells were seeded at an appropriate density in a 10 cm dish 24 hours before infection. On the second day of infection, the growth medium was changed, and virus supernatants were collected after 48 and 72 hours. A combination of a virus supernatant and polybrene (final concentration of 8 µg/mL) was used for various cell models. Subsequently, the plate was incubated for 24 hours. After incubation, the medium was removed, and a fresh medium containing puromycin was added. Approximately 72 hours after infection, cells were further split, and the selection was continued till all control cells were dead.
Comet assay
Cells were seeded at a density of 2×105 cells/well in a 6 cm well dish. After treatment with cisplatin or M3814 or with a combination of cisplatin and M3814 for 72 hours, cells were trypsinized, and DNA damage was analyzed using the Trevigen CometAssay kit (Trevigen, Gaithersburg, Maryland, USA). The alkaline comet assay was used to assess DNA damage in all cells, including PRKDC-knockdown cells and drug-treated cells, according to manufacturer’s instructions. Cells that were embedded on a glass slide in agarose were stained with SafeView nucleic acid stains (Applied Biological Materials, Richmond, Canada). Cells were observed using a fluorescence microscope for quantification; comets were analyzed as 100 cells per replica, and the DNA tail intensity was calculated using OpenComet, a plug-in processing platform in ImageJ (National Institutes of Health, USA).
CRSPER/Cas9 method
To generate MsPrkdc-knockout cells with the CRISPR/Cas9 system, the guide RNA (gRNA) sequence of mouse PRKDC was obtained from Optimized CRISPR Design (http://crispr.mit.edu). The following is the sequence list: gRNA1: CAGTAGCCAACACCGTACGC and gRNA2: AGGGAACCGGCGTACGGTGT. The gRNA/Cas9 expression vector was obtained from Dr Tsai-Yu Tzeng, Cancer Progression Research Center, Taiwan. For a surrogate reporter, oligonucleotides, including target sequences (TGGCGGAGGAGGGAACCGGCGTACGGTG
TTGGCTACTGCAGCTGCAGG), were synthesized (Genomics, Taiwan) and annealed in vitro by using a thermocycler (95°C for 5 min and then ramped down to 25°C at 5°C per min). The annealed oligonucleotides were ligated into reporter vectors digested with BgIII and KpnI. The gRNA1 or gRNA2 expression plasmid with a surrogate reporter was transfected by the X-tremeGene HP DNA Transfection Reagent (Roche) in CT26 cells. Red fluorescent protein (RFP)-positive cells were sorted through BD FACSAria flow cytometry. A total of 5000 or 10,000 cells were seeded in 10 cm dishes and incubated with 5% CO2 at 37°C until colony formation. Colonies were trypsinized using trypsin-EDTA, and cells were amplified in 24-well plates. The phenotype and genotype of cells were examined through western blot and sequencing (VYM Genome Research Center, Taiwan), respectively.
Animals
To evaluate tumorigenicity by treatment with various inhibitors, 1×106 CT26/CT26 PRKDC-knockout cells were resuspended in 100 µL of PBS and subcutaneously injected into male BALB/c mice (6 week old, 20–25 g of body weight). When the tumor attained a size of 0.5 cm, mice were divided into groups and randomized to different treatments as follows: sham group (normal saline, two times per week, intraperitoneal injection); anti PD-L1 antibody (200 µg, two times per week, intraperitoneal injection); M3814 (DNA-dependent protein kinase (DNAPK) inhibitor, 0.5 mg/kg, every day, tube feeding); cisplatin (1 mg/kg, two times per week, intraperitoneal injection); and various combination approaches, including M3814+anti PD-L1 antibody and M3814+anti PD-L1 antibody+cisplatin. The tumor volumes and body weights of mice were measured once a week, and tumor masses were harvested after 5 weeks.
Compounds
InVivoMAb anti-mouse PD-L1 (B7-H1) was purchased from BioCell (catalog# BE0101). M3814 was obtained from Merck (Darmstadt, Germany).
Statistical analysis
Differences between the mutation load in patients harboring or not harboring PRKDC mutations and those with MSI-H or MSS/MSI-L were determined using one-sided Student t-test. Enriched proportions from patients with MSI-H and PRKDC mutations were evaluated using a hypergeometric test. A p value less than 0.05 was considered significant. All statistical analyses were performed using R or SPSS V.17.0.
Results
Response rate of immune checkpoint blockades in patients harboring PRKDC mutations
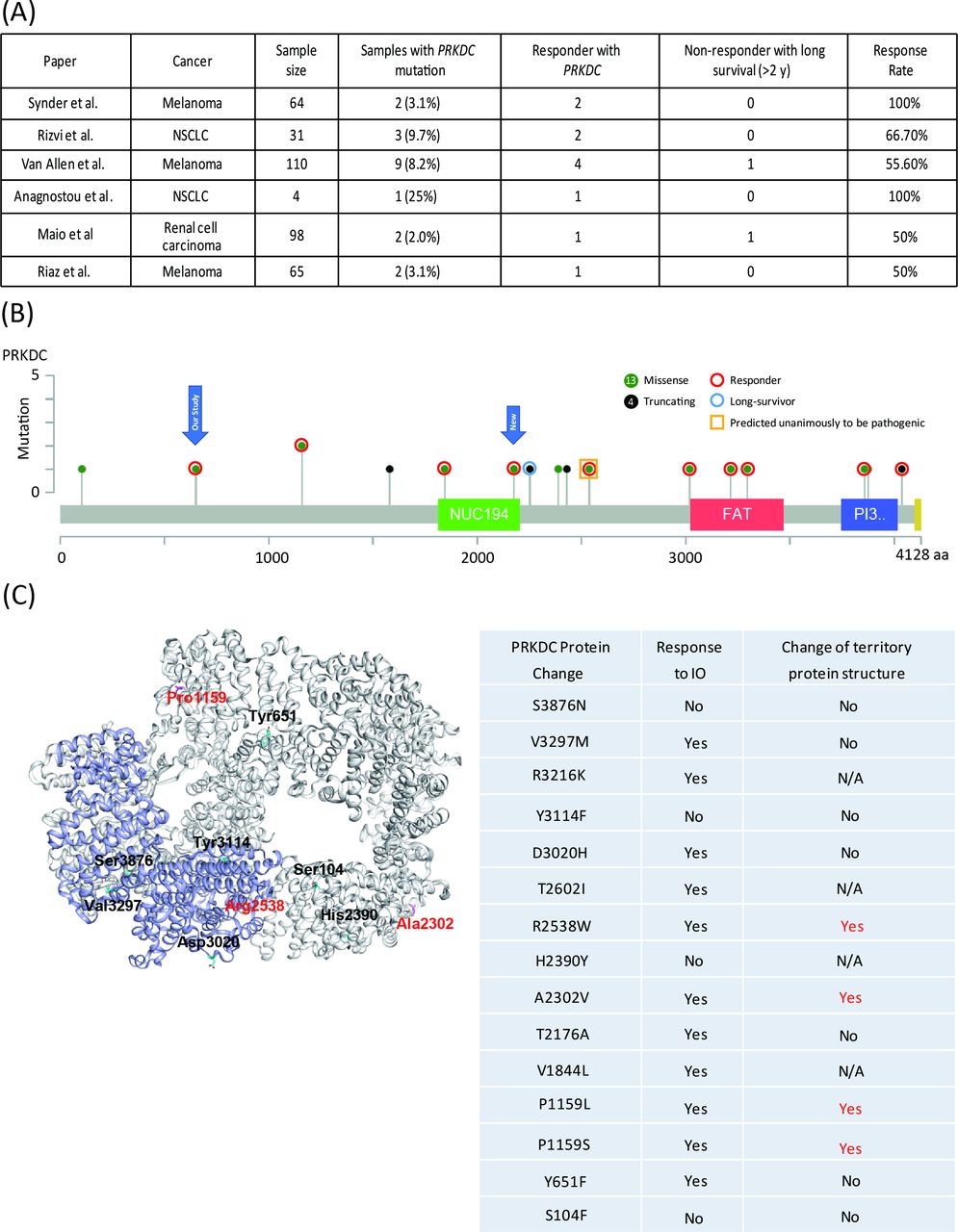
First, we identified PRKDC mutations in two responders to immune checkpoint therapy through target sequencing (online supplementary table S1) and mutation load through whole-exome sequencing. One responder had hepatocellular carcinoma, and the other had gastric cancer. To evaluate the predictive capability of PRKDC mutations in other cancers, we further analyzed six studies that provided details on the genetic landscape and immunotherapy response.8 22–26 In melanoma, the frequency of PRKDC mutations was 3.1%–8.2%, and the response rate to the anti-CTLA4 antibody was 53.8% (figure 1A and table 1). In lung cancer and renal cell carcinoma, the frequencies of PRKDC mutation occurrence were 9.7% and 2.0%, respectively, and the rates of response to the anti-PD-1 antibody were 75% and 50%, respectively (figure 1A and table 1).
Supplemental material
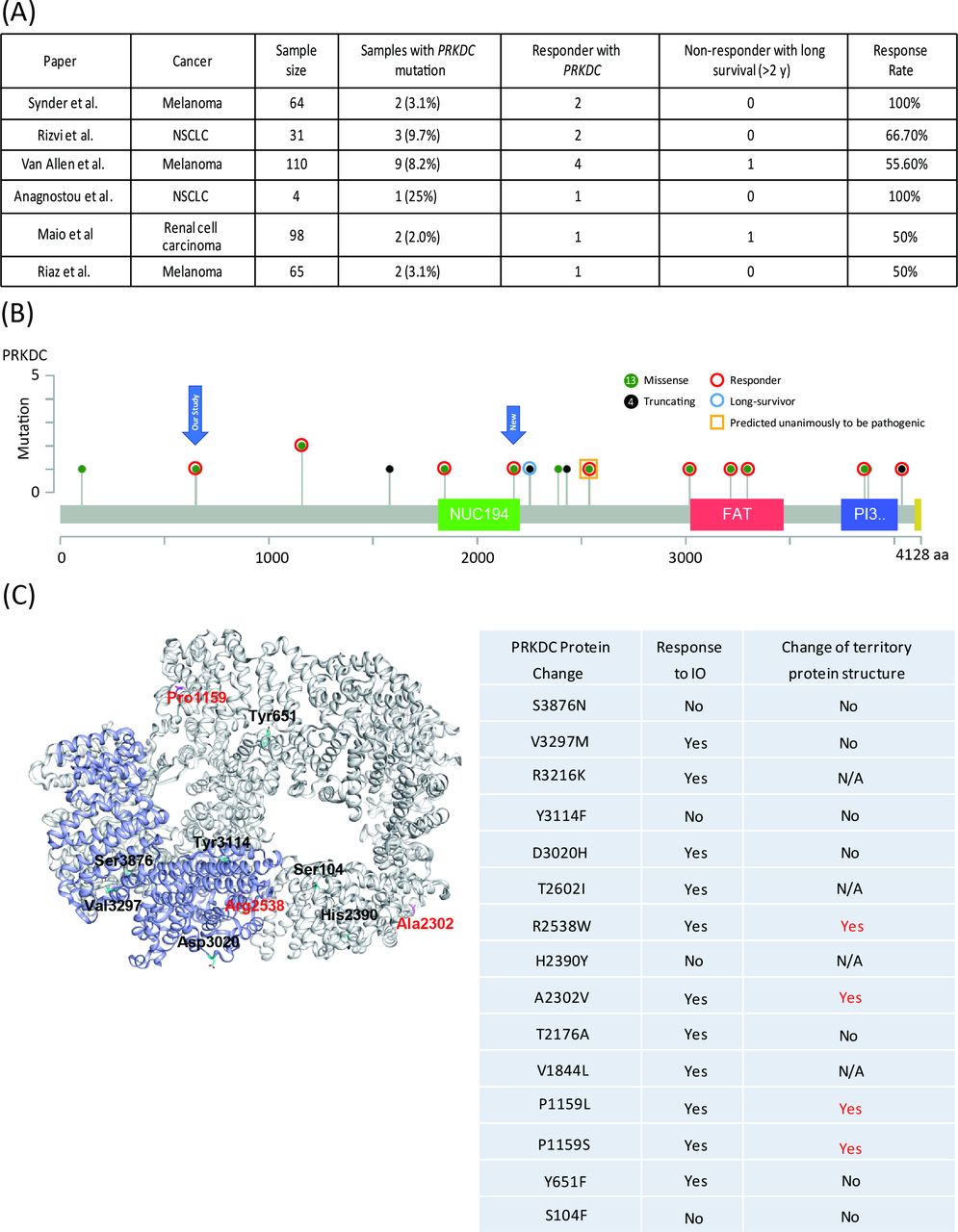
Clinical outcomes of immunotherapy in patients with melanoma or lung cancer with PRKDC mutations. (A) Response rate and prevalence of PRKDC mutations in data cohorts from Snyder et al,22 Rizvi et al,8 Van Allen et al,23 Anagnostou et al,26 Miao et al,24 and Riaz et al.25 (B) Responders or non-responders with mutation loci on the PRKDC protein. Red circles denote a responder; the blue circle denotes long survival; the yellow bar denotes predicted pathogenic mutation; green-filled circles denote a missense mutation; and black-filled circles denote a truncating mutation. (C) PRKDC tertiary protein structure. Red color denotes mutation loci with destabilizing PRBC protein structure. N/A, not applicable; NSCLC, non-small-cell lung carcinoma; PRKDC, protein kinase, DNA-activated, catalytic polypeptide.
Mutation types and locations in atients harboring PRKDC mutations
PRKDC mutation loci in patients who responded to immunotherapy
We subsequently examined the loci of PRKDC mutations from the collected data. As shown in figure 1B, 15 were missense mutations and six were truncating mutations. Among these mutations, seven (all missense) occurred in functional domains, among which five were responders (figure 1B). Among the six patients with truncating mutations, two were responders and one was a non-responder but with long-term survival for more than 2 years. Two of the 15 missense mutations were unanimously predicted to be pathogenic by all three tools, namely, Grantham,28 PolyPhen,29 and SIFT,30 and these mutations all belonged to responders. To further investigate the correlation between PRKDC missense mutations and the protein structure, we used FoldX energy calculations (figure 1C), which showed free energy changes that may affect the stability of PRKDC protein. All four patients with a destabilizing PRKDC protein structure responded to immune therapy (100%). Collectively, these results indicate that responders with PRKDC mutations likely contain dysfunctional DNA-PKcs proteins.
PRKDC mutation is significantly associated with high mutation loads in various cancer types
Recent findings suggest that high mutation load is a predictive biomarker of response to immune checkpoint inhibitors.8 We evaluated the correlation between PRKDC mutations and mutation load in various cancer types by using publicly available datasets. As shown in figure 2A, Van Allen et al showed that a significantly higher mutation load was observed in a melanoma bearing PRKDC mutation compared with that in a tumor with WT PRKDC.23 We found that PRKDC mutation is significantly associated with a high mutation load in cervical squamous cell carcinoma, endocervical adenocarcinoma, colon adenocarcinoma, head and neck squamous cell carcinoma, lung adenocarcinoma, gastric adenocarcinoma, and UCEC (figure 2B; p=0.008, p=0.0108, p=0.0166, p=0.0183, p<0.001, and p<0.001, respectively).

Association between PRKDC mutation and tumor mutation load. (A) Mutation loads of patients harboring PRKDC mutation and WT from data of Van Allen et al; (B) mutation loads of patients harboring PRKDC mutation or WT in TCGA datasets; (C) upper panel shows SNU-1 cell line with or without knockdown-PRKDC. Lower panel shows the change of DNA breaks by comet assay. (D) Change of tumor mutation burden in several conditions. MT, mutant; PRKDC, protein kinase, DNA-activated, catalytic polypeptide; RT, radiation; SCC, squamous cell carcinoma; TCGA, The Cancer Genome Atlas; UCEC, uterine corpus endometrial carcinoma; WT, wild type.
Knockdown of PRKDC increases mutation loads and DNA damage in the gastric cancer cell line
To evaluate whether decreased PRKDC expression might increase DNA damage and tumor mutations, we first established the PRKDC knockdown SNU-1 gastric cancer cell line and HT29 colon cancer cell line (figure 2C, upper panel, and online supplementary figure S2). The comet assay (single-cell alkaline gel electrophoresis), which measures single-strand and double-strand breaks of DNA, was performed to observe the intensity of DNA damage. The intensity of the comet tail relative to the head reflects the number of DNA breaks in a particular cell. Compared with SNU-1 cells exposed to radiation or HT29 cells exposed to cisplatin, long comet tails extending toward the anode were observed for PRKDC knockdown (figure 2C and online supplementary file 1, lower panel; p=0.03 and p=0.01, respectively).
Several conditions were next performed as follows: sham group plus radiation (SNU-1 cell line treated with 2 Gy radiation), knockdown-PRKDC, and knockdown-PRKDC plus radiation and knockdown-PRKDC group plus cisplatin (1 µM for 3 days). All groups were cultured for more than 30 generations. Tumor mutation burden was evaluated through whole-exome sequencing (online supplementary material 1). The results demonstrated that the tumor mutation in the sham group and knockdown-PRKDC group was similar (68 mutations/megabase); tumor mutation was higher in the sham plus radiation group (70 mutations/megabase) and the highest in the knockdown-PRKDC plus radiation or cisplatin group (78 and 86 mutations/megabase, respectively; figure 2D). These results indicated that the knockdown of PRKDC enhanced DNA damage and increased mutation burden when combined with other DNA damage agents such as cisplatin or radiation.
PRKDC mutation is correlated with MSI-H in various cancer types
Because the prevalence rate and the number of PRKDC mutations are the highest in gastric and colorectal cancers (10.9% and 5.8%, respectively; figure 2B), we first selected gastric cancer and colorectal cancer as a model to investigate the relationship between PRKDC mutation and other predictive biomarkers. First, we examined MSI status in gastric cancer. As shown in figure 3A, 78 (19.7%) patients with gastric cancer had MSI-H. Among the 43 patients with PRKDC mutations, 33 patients (76.7%) had MSI-H, and we additionally identified 10 patients without MSI-H. Therefore, compared with WT PRKDC, PRKDC mutation was highly associated with MSI-H (13% vs 76.7%, respectively; p<0.0001). In patients with colorectal cancer, PRKDC mutation was associated with MSI-H (13.5% vs 46.2%, respectively; p=0.0051; figure 3B). To validate the correlation between MSI-H and PRKDC mutation, we selected 35 patients with gastric cancer (15 patients with MSI-H and 19 patients with MSS) to evaluate PRKDC mutations (online supplementary table S2). As shown in figure 3C, nine patients with gastric cancer had PRKDC mutation. Among these, eight patients (88.9 %) also had MSI-H. Compared with WT PRKDC, PRKDC mutation was highly associated with MSI-H (28% and 88.9%, respectively; p=0.002). In addition, in patients with cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), head and neck squamous cell carcinoma (HNSC), and UCEC, PRKDC mutation were also associated with MSI-H (p=0.02, p=0.01, and p<0.01, respectively; figure 3D). Furthermore, as shown in figure 3E,F, patients with gastric cancer who harbored PRKDC mutations demonstrated significantly higher gene expression in three inhibitory immune checkpoints (PDL1, TIM3, and LAG3; p=0.0016, p=0.0142, and p=0.0017, respectively), interferon-γ gene signatures (IFNG, CXCL9, and CXCL10; p=0.0034, p=0.0118, and p<0.0001, respectively), and cytolytic activity markers (GZMA and PRF1; p=0.0001 and p<0.0102, respectively), compared with patients with MSS. As expected, patients with MSI-H also demonstrated a higher gene expression in these genes than did patients with MSS (p<0.0001, p=0.0202, p=0.001, p<0.0001, p=0.0012, p=0.0008, p=0.0001, and p=0.0022, respectively).

Comparison of PRKDC mutation and MSI status, and immune signatures. (A–D) Proportions and enrichment between patients with PRKDC mutations and MSI-H in the dataset of patients with TCGA gastric cancer, colorectal cancer, CESC, HNSC, UCEC, and 34 patients with gastric cancer; significance was evaluated by hypergeometric test. (E–G) Expression of a set of cytolytic activity, inhibitory immune checkpoint genes, and interferon-γ gene signatures by RNA sequencing between patients with gastric cancer with PRKDC mutation, MSI and MSS. Significance was evaluated by two-sided Wilcoxon rank-sum tests. CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; HNSC, head and neck squamous cell carcinoma; LUAD, lung adenocarcima; MSI, microsatellite instability; MSI-H, microsatellite instability—high; MSI-L, microsatellite instability—low; MSS, MT, mutant; PRKDC, protein kinase, DNA-activated, catalytic polypeptide; TCGA, The Cancer Genome Atlas; UCEC, uterine corpus endometrial carcinoma; WT, wild type.
Knockout PRKDC enhanced anti-PD-L1 antibody efficacy in a CT26 animal model
To evaluate the role of PRKDC as a predictive biomarker, we first performed whole-genome sequencing of the CT26 cell line (Sequence Read Archive submission: SUB5457457) and confirmed the absence of PRKDC mutation. Then, we established the PRKDC-knockout CT26 cell line (figure 4A, left panel). In the meantime, we performed the expression levels of γH2A.X, a DNA damage marker, in CT26 PRKDC-knockout cells, to examine whether knockout PRKDC will enhance the DNA damage. As the result, higher γH2A.X was found in CT26 PRKDC knockout cells than parental cells, and even become higher after passaged more 10 generations (figure 4A, left panel). We treated mice with the anti-PD-L1 antibody (200 µg, two times per week, intraperitoneal injection) after they were xenografted with 1×106 CT26/PRKDC-knockout CT26 cells. The tumor volume and body weight of mice were monitored once a week. We first observed that treatment with the anti-PD-L1 antibody suppressed the growth of CT26 cells, although not significant, in vivo compared with the vehicle group (figure 4A; right panel, 4C and 4D). We observed that treatment with the anti-PD-L1 antibody significantly suppressed the growth of PRKDC-knockout CT26 cells in vivo (figure 4C, p=0.007). No apparent changes in body weight were detected in either group of mice during the treatment period (figure 4B). This result demonstrated that knockout of PRKDC enhanced the efficacy of immune therapy.

PRKDC knockout increased immunotherapy response in vivo. (A) Left panel shows CT26 with or without knockout PRKDC and γH2A.X expression levels were performed by western blotting using CT26 parental cell, CT26 PRKDC-knockout cell (short-term) and CT26 PRKDC- knockout cell passaged more 10 generations (long-term). Representative photographs in the right panel of tumor size (n=4 per treatment group). (B) no significant change in body weight in each group. (C) Tumor weight showed consistency with tumor volume; CT26 PRKDC-knockout mice combined with anti-PD-L1 had lower tumor weight than that of mice in the sham group. (D) Anti-PD-L1 led to a significant decrease in CT26 cell growth in vivo, compared with the sham group (p=6.09×106). A combination of CT26 PRKDC-knockout and anti-PD-L1 inhibited tumorigenicity, compared with the sham group (p<0.001). (A–D) The statistical significance was analyzed using unpaired t-test. NS, not significant; PD-L1, programmed cell death protein one pathway; PRKDC, protein kinase, DNA-activated, catalytic polypeptide.
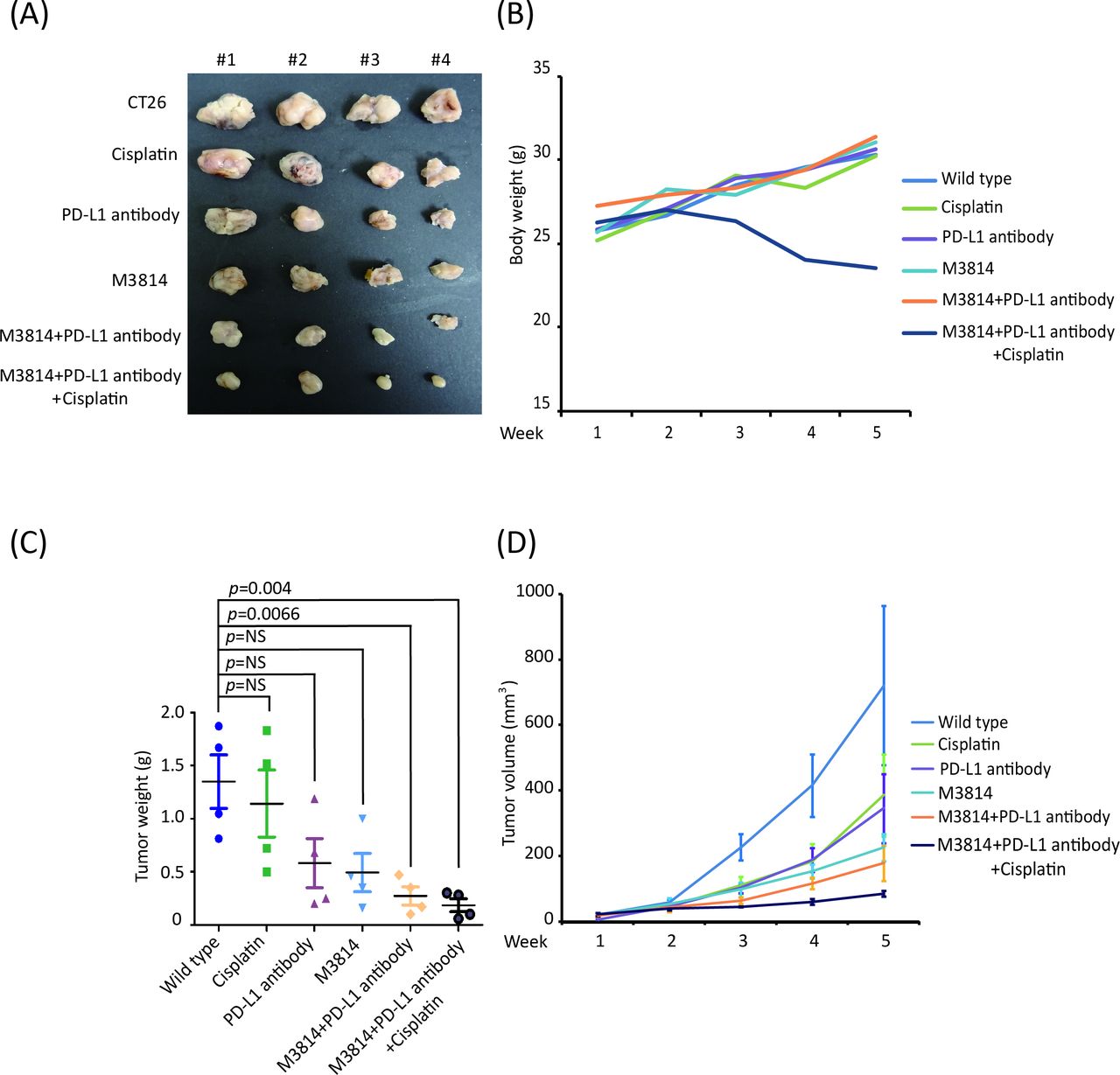
DNA-PK inhibitor enhanced anti-PD-L1 antibody efficacy in the CT26 animal model
PRKDC encodes a catalytic subunit of DNA-PKcs.15 16 Therefore, we hypothesized that the DNA-PK inhibitor can also enhance the efficacy of the anti-PD-L1 antibody. To evaluate the effectiveness and safety of DNA-PK inhibitor (M3814) treatment in vivo, we divided mice into several groups: sham group; anti-PD-L1 antibody; M3814 (DNAPK inhibitor), cisplatin, M3814+anti-PD-L1 antibody, and M3814+anti-PD-L1 antibody+cisplatin. We observed that M3814 +anti-PD-L1 antibody and M3814+anti-PD-L1 antibody+cisplatin treatment significantly suppressed the growth of CT26 cells in vivo compared with the sham group (figure 5A,C,D; p=0.0066 and 0.004, respectively). However, apparent changes in body weight were detected in the M3814+anti-PD-L1 antibody+cisplatin group during the treatment period, indicating that the triple combination therapy was toxic to these animals (figure 5B). Taken together, these results demonstrate that M3814 enhanced the efficacy of the anti-PD-L1 antibody in the animal model of CRC.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
DNAPK inhibitor as the potential moderator for immunotherapy to suppress tumor growth in vivo. (A) Representative photographs of tumor size (n=4 per treatment group). (B) No significant change in body weight in each group; expect the M3814 plus anti-PD-L1 antibody plus cisplatin group. (C) Tumor weight showed consistent result with tumor volume; (D) DNAPK inhibitor combined with anti-PD-L1 or anti-PD-L1 plus cisplatin could suppress tumor growth rate in vivo. (A–D) The statistical significance was analyzed using unpaired t-test. NS, not significant; PD-L1, programmed cell death protein one pathway. DNAPK, DNA-dependent protein kinase.
Discussion
Although immune checkpoint inhibitors may be effective in treating cancer, their response rate remains relatively low. The response rates in cohorts of unselected patients with melanoma who were administered the anti-CTLA4 antibody and patients with lung cancer who were administered the anti-PD-1 antibody were 10.9% and 19%, respectively.1 3 These response rates could be dramatically increased with patient selection; in patients selected by colon cancer by MSI status, the response rate increased from 0% to 40%.9 In this study, we found that PRKDC may be a potential candidate marker that could guide patient selection. We retrospectively analyzed 19 patients with lung cancer or melanoma or renal cell carcinoma who harbored a PRKDC mutation and were administered immune checkpoint blockade therapy. The response rate in these patients was 53.8% in melanoma, 75% in lung cancer, and 50% in renal cell carcinoma. These response rates were consistent with those reported in a previous study on MSI, of which patients were also selected.9
Defective DNA repair is a common hallmark of cancer. Recently, Chae et al analyzed a comprehensive list of 193 DNA repair genes using large databases including COSMIC and TCGA.35 PRKDC was identified as one of the top ten most frequently mutated DNA repair genes in common cancers including lung, breast, liver, colorectal, and skin cancer. Furthermore, Chae et al reported that all patients with colorectal cancer who harbor PRKDC mutation have a significantly higher mutation burden.36 Our study showed consistent results, where a significantly higher mutation load was associated with PRKDC mutation compared with WT PRKDC in several tumor types. Furthermore, we observed that mutation loads and DNA breaks increased in gastric cancer cell lines with PRKDC knockdown and when treated with DNA-damaging agents (radiation or cisplatin). Moreover, we demonstrated that PRKDC mutation was significantly associated with other immune biomarkers, including MSI, inhibitory immune checkpoints (PD-L1, TIM3, and LAG3),12 IFN-γ gene signatures,12 and cytolytic activity.12 MSI-H is associated with defective DNA MMR. Therefore, instability at coding microsatellites in target genes causes frameshift mutations and the functional inactivation of affected proteins, including PRKDC.36 37 A previous study reported that frameshift mutations in PRKDC were found in 24.3% of patients with MSI-H gastric cancer and suggested that (A)10 frameshift mutations in PRKDC are a target in MSI-H gastric cancers.38 It might partially explain why PRKDC mutation is highly correlated with MSI-H. These results suggested that PRKDC mutation might be a novel positive biomarker of immunotherapy across various tumor types.
We found that most PRKDC mutations occurred in functional domains or were truncating mutations in responders. Two other responders had a predicted pathogenic mutation (figure 1B). For 14 PRKDC missense mutations, we further used FoldX energy calculations and identified four patients with destabilizing PRKDC proteins; all patients responded to immune therapy. Most importantly, we demonstrated that knockout PRKDC enhanced the efficacy of the anti-PD-L1 antibody in the animal model. These results suggested that cancers that harbor loss-of-function PRKDC mutations, which lead to dysfunctional or impaired protein product, may benefit from immune therapy.
According to the COSMIC database, the frequency of PRKDC mutations is very low (2.1%) in the five most common cancers.35 Therefore, drugs that can inhibit DNA-PK protein activity might have the same role as PRKDC mutations do. M3814 is an orally administered, highly potent, and selective inhibitor of DNA‐PK.39 In our cell line study, we observed that cell lines treated with M3814 alone or a combination of M3814 and cisplatin showed increased DNA breaks (online supplementary figure S1). Furthermore, the animal study demonstrated that M3814 alone or in combination with cisplatin enhanced the efficacy of the anti-PD-L1 monoclonal antibody. However, mice body weight decreased when treated with a combined therapy of three treatments, suggesting that toxicity could be a concern. Currently, a phase I study is ongoing to evaluate the recommended phase II dose and the maximum tolerated dose of M3814 combined with avelumab with or without radiation in patients with advanced solid tumors.
In conclusion, a systematic analysis of the cancer genome has generated new scenarios for cancer treatment. Given the correlation among the mutation load, MSI-H, and PD-L1 expression plus enhanced interferon-γ gene signatures, PRKDC mutations can be potential candidates to guide patient selection for immunotherapy. Further animal studies demonstrated that a loss-of-function PRKDC mutation or DNA-PK inhibitor can enhance the efficacy of immune therapy. These findings have implications in patient stratification in clinical settings and could benefit in developing novel clinical trials.
Acknowledgments
The authors acknowledge the Cancer Progression Research Center-Genome Editing Core Facility of Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education.
References
Footnotes
KTT and C-NY contributed equally.
Contributors M-HuC designed and drafted the study. KTT and W-LF analyzed and interpreted the patient data regarding the gastric cancer. Y-CW, DS-SH, C-EW and J-HC used software to analyze genomic data regarding PRKDC mutation. C-NY and Y-CY performed the histological examination of the gastric cancer. Y-CC, J-IL, PM-HC, M-HaC and M-LL participated in the bench study, including cell line and animal study. S-JC, YC and MH drafted the work and also substantively revised it. All authors read and approved the final manuscript.
Funding This work was supported by Taiwan Cancer Clinic Foundation and Melissa Lee Cancer Foundation. We like to thank Academia Sinica for their support. Additional support was provided by the Taipei Veterans General Hospital (V108C-022 and VTA108-A-5-2 to M-HuC and V106D29-001-MY3 to YC), the Ministry of Science and Technology (MOST104-2314-B-075-064-MY2 to M-HuC, MOST107-2314B-182A-134-MY3, NMRPG3H6211, and CMRPG3I0231 to C-NY, MOST105-0210-01-13-01 to MH).
Competing interests KTT, J-HC and S-JC are employees of ACT genomics. DS-SH is an employee of Asclepiumm Taiwan Com.
Patient consent for publication Not required.
Ethics approval The institutional review board of Taipei Veterans General Hospital and Academia Sinica approved this study (2011-04-026GB). All animal experiments were performed in accordance with the protocol approved by the Academia Sinica Institutional Animal Care and Utilization Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as supplementary information. All data generated or analyzed during this study are available in a public.