Article Text

Abstract
Background Anti(α)-programmed cell death-1 (PD-1)/programmed death-ligand 1 (PD-L1) monotherapy fails to provide durable clinical benefit for most patients with carcinoma. Recent studies suggested that strategies to reduce immunosuppressive cells, promote systemic T-cell responses and lymphocyte trafficking to the tumor microenvironment (TME) may improve efficacy. N-809 is a first-in-class bifunctional agent comprising the interleukin (IL)-15 superagonist N-803 fused to two αPD-L1 domains. Thus, N-809 can potentially stimulate effector immune cells through IL-15 and block immunosuppressive PD-L1. Here, we examined the antitumor efficacy and immunomodulatory effects of N-809 versus N-803+αPD-L1 combination.
Methods The ability of N-809 to block PD-L1 and induce IL-15-dependent immune effects was examined in vitro and in vivo. Antitumor efficacy of N-809 or N-803+αPD-L1 was evaluated in two murine carcinoma models and an extensive analysis of immune correlates was performed in the tumor and tumor-draining lymph node (dLN).
Results We demonstrate that N-809 blocks PD-L1 and induces IL-15-dependent immune effects. N-809 was well-tolerated and reduced 4T1 lung metastasis, decreased MC38 tumor burden and increased survival versus N-803+αPD-L1. Compared with N-803+αPD-L1, N-809 enhanced natural killer (NK) and CD8+ T-cell activation and function in the dLN and TME, relating to increased gene expression associated with interferon and cytokine signaling, lymphoid compartment, costimulation and cytotoxicity. The higher number of TME CD8+ T cells was attributed to enhanced infiltration, not in situ expansion. Increased TME NK and CD8+ T-cell numbers correlated with augmented chemokine ligands and receptors. Moreover, in contrast to N-803+αPD-L1, N-809 reduced immunosuppressive regulatory T cells (Treg), monocytic myeloid-derived suppressor cells (M-MDSC) and M2-like macrophages in the TME.
Conclusions Our results suggest that N-809 functions by a novel immune mechanism to promote antitumor efficacy. Foremost, N-809 enhances intratumoral lymphocyte numbers by increasing trafficking via altered chemokine levels in the TME and chemokine receptor expression on CD8+ T cells and NK cells. In addition, N-809 reduces immunosuppressive and pro-tumorigenic immune cells in the TME, including Treg, M2-like macrophages and M-MDSC. Overall, these novel effects of N-809 promote an inflamed TME, leading to lower tumor burden and increased survival. These results provide mechanistic insight and rationale supporting the potential clinical study of N-809 in patients with carcinoma.
- tumor microenvironment
- CD8-positive T-lymphocytes
- immunotherapy
- natural killer T-Cells
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
While the use of single-agent immunotherapies, in particular immune checkpoint inhibitors, can produce durable and effective responses in some patients, clinical benefit for most patients with carcinoma is minimal.1 This is due to multiple factors, including tumor heterogeneity, the immunosuppressive nature of the tumor microenvironment (TME) and innate or acquired resistance to treatment.1 In order to improve patient response rates, immunotherapy combinations are used to target distinct components of the tumor/immune system interaction. Recent studies have demonstrated that decreasing the number of immunosuppressive cells, systemic activation of effector immune cells and altered lymphocyte migration to the TME, not in situ expansion, mediate optimal antitumor responses.2–6
The combination of immunotherapies that decrease immunosuppression and activate immune effector cells may improve antitumor efficacy and induce the aforementioned immune effects. Anti(α)-programmed cell death-1 (PD-1)/programmed cell death-ligand 1 (PD-L1) antibodies decrease immune suppression by preventing the interaction of PD-L1 expressed on tumor or suppressive immune cells with PD-1 on natural killer (NK) and T cells. PD-1 ligation reduces NK and T-cell maturation, proliferation and effector function.6 In addition, αPD-L1 antibodies can decrease the number of and/or reprogram immunosuppressive immune cells like regulatory T cells (Treg) and M2 macrophages.4 6 M2 macrophages are emerging as an important suppressive cell type, and therapies that skew these cells to an antitumor M1 phenotype are of particular interest. Overall, PD-1/PD-L1-targeted antibodies have increased patient survival in carcinomas with high mutational burden such as melanoma,7 lung cancer8 and bladder cancer,9 but most patients with carcinoma fail to respond to PD-1/PD-L1 blockade.
Interleukin (IL)-15, on the other hand, is an immune-activating cytokine that promotes the systemic expansion and activation of NK and CD8+ T cells.10 Clinically, administration of recombinant IL-15 (rIL-15) promoted hyperproliferation and activation of NK and CD8+ T cells.11 12 However, the high doses required to induce immune changes resulted in dose-limiting toxicities with no clinical response observed.11 12 To promote patient responses with less toxicity, new IL-15 agents were developed. N-803 is an IL-15 superagonist (IL-15 N72S/IL-15RαSushi complex) that enhances CD8+ T-cell and NK cell activation, proliferation, cytotoxicity and survival versus rIL-15, leading to improved antitumor responses preclinically.10 13 While N-803 significantly expanded patient NK and CD8+ T cells and was well-tolerated, it demonstrated limited clinical efficacy.11 14 However, N-803 in combination with an αPD-L1/PD-1 antibody showed clinical promise. A recent phase Ib study in non-small cell lung cancer reported that N-803 and αPD-1 antibody nivolumab was well-tolerated, and 29% of patients, including 3/11 (27%) patients previously treated but progressing with αPD-1, exhibited an objective response.15 Similarly, the combination of N-803+αPD-L1 demonstrated improved antitumor efficacy in murine carcinoma models, including αPD-L1-refractory tumors versus either monotherapy.16 This efficacy was attributed to improved systemic CD8+ T-cell and NK cell responses.16 However, the effect of N-803+αPD-L1 on lymphocyte trafficking, an emerging paradigm important for efficacy,2 3 5 was not explored.
Other promising treatment strategies include bifunctional molecules, a novel class of immunotherapeutics that allows for interaction with two targets using a single agent. Bifunctional molecules have the potential to increase localization of an agent to the TME or immune cell of choice, leading to enhanced local effects, reduced systemic toxicity and/or for dual suppression and/or activation of immune pathways.16–19 Preclinical studies demonstrated that some bispecific molecules increased antitumor efficacy versus a similar immunotherapy combination,20 thus a potential for clinical advantage. Recently, N-809, a bispecific fusion protein consisting of N-803 fused to two single chain αPD-L1 variable region (ScFv) domains, was described. N-809 was able to block human PD-L1 and stimulate human immune cells via the IL-15 superagonist in vitro.21 A preliminary study suggested that N-809 had antitumor efficacy in a murine tumor model.21 The mechanisms of action of N-809 and its therapeutic advantage relative to N-803+αPD-L1 combination were not examined.
Here, for the first time, we compare the antitumor efficacies of N-809 versus N-803+αPD-L1 in murine carcinoma models. In addition, we perform a comprehensive immune analysis in the tumor and periphery. We describe the novel immune-mediated mechanism of action of N-809 as compared with N-803+αPD-L1, including its effects on immunosuppressive immune cells and effector cell activation and migration.
Methods
Reagents
Murine N-809 and N-803 (previously ALT-803) were generously provided by ImmunityBio under a Cooperative Research and Development Agreement with the National Cancer Institute. Murine N-809 consists of N-803, an IL-15 N72D/IL-15RαSushi superagonist complex containing the Fc-domain of murine IgG2a fused to two anti-PD-L1 ScFv domains. αPD-L1 (10F.9G2) and CD8 (2.43) antibodies were from BioXcell. The NK depletion antibody (anti-asialo-GM1) was from Wako Chemicals. FTY720 was from Sigma.
Mice
Female Balb/c and C57BL/6 mice aged 6–10 weeks were obtained from NCI Frederick Cancer Research Facility and maintained under specific pathogen-free conditions in accordance with Association for Assessment and Accreditation of Laboratory Animal Care guidelines.
Murine tumor cell lines and tumor studies
4T1 triple negative (TN) breast carcinoma and Yac-1 lymphoma cells were obtained from American Type Culture Collection. MC38 colon carcinoma cells are previously described.22 All cell lines were cultured according to providers’ instructions, determined as mycoplasma free by MycoAlert Mycoplasma Detection Kit (Lonza) and used at low passage number.
For antitumor studies, 4T1 tumor cells (5×104, subcutaneously) were orthotopically implanted into the mammary fat pad of female Balb/c mice. MC38 (3×105, subcutaneously) tumor cells were implanted into the right flank of female C57BL/6 mice. Tumors were measured biweekly using digital calipers, and volumes were determined as (length2×width)/2. Mice were randomized based on tumor size and treatment initiated when tumors reached 50–100 mm3. Unless otherwise stated, mice received two doses of N-809 (subcutaneously) or two doses of N-803 (0.3 µg, subcutaneously) plus αPD-L1 (200 µg, intraperitoneally). Quantification of 4T1 lung metastasis was performed as previously described.23
Isolation of immune cells
For all N-809 studies, unless otherwise stated, immune cells in the lymph nodes, spleen and tumors were isolated 2 days after the final treatment as previously described.16 Cell counts were performed using 123count eBeads (Thermo Fisher Scientific).
Flow cytometry and antibodies
Antibody labeling of cells for flow cytometry (1–10×106 immune cells) was performed using the BD Cytofix/Cytoperm Kit (BD Biosciences) according to the manufacturer’s instructions. Antibodies (online supplementary table S1) and matched isotypes were obtained from the listed manufacturers. Live/Dead Fixable Dead Cell Stain was from Invitrogen. Flow cytometry (≥1×105 events) was performed on a BD LSRFortessa flow cytometer (Beckton Dickinson) and analyzed with FlowJo FACS Analysis Software V.9.9.6 (Treestar). Cell populations were identified as listed (online supplementary table S2). Expression of phenotypic proteins was determined by subtracting the respective isotype, set between 1% and 5% of the population.
Supplemental material
CD8+ T-cell restimulation
Isolated immune cells were stimulated with αCD3 (2C11, BD Biosciences) +α CD28 (37.51, BD Biosciences) as previously described.16 Frequency of interferon gamma (IFNγ)+ and/or tumor necrosis factor alpha (TNFα)+ cells were calculated by subtracting the non-stimulated controls.
NK cell cytotoxicity assay
NK cell killing of Yac-1 targets was determined as previously described.16 24
RNA extraction and NanoString analysis
Tumor fragments were preserved in RNAlater (Thermo Fisher Scientific) and stored at −80°C. RNA was extracted using the RNeasy Mini Plus Kit (Qiagen) following the manufacturer’s protocol. RNA purity was assessed on the Nanodrop One Spectrophotometer (Thermo Fisher Scientific) and Agilent Bioanalyzer (Agilent). RNA analysis was performed using the PanCancer Mouse IO 360 Panel and data analyzed using the nSolver Software and nCounter Advanced Analysis Software (NanoString). Heatmaps were generated using the Morpheus Software (Broad Institute) for the fold change of a given treatment over phosphate-buffered saline (PBS) calculated by NanoString analyses.
Statistics
Statistical analyses were performed in Prism V.7.0a or V.8.2 (GraphPad Software). Unless otherwise stated, data presented in bar graphs or scatter plots were analyzed using one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons. Two-way ordinary ANOVA was used to analyze tumor growth curves. Survival was analyzed using log-rank (Mantel-Cox) test. Outliers were identified using Robust regression and Outlier removal (ROUT test). Statistical significance was set at p<0.05.
Additional materials and methods are described in online supplementary additional files.
Supplemental material
Results
N-809 binds to PD-L1 and has IL-15 activity in vivo and in vitro
First, we validated PD-L1 binding ability and IL-15 activity of N-809. In vitro, N-809 bound to murine PD-L1 in a cell-free system (figure 1A) and on murine MC38 colon tumor cells (figure 1B) with similar affinity to a commercially available αPD-L1. In vivo administration of N-809 reduced detectable surface expression of PD-L1 on CD45+ cells in MC38 primary tumor, draining and non-draining lymph nodes (LN) and spleen (figure 1C).
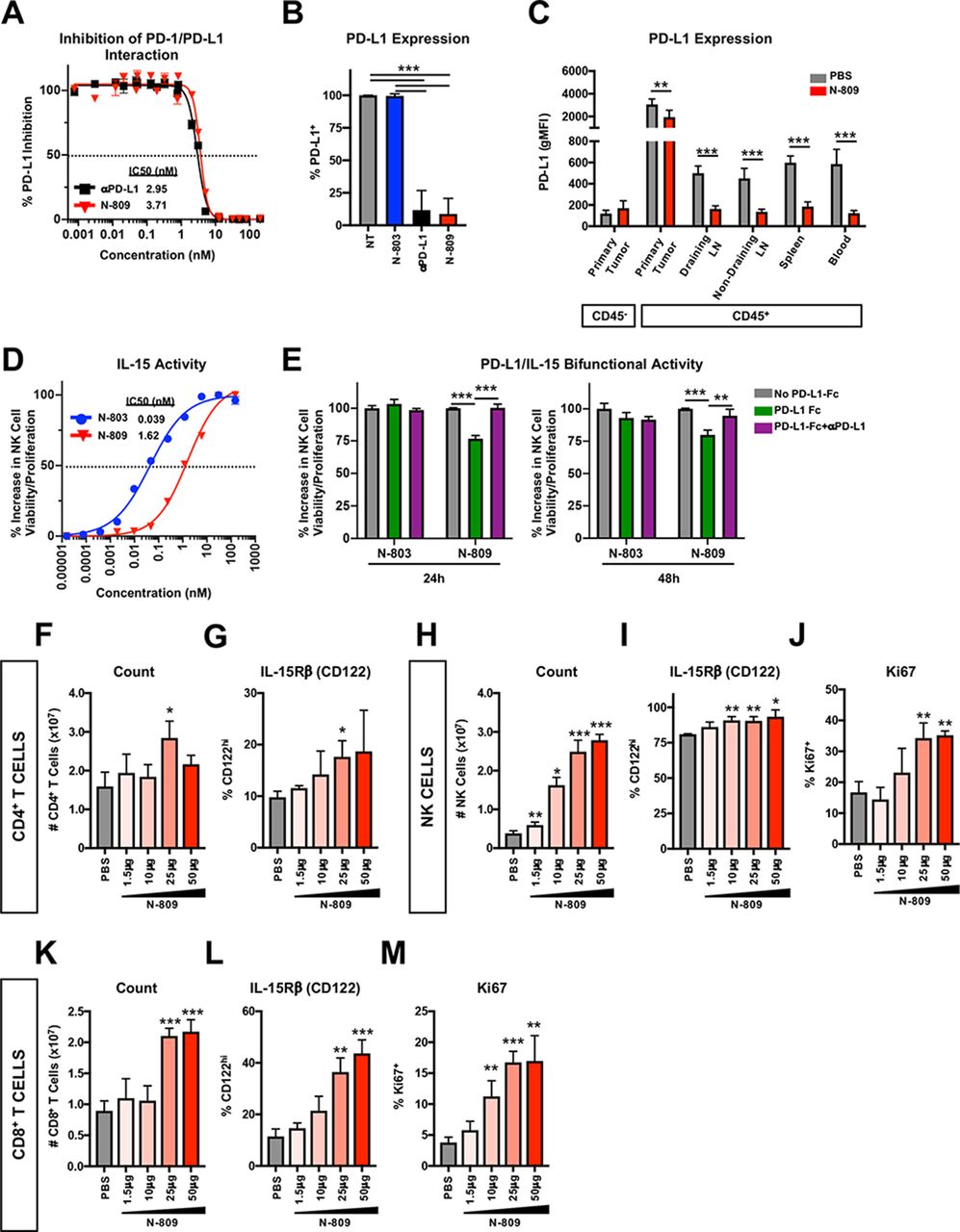
N-809 binds to programmed death-ligand 1 (PD-L1) and displays interleukin (IL)-15 immune-related activity in vitro and in vivo. (A) Plate bound PD-L1-Fc was incubated with anti-PD-L1 (αPD-L1) or N-809 prior to the addition of biotinylated programmed cell death-1 (PD-1). The amount of biotinylated PD-1 was quantified for % PD-L1 inhibition (inset: IC50). Data are combined from two independent experiments. (B) MC38 tumor cells were treated with 100 nM interferon (IFN)γ. After 24 hours, 1×106 cells were incubated for 30 min at 37°C with 1 nM N-803, αPD-L1 or N-809. Unoccupied surface PD-L1 was detected by flow cytometry. Data are combined from two to three independent experiments. (C) 3x105 MC38 tumor cells were implanted into the flank of female C57BL/6 mice. When tumor volumes reached ~50 mm3, mice were treated at days 10 and 14 with 100 μg N-809 (subcutaneously). PD-L1 expression was detected by flow cytometry on CD45− and CD45+ cells 2 days after treatment. All statistics shown are two-tailed t-test of phosphate-buffered saline (PBS) vs N-809 in the respective immune compartment. Data are from one experiment, n=6 mice/group. (D) Splenic natural killer (NK) cells were incubated with N-803 and N-809 for 48 hours and viability/proliferation was measured using CellTiter-Glo (inset: IC50). Data are representative of five independent experiments. (E) Plate bound PD-L1-Fc was unblocked or blocked with αPD-L1. 200 nM N-803 or N-809 was added for 30 min prior to measurement of splenic NK cell viability/proliferation using CellTiter-Glo. Data are from one experiment. (F–M) Naïve Balb/c mice were treated with N-809 (subcutaneously) on days 0 and 4. Splenic CD4+ T cells (F, G), NK cells (H–J) and CD8+ T cells (K–M) were examined by flow cytometry 3 days after treatment. Graphs show CD4+ T-cell number (F) and IL-15Rβ (CD122) expression (G), NK cell number (H) and IL-15Rβ (I) or Ki67 (J) expression, and CD8+ T-cell number (K) and IL-15Rβ (L) or Ki67 (M) expression. All statistics shown are two-tailed t-test vs PBS. Data are from one experiment, n=5 mice/group. All graphs show mean±SD. *P<0.05, **p<0.01, ***p<0.001.
IL-15 is a NK cell prosurvival and proliferation-inducing factor.25 To determine IL-15 activity of N-809 in vitro, splenic NK cells were incubated with N-809 or N-803 and viability/proliferation was determined. Untreated NK cells did not survive (figure 1D). N-809 significantly enhanced NK cell viability/proliferation, although 41-fold less than N-803 (figure 1D). When bound to murine PD-L1, N-809 retained 75% of its IL-15 activity (figure 1E). N-809 also demonstrated IL-15-related activity in vivo, as shown by minor changes to CD4+ T-cell number (figure 1F) and IL-15Rβ expression (figure 1G) and major dose-dependent alterations to NK and CD8+ T cells. CD4+ T cells are known to be less responsive than NK and CD8+ T cells to IL-15.25 N-809 greatly enhanced NK cell number (figure 1H) and expression of IL-15Rβ (figure 1I) and Ki67 (figure 1J). Similar effects were observed on CD8+ T cells (figure 1K–M). Together, these data demonstrate that the αPD-L1 and IL-15 components of N-809 are functional in vitro and in vivo.
Subcutaneous administration of N-809 induces antitumor efficacy against murine breast and colon carcinomas
Clinically, αPD-L1 is administered intravenously9 while experimentally given intravenously or intraperitoneally.15 16 N-803 is given subcutaneously both in the clinic and experimentally.14–16 26 Since N-809 consists of αPD-L1 and N-803, we determined whether route of administration impacted antitumor efficacy. Subcutaneous and intraperitoneal administration of N-809 to 4T1 tumor-bearing mice significantly reduced lung metastasis burden by 36% and 35%, respectively (online supplementary figure S1a-c). As route of administration did not affect N-809 efficacy and N-803 subcutaneously is better tolerated clinically,14 26 N-809 was administered subcutaneously.
We also determined the optimal dose of N-809 for antitumor efficacy in the highly aggressive 4T1 TN breast and MC38 colon carcinoma models. N-809 did not greatly affect primary 4T1 tumor growth at any dose (online supplementary figure S1a,d), but 50 µg N-809 induced the greatest reduction in 4T1 lung tumor burden compared with 25 and 100 µg N-809 (online supplementary figure S1b,c,e,f). In contrast, 100 µg N-809 best decreased tumor burden and increased median overall survival (mOS) in MC38 tumor-bearing mice (online supplementary figure S1g-i). Thus, N-809 was used at 50 µg for 4T1 tumor studies and 100 µg for MC38 tumor studies.
N-809 improves antitumor efficacy versus N-803+αPD-L1 combination
Next, we compared the antitumor efficacy of N-809 versus N-803+αPD-L1 in murine 4T1 and MC38 tumor models, which are resistant to either αPD-L1 or N-803 monotherapy, respectively.16 Due to significant treatment-related mortality of mice with high-dose N-803 (online supplementary figure S1j,k), we were unable to use equimolar doses of N-809 and N-803+αPD-L1 (online supplementary table S3). Instead, N-809 was compared with 0.3 µg (15 µg/kg) N-803 and 200 µg (10 mg/kg) αPD-L1. These doses are similar to those used in the clinic9 14 15 and have not been previously evaluated in murine tumor models.
N-809 and N-803+αPD-L1 were well tolerated in both tumor models, as neither 4T1 nor MC38 tumor-bearing mice significantly lost weight after treatment (online supplementary figure S2a,c). After N-809 treatment, 4T1 and MC38 tumor-bearing mice had increased inflammation in the lung (online supplementary figure S2b) or kidney, liver and lung (online supplementary figure S2d), respectively. However, these effects were transient and displayed no evidence of morbidity (online supplementary figure S2d).
We have previously demonstrated that N-803 increases systemic PD-L1 expression on CD45+ cells, in particular monocytic (M-MDSC) and granulocytic (G-MDSC) myeloid-derived suppressor cells.16 Furthermore, N-803 greatly enhances macrophage PD-L1 expression (online supplementary figure S3a). N-809 reduced detectable surface PD-L1 expression less than N-803+αPD-L1 on CD45− and CD45+ cells in the primary tumor and periphery of 4T1 (figure 2A) and MC38 (figure 2B) tumor-bearing mice, suggesting lower PD-L1 blockade by N-809. However, N-809 induced greater antitumor efficacy versus N-803+αPD-L1. N-803+αPD-L1 significantly reduced 4T1 lung tumor burden by 36% vs PBS, whereas N-809 decreased lung metastasis by 60% (figure 2C–E). In addition, N-809 significantly reduced MC38 tumor burden (figure 2F,G) and increased mOS from 24 to 29 days (figure 2H) versus N-803+αPD-L1. N-809 cured 8% of MC38 tumor-bearing mice (figure 2G), and mice did not develop palpable tumors after rechallenge (figure 2I,J). Together, these data demonstrate that N-809 is well-tolerated and has increased antitumor efficacy versus N-803+αPD-L1.

N-809 decreases 4T1 lung metastasis and MC38 tumor burden and improves survival vs N-803+αPD-L1. (A) 5x104 4T1 tumor cells were orthotopically implanted into female Balb/c mice. When tumor volumes reached ~50 mm3, mice were treated at days 9 and 13 with 0.3 μg N-803 (subcutaneously)+200 μg αPD-L1 (intraperitoneally) or 50 μg N-809 (subcutaneously). Programmed death-ligand 1 (PD-L1) expression was detected by flow cytometry on CD45− and CD45+ cells. Data are from one experiment, n=5 mice/group. (B) MC38 tumors were implanted as in figure 1 and mice were treated at days 8 and 12 with 0.3 µg N-803 (subcutaneously)+200 µg αPD-L1 (intraperitoneally) or 100 µg N-809 (subcutaneously). PD-L1 expression was detected by flow cytometry on CD45− and CD45+ cells. Data are combined from two independent experiments, n=5 mice/group per experiment. (C–E) 4T1 tumors were implanted and mice were treated as in (A). Graphs show tumor volumes at day 27 post-tumor implant (C) and number of lung metastases in individual mice at day 28 post-tumor implant (D). (E) Table denotes the distribution of lung metastases number and % reduction in mean vs phosphate-buffered saline (PBS). Data are representative of two independent experiments, n=20–25 mice. (F–H) MC38 tumors were implanted and mice were treated as in (B). Graphs show primary tumor growth curves (F) and tumor volumes of individual mice at day 18 (inset: % cured mice) (G). Graphs show mean±SD. (H) Survival curves (inset: median overall survival (mOS)) show % survival. Data are representative of two independent experiments, n=13 mice. (I–J) At least 1 month after tumor resolution (days 52–92), cured mice and paired naïve C57BL/6 were implanted with 3×105 MC38 tumor cells. (I) Graph shows primary tumor growth curves. Graphs show mean±SD. (J) Survival curves (inset: # mice with memory response) show % survival. Data are combined from two independent experiments, n=3–5 mice per experiment. *P<0.05, **p<0.01, ***p<0.001.
N-809 induces a systemic and intratumoral immunostimulatory environment
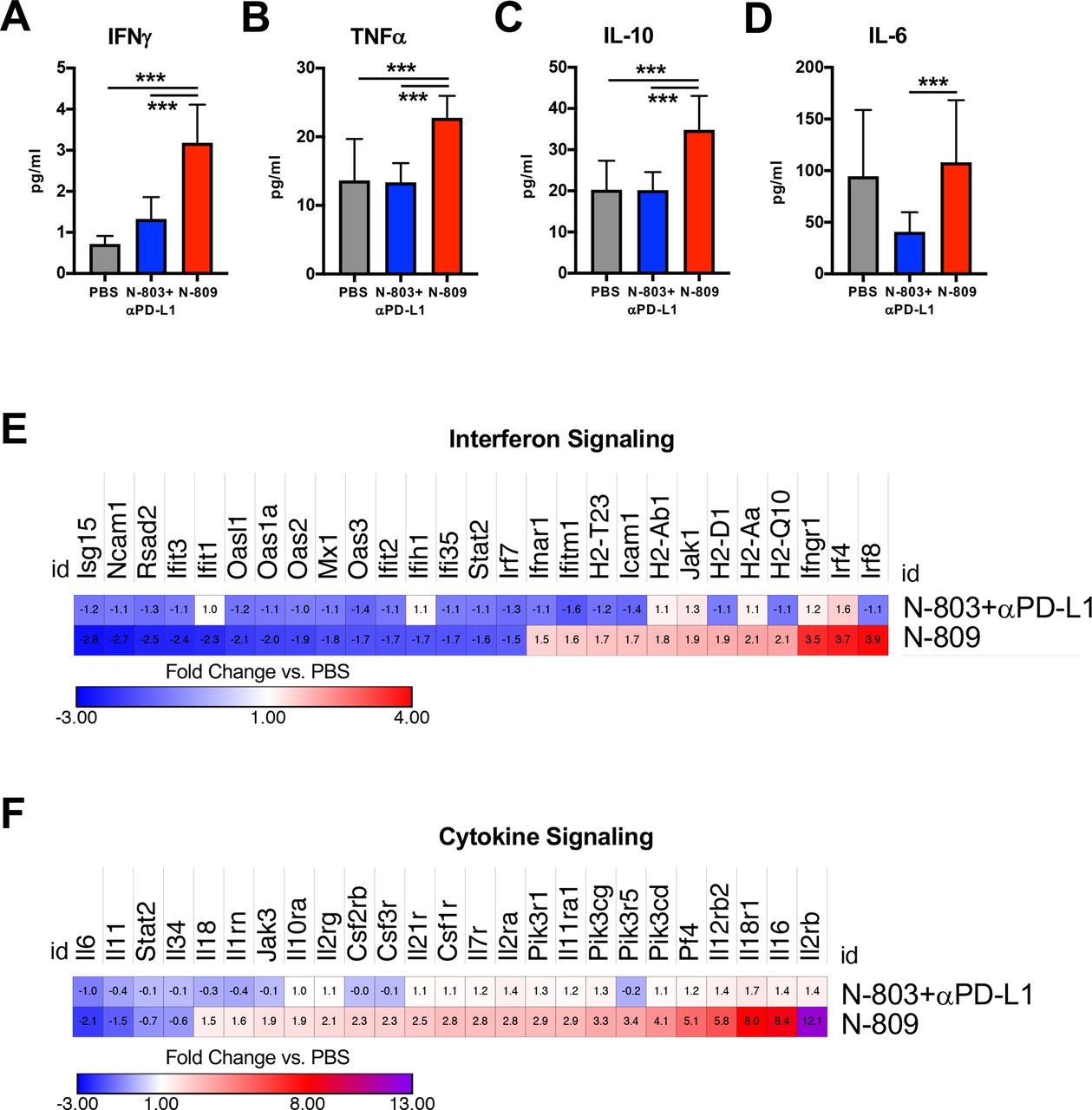
In order to dissect the mechanism of action of N-809, we first examined serum cytokine levels in MC38 tumor-bearing mice. Immunostimulatory cytokines IFNγ (figure 3A) and TNFα (figure 3B) were significantly elevated by N-809 compared with PBS and N-803+αPD-L1. N-809 also elevated IL-10 (figure 3C), an immunosuppressive cytokine which counteracts high levels of IFNγ and TNFα,27 but did not increase serum IL-6 (figure 3D), a known driver of IL-15-associated toxicity.11 26 N-803+αPD-L1 did not increase serum cytokine levels compared with PBS (figure 3A–D).

N-809 induces immunostimulatory cytokines in serum and increases cytokine signaling gene signatures in the tumor microenvironment (TME). MC38 tumors were implanted and mice treated as in figure 2. Primary tumor and serum were harvested 2 days after the last treatment. (A–D) Serum was analyzed for level of interferon (IFN)γ (A), tumor necrosis factor (TNF)α (B), interleukin (IL)-10 (C) and IL-6 (D). All graphs show mean±SD. Data are combined from two to three independent experiments, n=5 mice/group per experiment. (E, F) RNA from the primary tumor was extracted and analyzed by NanoString. Heatmaps show fold change in gene expression related to IFN signaling (E) or cytokine signaling (F) over phosphate-buffered saline (PBS) (genes N-809 altered >1.5-fold). Data are from one experiment, n=3–4 mice/group. ***P<0.001.
Intratumoral cytokine signaling was examined by NanoString. Overall, N-809 induced greater changes to genes involved in interferon (figure 3E) and cytokine (figure 3F) signaling than N-803+αPD-L1. Notably, N-809 increased expression of IFNγ targets (MHC class I genes H2-Aa and H2-Q10) and genes associated with positive regulation of IFNγ signaling (Jak1 (JAK1), Ifngr1 (IFNγRI)). Many downregulated genes were type I IFN-related, including Irf7, Ifit1-3, Isg15, Rsad2, Oasl1, Oas1a, Oas2-3 (figure 3E). N-809 also increased gene expression related to immunostimulatory cytokines IL-2 (Il2ra (IL-2Rα/CD25)), IL-12 (Il12ra (IL-12Rα), Il12rb2 (IL-12RβII)) and IL-18 (Il18r1 (IL-18RI)) (figure 3F). Il2rb, a component of the IL-15 receptor (CD122/IL-15Rβ), had the greatest upregulation of all cytokine-associated genes after N-809 treatment (figure 3F). Together, these results suggest that N-809 induces an immunostimulatory environment both systemically and in the TME.
N-809 enhances the ratio of effector to suppressive immune cells in the TME and draining LN
Changes to cytokine levels and signaling suggested major alterations to the immune compartment by N-809, especially compared with N803+αPD-L1. Supporting this, NanoString analysis of the tumor (figure 4A) showed an enrichment of genes associated with the lymphoid compartment (figure 4B), cytotoxicity (figure 4C), costimulation (figure 4D) and the myeloid compartment (figure 4E) by N-809, not N-803+αPD-L1. The most upregulated genes by N-809 were related to NK and CD8+ T-cell cytotoxicity and infiltration, including Grzma (granzyme A), Prf1 (perforin) and Grzme (granzyme E), Cxcr3 (CXCR3), Tbx21 (T-bet), Lck (LCK) and Cd3e (CD3ε) (figure 4A).

N-809 increases the ratio of immune effector to suppressive cells within the tumor microenvironment (TME) and periphery. MC38 tumors were implanted and mice treated as in figure 2. Primary tumor and draining lymph nodes (LN) were harvested 2 days after the last treatment. (A–E) RNA from the primary tumor was extracted and analyzed. (A) Heatmap of the genes from pathway scores for lymphoid compartment (B), cytotoxicity (C), costimulation (D) and myeloid compartment (E) that N-809 altered more than two-fold. Data in heat map are represented as fold change in gene expression over phosphate-buffered saline (PBS). Data are from one experiment, n=3–4 mice/group. (F–N) The number of natural killer (NK) cells (F), CD8+ T cells (G), regulatory T cells (Treg) (H) and CD8+ T cell to Treg ratio (I), or number of monocytic myeloid-derived suppressor cells (M-MDSC) (J), total macrophages (K), M1-like macrophages (L), M2-like macrophages (M) and M1-like to M2-like macrophage ratio (N) were determined in the primary tumor (top) or draining LN (bottom) by flow cytometry. Data are combined from two to three independent experiments, n=5 mice/group per experiment. All graphs show mean±SD. *P<0.05, **p<0.01, ***p<0.001.
In order to confirm and expand these findings, we performed a comprehensive analysis of immune subsets in the TME by flow cytometry. Corroborating the gene expression results, NK (figure 4F) and CD8+ T-cell (figure 4G) numbers were significantly enhanced after N-809 treatment while the number of Treg (figure 4H) and M-MDSC were decreased (figure 4J). Reduction in total macrophage numbers (figure 4K) was due to decreased immunosuppressive M2-like (figure 4M) but not pro-inflammatory M1-like (figure 4L) macrophages. N-809 did not alter CD4+ T-cell (online supplementary figure S3b) and G-MDSC (online supplementary figure S3c) numbers. Overall, N-809 promoted an inflamed TME by increasing the proportion of immune effector to suppressive cells, as shown by increased CD8+ T cell:Treg (figure 4I) and M1-like:M2-like macrophage (figure 4N) ratios. Other than reducing total macrophage numbers (figure 4K), although lesser than N-809, N-803+αPD-L1 did not affect TME immune cell numbers (figure 4F–N).
We also evaluated immune cell populations in the draining (d) LN. N-809 significantly enhanced NK cell (figure 4F), CD8+ T-cell (figure 4G) and CD4+ T-cell (online supplementary figure S3b) numbers. In contrast to the TME, Treg (figure 4H), M-MDSC (figure 4J) and total macrophage numbers (figure 4K) increased in the dLN with N-809. The expanded total macrophage number (figure 4K) was due to increased M1-like (figure 4L), not M2-like (figure 4M), macrophages. G-MDSC were not affected by treatment (online supplementary figure S3c). N-809 increased the ratio of immune effector to suppressive cells in the dLN, as shown by enhanced CD8+ T cell:Treg (figure 4I) and M1-like:M2-like macrophage (figure 4N) ratios. N-803+αPD-L1 treatment did not affect immune cell numbers in the dLN (figure 4F–N). These data demonstrate that N-809, not N-803+αPD-L1, enhances the ratio of immune effector to suppressive cells within the TME and periphery.
NK and CD8+ T cells are responsible for antitumor efficacy of N-809
Given the increased number of NK and CD8+ T cells with N-809 treatment (figure 4F,G), we next performed depletion studies to determine whether these cell types were required for N-809 efficacy. Depletion of either CD8+ T cells (figure 5A) or NK cells (figure 5B) completely abrogated the antitumor efficacy of N-809 (figure 5C–E). Depletion of both CD8+ T cells and NK cells (figure 5A,B) did not further enhance this effect (figure 5C–E). Thus, CD8+ T cells and NK cells are required for the antitumor efficacy of N-809.
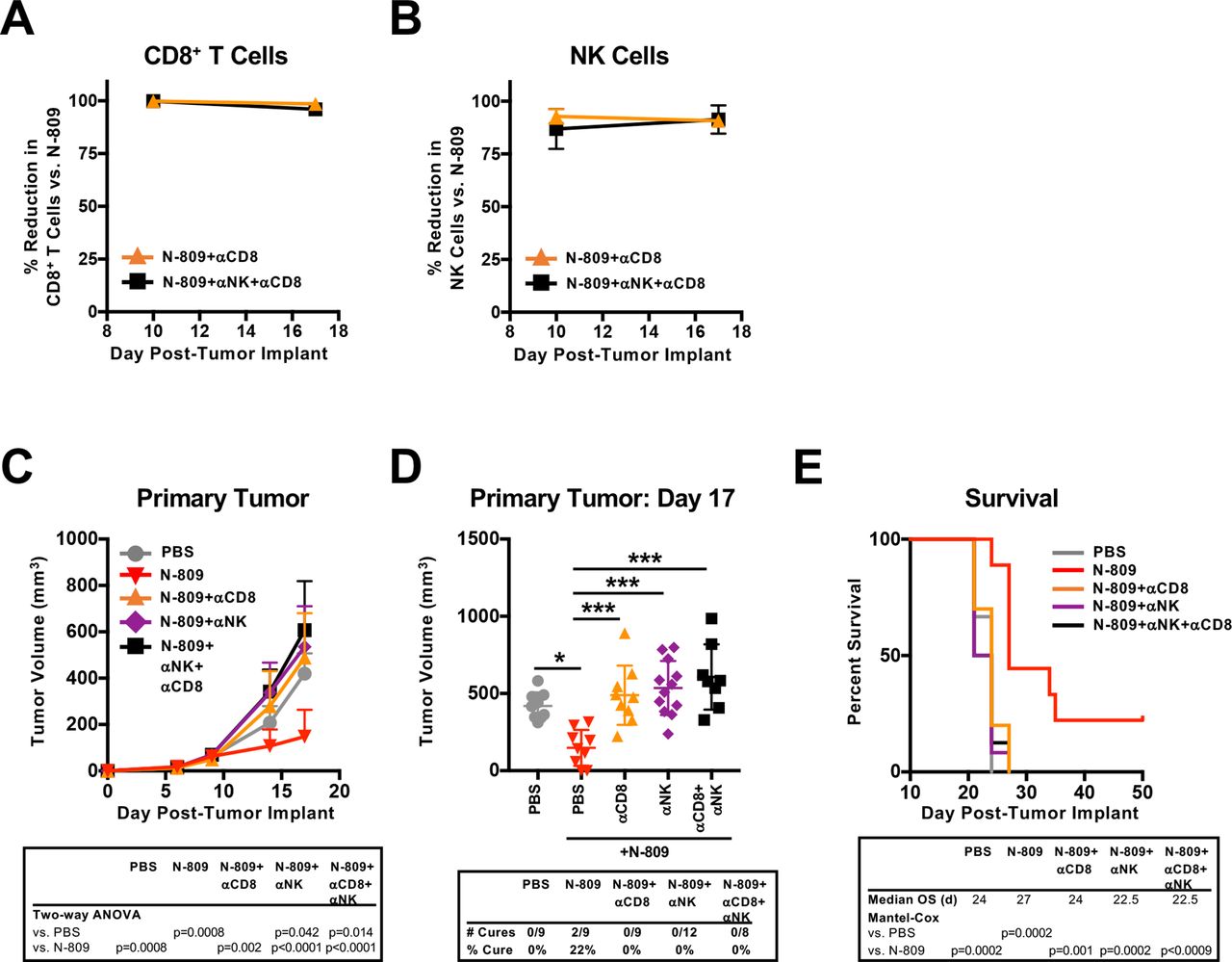
CD8+ T cells and natural killer (NK) cells are required for the antitumor efficacy of N-809. MC38 tumors were implanted as in figure 1 and mice were treated at days 9 and 13 with 100 µg N-809 (subcutaneously). CD8-expressing cells and NK cells were depleted on days 8, 10, 12, 16 and 19 using 100 µg anti-CD8 and/or 25 µL anti-asialo-GM1 (intraperitoneally). (A, B) Blood was obtained on days 10 and 17 to determine efficiency of CD8+ T-cell (a) or NK cell (B) depletions. Graphs show mean±SD. (C, D) Primary tumor growth curves (inset: two-way analysis of variance (ANOVA)) (C) and tumor volumes of individual mice at day 17 (inset: % cured mice) (D) show mean±SD. (E) Survival curves (inset: mOS) show % survival. Data are from one experiment, n=8–12 mice. *P<0.05, ***p<0.001.
N-809 promotes NK cell activation in the TME and improves NK killing
Next, we performed a comprehensive analysis of NK and CD8+ T-cell phenotype and function to further interrogate the mechanism of action of N-809.
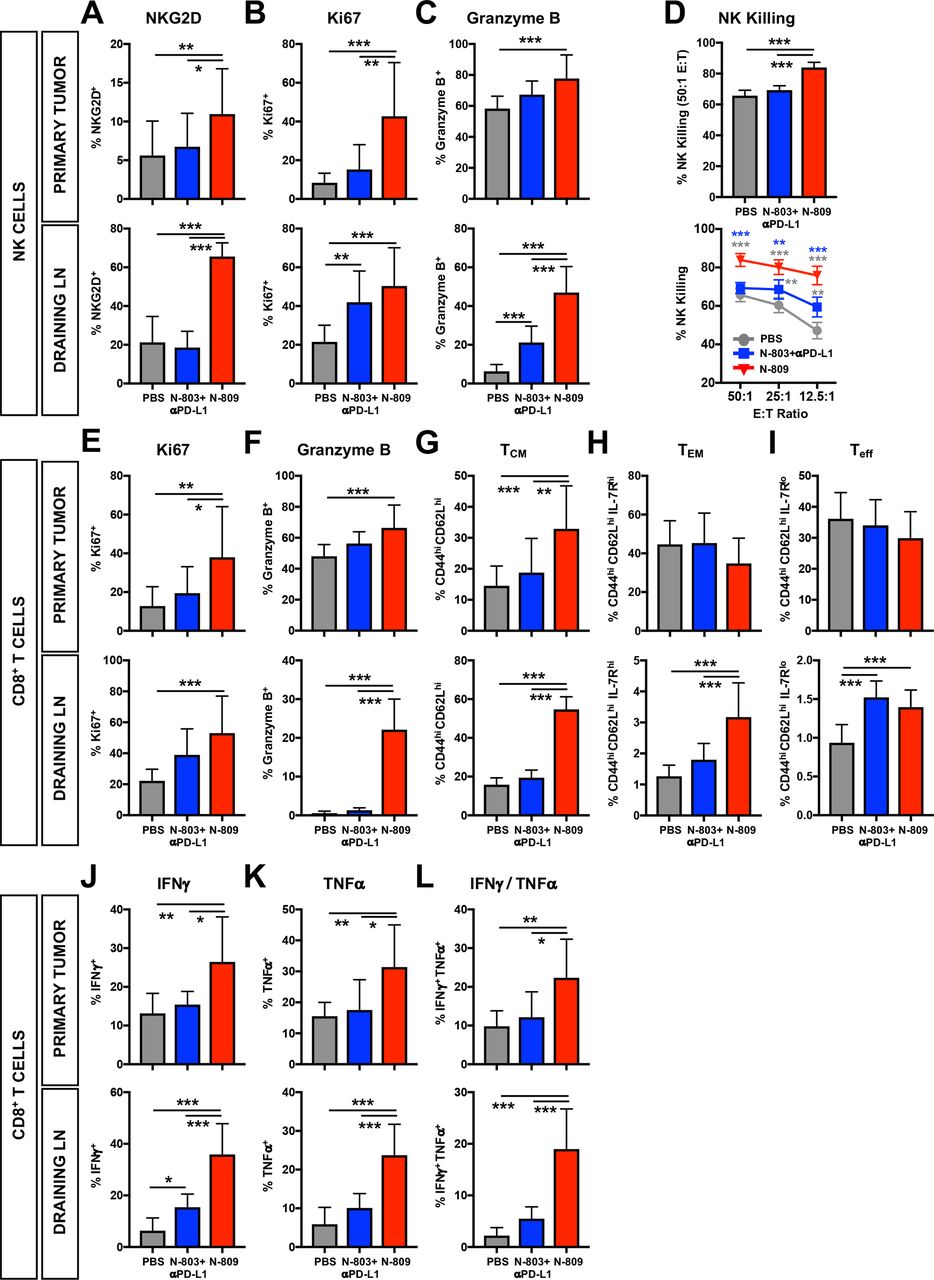
In the TME, N-809 significantly increased NK cell expression of activation-induced NKG2D (figure 6A), proliferation-associated Ki67 (figure 6B) and cytolytic molecule granzyme B (figure 6C) versus PBS and N-803+αPD-L1. Similar effects were observed in the dLN (figure 6A–C). In contrast to the TME, where N-803+αPD-L1 did not affect NKG2D, Ki67 or granzyme B levels, the combination significantly enhanced NKG2D (figure 6A) and granzyme B (figure 6C) in the dLN, although less than N-809 (figure 6A–C). The function of NK cells markedly improved with N-809 versus PBS or N-803+αPD-L1, as shown by increased NK killing of Yac-1 target cells at all effector:target (E:T) ratios (figure 6D). N-803+αPD-L1 also enhanced NK cell killing, but it was significantly reduced versus N-809 (figure 6D). Similar effects on NK-mediated lysis were observed in 4T1 tumor-bearing mice (online supplementary figure S3d). Thus, N-809 promotes greater activation and function of NK cells than N-803+αPD-L1.

N-809 induces an activated natural killer (NK) and CD8+ T-cell phenotype and enhances function. MC38 tumors were implanted and mice treated as in figure 2. NK (A–D) and CD8+ T cells (E–l) were examined by flow cytometry. (A–C) Graphs show frequency of NKG2D+ (A), Ki67+ (B) and granzyme B+ (C) NK cells in the primary tumor (top) and draining lymph nodes (LN) (bottom). (D) Purified splenic NK cells were co-cultured with 111In-labeled Yac-1 target cells at the designated E:T ratios for 18 hours. 111In release was measured to determine cytotoxic function. (E–I) Graphs show frequency of CD44hiKi67+ (E), CD44higranzyme B+ (F), CD44hiCD62Lhi TCM (G), CD44hiCD62LloIL-7Rhi TEM (H) and CD44hiCD62LloIL-7Rlo Teff (I) CD8+ T cells in the primary tumor (top) and draining LN (bottom). (J–L) Immune cells were stimulated with 1 μg/mL αCD3 + 1 μg/mL αCD28 for 4 hours. Graphs show frequency of total interferon (IFN)γ+ (J), total tumor necrosis factor (TNF)α+ (K) and IFNγ/TNFα-double producing (L) CD44hi CD8+ T cells in the primary tumor (top) and draining LN (bottom) as determined by flow cytometry. All graphs show mean±SD. Data combined from two to three independent experiments, n=5 mice/group per experiment. *P<0.05, **p<0.01, ***p<0.001.
N-809 induces CD8+ T-cell activation and enhances cytokine production in the TME
Similar to the effect on NK cells, N-809 enhanced expression of Ki67 (figure 6E) and granzyme B (figure 6F) on activated intratumoral CD8+ T cells as compared with PBS and N-803+αPD-L1. Additionally, central memory (TCM) (figure 6G), but not effector memory (TEM) (figure 6H) or effector (Teff) (figure 6I) CD8+ T cells, were expanded. N-809 did not affect CD8+ T-cell clonality in the tumor (online supplementary figure S3e,f). Similar to the TME, N-809 enhanced Ki67 (figure 6E) and granzyme B (figure 6F) expression on activated CD8+ T cells in the dLN. In contrast to the tumor, N-809 increased the proportion of TCM (figure 6G), TEM (figure 6H) and Teff (figure 6I) CD8+ T cells. N-803+αPD-L1 did not significantly alter CD8+ T-cell phenotype in the tumor or dLN (figure 6E–I).
Correlating with the development of a more active CD8+ T-cell phenotype, N-809 increased the proportion of activated CD8+ T cells in the TME and dLN that produced IFNγ (figure 6J), TNFα (figure 6K) and both IFNγ and TNFα (figure 6L) versus PBS and N-803+αPD-L1. IFNγ production in the tumor did not change with treatment (online supplementary figure S3g), but N-809 significantly increased TNFα production on a per cell basis in the dLN (online supplementary figure S3h). N-803+αPD-L1 only increased the proportion of CD8+ T cells producing IFNγ in the dLN, but not to the level induced by N-809 (figure 6J). N-809, but not N-803+αPD-L1, also enhanced CD8+ T-cell function in the lungs of 4T1 tumor-bearing mice, as shown by the increased proportion of IFNγ-producing (online supplementary figure S3i), TNFα-producing (online supplementary figure S3j) and IFNγ/TNFα-producing (online supplementary figure S3k) CD8+ T cells. Together, these data suggest that N-809 induces activation and increases function of CD8+ T cells greater than N-803+αPD-L1.
N-809 drives CD8+ T-cell and NK cell migration into the TME through regulation of chemokine levels and chemokine receptor expression
N-809 promotes the accumulation of a large number of active, functional CD8+ T cells and NK cells within the TME (figures 4F,G and 5). This occurs through proliferation within the tumor or migration of de novo CD8+ T cells and NK cells from the periphery. In order to distinguish between these possibilities, we administered FTY720 to mice treated with N-809. FTY720 is a sphingosine-1 phosphate receptor inhibitor that sequesters lymphocytes within secondary lymphoid organs and prevents migration in the blood.28 CD8+ T cells are more sensitive to FTY720 than NK cells,29 30 as demonstrated by a significant reduction in CD8+ T cells (figure 7B), but not NK cells (figure 7A), in the blood after FTY720 treatment. Thus, only CD8+ T-cell expansion versus migration could be evaluated. N-809-induced increase of CD8+ T-cell number in the TME was completely abrogated by FTY720 administration (figure 7C), suggesting that N-809 promotes lymphocyte trafficking of CD8+ T cells into the tumor, not in situ expansion.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
N-809 increases chemokine levels and chemokine receptors related to CD8+ T-cell and natural killer (NK) cell migration. (A–C) MC38 tumors were implanted as in figure 1 and mice were gavaged daily beginning at day 9 with 25 μg FTY720. At days 10 and 14, mice were treated with 100 µg N-809 (subcutaneously). Immune cells were examined by flow cytometry. Graphs show numbers of NK cells (A) or CD8+ T cells in the blood (B) or in the primary tumor (C). Data are combined from two independent experiments, n=5–6 mice per group. (D–L) MC38 tumors were implanted and mice treated as in figure 2. All analysis was performed 2 days after the last treatment. (D) RNA from the primary tumor was extracted and analyzed by NanoString. Heatmaps show fold change in gene expression related to chemokine signaling over phosphate-buffered saline (PBS) (genes N-809 altered >1.5-fold). Data are from one experiment, n=3–4 mice/group. (E–H) Graphs show intratumoral concentrations of CXCR3 ligands CXCL9 (E) and CXCL10 (F) and CCR5 ligands CCL4 (G) and CCL5 (H). Data are combined from two to three independent experiments, n=5 mice/group per experiment. (I–L) NK cells (I, J) and CD8+ T cells (K, L) were examined by flow cytometry in the draining lymph nodes (LN) (top) and spleen (bottom). Graphs show CXCR3+ (I) or CCR5+ (J) NK cells and CXCR3+ (K) or CCR5+ (L) total or CD44hi CD8+ T cells. Data are combined from one to two independent experiments, n=5 mice/group per experiment. All graphs show mean±SD. *P<0.05, **p<0.01, ***p<0.001.
To investigate the effect of N-809 on CD8+ T-cell and NK cell trafficking, intratumoral gene expression associated with chemokine signaling was evaluated by NanoString. N-809 had a greater overall effect on genes related to chemokine signaling than N-803+αPD-L1 (figure 7D). N-809 significantly enhanced the expression of genes associated with migration of CD8+ T cells and NK cells, including Cxcr3 (CXCR3), Ccr5 (CCR5) and Ccl5 (CCL5), with Cxcr3 being the most upregulated gene in the TME (figure 7D).
To confirm these findings, we determined chemokine levels in the TME and chemokine receptor expression on NK and CD8+ T cells. Particular focus was placed on NK and CD8+ T-cell-expressed chemokine receptors CXCR3 and CCR5 and their respective ligands CXCL9/10 and CCL4/5, as they are the main drivers of NK and CD8+ T-cell migration to inflamed tissues and tumors.31–33 In the TME, N-809 significantly increased the level of CXCR3 ligand CXCL9 (figure 7E) but not CXCL10 (figure 7F). N-809 also increased CCR5 ligands CCL4 (figure 7G) and CCL5 (figure 7H). N-809 greatly upregulated expression of CXCR3 and CCR5 on NK (figure 7I,J) and CD8+ T cells (figure 7K,L) in the dLN and spleen. There was no expression of CXCR3 or CCR5 on NK or CD8+ T cells in the tumor (online supplementary figure S4). Correlating with its inability to increase immune cell numbers in the tumor, N-803+αPD-L1 did not affect chemokine levels in the TME (figure 7E–H) or chemokine receptor expression on NK or CD8+ T cells (figure 7I–L). These data suggest that N-809 increases effector cell numbers in the tumor through chemokine-induced CD8+ T-cell and NK cell trafficking to the TME.
Discussion
αPD-1/PD-L1 therapies can reduce immunosuppression within the TME by blocking the αPD-1/αPD-L1 interaction of T cells and tumor or immunosuppressive cell types.6 These therapies can also skew immunosuppressive cells like macrophages toward a more inflammatory, antitumor phenotype to support tumor rejection.4 The effect of αPD-1/PD-L1 therapies on effector cell populations has recently undergone a paradigm shift with the demonstration that they do not primarily mediate their efficacy through reinvigoration of T cells in the TME. Instead, migration of novel, peripherally activated CD8+ T-cell clones is required for improved antitumor efficacy.2 3 5 Together, these observations suggest that immunotherapy strategies that reduce immunosuppressive cell populations, promote systemic T-cell responses and mediate lymphocyte trafficking to the TME may surpass the limited clinical efficacy of αPD-1/PD-L1 monotherapy in patients with carcinoma.
Bispecific/bifunctional immunotherapeutic agents simultaneously interact with two targets, allowing for specific localization of the agent, increased local effects, reduced toxicity and/or dual effects on distinct biological mechanisms.17–19 Thus, bifunctional immunotherapeutics may improve clinical outcome for patients with carcinoma using a single agent versus multiagent combination therapy strategies. In the studies presented here, we evaluated N-809, a novel bifunctional molecule composed of the IL-15/IL-15Rα superagonist N-803 fused to anti-PD-L1 domains.21 IL-15/IL-15Rα complexes have been fused with antibody domains before.34–36 To our knowledge, N-809 is the first bifunctional agent that pairs an IL-15/IL-15Rα complex and a checkpoint inhibitor and thus the potential to provide immune-activating signals while preventing suppression.
N-809 was administered subcutaneously, as N-803 is better tolerated when given subcutaneously versus intravenously.14 15 26 Recent studies demonstrated that subcutaneous administration of antibodies, including αPD-L1, does not reduce efficacy and may improve tolerability versus intravenous delivery.37 38 N-809 was well-tolerated when given subcutaneously, with no visible toxicity or loss of body weight. The observed transient, non-lethal, dose-dependent increase in inflammation in multiple organs correlated with an increased level of serum IFNγ, a known driver of IL-15 superagonist-mediated toxicity.26 39 40 Increasing the interval between N-809 doses, similar to the once per week clinical N-803 dosing,15 may ameliorate these effects. In contrast to N-809, N-803 is extremely toxic at high doses, so we were unable to evaluate equimolar doses of N-809 and N-803+αPD-L1. Thus, we compared N-809 with the clinically relevant dose of N-803+αPD-L1,9 14 15 a dose not previously evaluated in murine models.16 This difference in tolerability may be attributed to the reduction in IL-15 activity of N-809 versus N-803. However, it also suggests that PD-L1-targeting of an IL-15 superagonist reduces toxicity, as previously described.41 42 Extrapolating our findings, it may be possible that the clinically tolerated dose of N-809 is higher than N-803, leading to increased efficacy of N-809 in patients. The studies described here provide the rationale for such clinical investigation.
N-809 was superior to N-803+αPD-L1 in reducing 4T1 and MC38 tumor burden and increasing survival of MC38 tumor-bearing mice. The greater antitumor efficacy of N-809 versus N-803+αPD-L1 was unexpected, especially since N-809 had reduced PD-L1 occupancy in the tumor and peripheral immune compartment. This suggests that the N-803 component is the main driver of N-809 antitumor efficacy. Less PD-L1 blockade by N-809 versus N-803+αPD-L1 was most likely due to the reduced relative dose of αPD-L1 antibody administered (100 µg N-809 vs 200 µg αPD-L1). Additionally, it could be attributed to an increase in global PD-L1 expression by the IL-15 superagonist component since N-803 is known to upregulate PD-L1 expression.16 Thus, one can speculate that the superior antitumor efficacy of N-809 may arise from increased retention of its IL-15/IL-15Rα in the TME, brought by the binding to PD-L1.
The improved efficacy of N-809 correlated with an inflamed gene expression signature in the TME characterized by increased costimulation, cytotoxicity and IFN and proinflammatory cytokine signaling. N-809 enhanced the number of activated NK and CD8+ T cells in the periphery and tumor, and significantly increased NK and CD8+ T-cell function. These results support the hypothesis that N-809 enhances intratumoral lymphocyte numbers by increasing trafficking via altered chemokine levels in the TME and chemokine receptor expression on CD8+ T cells and NK cells. In addition, chemical inhibition of T-cell migration reduced intratumoral CD8+ T-cell numbers after N-809 treatment. Moreover, N-809 greatly upregulated the chemokine receptors CXCR3 and CCR5 on peripheral CD8+ T cells and NK cells, which are important for efficient T-cell and NK cell tumor infiltration.31–33 CXCR3 and CCR5 ligands CXCL9, CCL4 and CCL5, but not other chemokines, were also increased in the TME. N-803+αPD-L1 did not alter chemokine receptor expression or chemokine levels, suggesting that the mechanism of action of N-809 may be unique. While IL-15 has been shown to promote transient, peripheral retention of immune cells,43 the effect of IL-15 immunotherapy on immune cell trafficking in cancer has not been described. Our results showing that the increase in CD8+ T-cell number in the TME was due to migration, not in situ expansion, mirrors a recent clinical study demonstrating that the enhanced number of intratumoral CD8+ T cells observed after αPD-1 therapy was due to the migration of de novo activated peripheral T cells.5 Additional studies have described similar results with αPD-L1 therapy in preclinical models.2 3
Given the clinical and preclinical studies with N-803+αPD-1/PD-L1 combination, it was expected for N-809 to increase the number of CD8+ T cells and NK cells.15 16 24 44 However, in contrast to previous studies with N-803+αPD-L1,16 N-809 treatment significantly reduced immunosuppressive and pro-tumorigenic immune cells in the TME, including Treg, M2-like macrophages and M-MDSC. Given the pro-inflammatory cytokine signature in the TME and serum, the decrease in Treg and M2-like macrophages could be due to reprogramming of these cells. αPD-L1 therapy has been shown to skew M2-like to M1-like macrophages.4 However, the number of non-Treg CD4+ T cells and M1-like macrophages did not concomitantly increase with the reduction of Treg and M2-like macrophages, suggesting that these suppressive cells underwent apoptosis or cell-mediated death. Treg versus conventional CD4+ T cells undergo increased apoptosis in the TME due to oxidative stress,45 and high levels of IFNγ can induce cell death by other mechanisms. Alternatively, N-809 can mediate antibody-dependent cellular cytotoxicity (ADCC) of PD-L1-expressing tumor targets by NK cells.21 N-809 enhanced NK cell activation and function, and PD-L1 expression increased on macrophages and M-MDSC upon IL-15 superagonist treatment. Thus, it is possible that PD-L1-expressing M2-like macrophages and M-MDSC underwent NK cell-mediated ADCC after N-809 treatment. While PD-L1 expression on macrophages prevents killing by effector cells,46 the lower level of MHC class I on M2 versus M1 macrophages may increase their susceptibility to NK-mediated killing.47 The exact effect of N-809 on immunosuppressive cell populations in the TME is currently under investigation. Regardless, the reduction in intratumoral Treg, M2-like macrophages and M-MDSC could support N-809 efficacy, as the presence of these cell populations is associated with worse effector responses and prognosis.48–50
Conclusions
For the first time, we demonstrate that a bifunctional molecule targeting PD-L1 and IL-15 induces significant tumor control in multiple murine carcinoma models resistant to N-803 or αPD-L1 monotherapy. N-809 functions by increasing intratumoral NK and CD8+ T cells, most likely through migration of peripherally activated cells into the TME, not in situ proliferation. The antitumor efficacy of N-809 was further associated with significant reduction in tumor Treg and M2-like macrophages. Notably, this antitumor efficacy and novel immune mechanism was not observed with N-803+αPD-L1, an immunotherapy combination in the clinic. Overall, we conclude that N-809 reduces tumor burden by inducing the development of an inflamed TME through reduction of immunosuppressive cell populations and altered trafficking of activated effector immune cells. This is of particular importance as non-inflamed tumors are unlikely to respond to immune checkpoint inhibitors alone or in combination. Thus, agents such as N-809 represent an unmet medical need for converting ‘cold’ tumors to inflamed ones. These results provide the rationale for the clinical use of N-809 for treatment of carcinomas, including for patients with cold tumors, or an innate or acquired resistance to αPD-L1 therapy.
Acknowledgments
The authors would like to thank Curtis Randolph for excellent technical assistance; Yanyu Wang, Sara Loftus and Ludmila Krymskaya (Clinical Support Laboratory, Frederick National Laboratory for Cancer Research, Leidos Biomedical Research) for their technical support; and Debra Weingarten for her assistance in the preparation of this manuscript.
References
Footnotes
JS and SRG contributed equally.
Contributors Designing of research studies: KMK, SG and JS. Conducting experiments: KMK, KCH, YO, SG. Acquiring data: KMK, KCH. Analyzing data: KMK, KCH, SG. Writing the manuscript: KMK, SG and JS. All authors read and approved the final manuscript.
Funding This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute (NCI), National Institutes of Health, and by a Cooperative Research and Development Agreement (CRADA) between the NCI and ImmunityBio.
Disclaimer The authors from the National Cancer Institute do not have any competing interests to disclose.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All in vivo experiments were approved by the NIH Intramural Animal Care and Use Committee (IACUC).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. The data generated and analyzed will be made available from the corresponding author on reasonable request.