Article Text

Abstract
Background Spartalizumab is a humanized IgG4κ monoclonal antibody that binds programmed death-1 (PD-1) and blocks its interaction with PD-L1 and PD-L2. This phase 1/2 study was designed to assess the safety, pharmacokinetics, and preliminary efficacy of spartalizumab in patients with advanced or metastatic solid tumors.
Methods In the phase 1 part of the study, 58 patients received spartalizumab, intravenously, at doses of 1, 3, or 10 mg/kg, administered every 2 weeks (Q2W), or 3 or 5 mg/kg every 4 weeks (Q4W).
Results Patients had a wide range of tumor types, most commonly sarcoma (28%) and metastatic renal cell carcinoma (10%); other tumor types were reported in ≤3 patients each. Most patients (93%) had received prior antineoplastic therapy (median three prior lines) and two-thirds of the population had tumor biopsies negative for PD-L1 expression at baseline. The maximum tolerated dose was not reached. The recommended phase 2 doses were selected as 400 mg Q4W or 300 mg Q3W. No dose-limiting toxicities were observed, and adverse events included those typical of other PD-1 antibodies. The most common treatment-related adverse events of any grade were fatigue (22%), diarrhea (17%), pruritus (14%), hypothyroidism (10%), and nausea (10%). Partial responses occurred in two patients (response rate 3.4%); one with atypical carcinoid tumor of the lung and one with anal cancer. Paired tumor biopsies from patients taken at baseline and on treatment suggested an on-treatment increase in CD8+ lymphocyte infiltration in patients with clinical benefit.
Conclusions Spartalizumab was well tolerated at all doses tested in patients with previously treated advanced solid tumors. On-treatment immune activation was seen in tumor biopsies; however, limited clinical activity was reported in this heavily pretreated, heterogeneous population. The phase 2 part of this study is ongoing in select tumor types.
Trial registration number NCT02404441.
- programmed cell death 1 receptor
- immunotherapy
- clinical trials as topic
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Programmed death-1 (PD-1) is an inhibitory receptor expressed on a variety of immune cells, including activated T cells, regulatory T cells, and B cells.1 2 Interaction between PD-1 and its ligands, PD-L1 or PD-L2, leads to downregulation of effector T cell responses and mediates immune tolerance.3 4 PD-1 and PD-L1 are commonly upregulated on tumor-infiltrating lymphocytes and a wide variety of tumor cells, respectively.1 5 6 Monoclonal antibodies (mAbs) targeting PD-1 can restore effector T cell function and antitumor activity7 and have shown clinical benefit in patients with advanced cancers.8 9 Spartalizumab (PDR001) is a humanized IgG4κ mAb that binds PD-1 with subnanomolar activity in vitro and blocks interaction with PD-L1/PD-L2 in cell-based assays. Spartalizumab has also demonstrated pharmacodynamic (PD) activity and a favorable toxicology profile in preclinical studies, outlined in the Results section; notable differences from other PD-1 antibodies have not been observed. This first-in-human phase 1/2 study was designed to investigate the safety, pharmacokinetics (PK), and efficacy of spartalizumab in patients with advanced or metastatic solid tumors. Here, we describe the results from the phase 1 part of the study.
Methods
Preclinical analyses
In vitro binding of spartalizumab to PD-1 was assessed using surface plasmon resonance (Biacore). PD-1 immunoglobulin was covalently bound as ligand to a CM-5 chip, and spartalizumab was passed over in serial dilutions at a rate of 50 µL/min.
Spartalizumab was tested for its ability to block the binding of PD-L1 and PD-L2 to PD-1 in a competitive flow cytometry binding assay. Murine 300.19 cells expressing PD-1 were incubated with solutions that contained a constant concentration of PE-labeled PD-L1-Fc or PD-L2-Fc and serial dilutions of spartalizumab at 4°C for 4 hours. Bound labeled PD-L1-Fc or PD-L2-Fc were then quantified using fluorescence-activated cell sorting (FACS), and half maximal inhibitory concentration (IC50) values were derived from best-fit competition curves generated with Prism GraphPad software.
Clinical study design
This was a phase 1/2, multicenter, open-label study (NCT02404441), designed and sponsored by Novartis Pharmaceuticals Corporation. The data cut-off date was October 5, 2018.
Study objectives
The primary objective for the phase 1 part of the study was to estimate the recommended phase 2 dose (RP2D) and/or maximum tolerated dose (MTD) for spartalizumab. Secondary objectives included characterization of the safety and tolerability, and the PK profile of spartalizumab, and evaluation of the preliminary efficacy of spartalizumab. Exploratory objectives included assessment of potential predictive biomarkers for efficacy.
Patient selection
Eligible patients had locally advanced and/or metastatic solid tumors that had progressed on standard therapy, were intolerant to therapy, or for whom no standard therapy exists. Patients were aged ≥18 years and had Eastern Cooperative Oncology Group (ECOG) performance status of ≤2. Patients were required to have measurable disease or non-measurable disease using Response Evaluation Criteria In Solid Tumors (RECIST) v1.1, to have tumor(s) amenable to biopsy, and to give consent to tumor biopsy at baseline and during therapy with study drug. Key exclusion criteria included symptomatic central nervous system (CNS) metastases or CNS metastases requiring local therapy, impaired cardiac function or clinically significant cardiac disease, a history of severe hypersensitivity reactions to mAbs or drug-induced pneumonitis, and active, known, or suspected autoimmune disease. Immunosuppressive medication was not permitted, and patients were not eligible if they had received prior PD-1– or PD-L1–directed therapy at any time, or systemic anticancer therapy, radiotherapy, or major surgery within 2 weeks prior to the start of treatment. Concomitant chemotherapy and/or radiotherapy were not permitted.
Treatment plan
The starting dose of spartalizumab was 1 mg/kg, administered intravenously, every 2 weeks (Q2W), with an option to amend to schedules of every 3 weeks (Q3W) or every 4 weeks (Q4W). Treatment was administered until unacceptable toxicity, progressive disease as per immune-related response criteria (irRC),10 or patient/physician decision. Dose escalation decisions were based on all available safety, PK, and PD data, guided by an adaptive Bayesian logistic regression model (BLRM) following the escalation with overdose control (EWOC) principle. Treatment was discontinued if more than two consecutive doses were missed due to toxicity; delays of up to 7 days to allow recovery from adverse events (AEs) were permitted.
Safety assessments
Dose-limiting toxicities (DLTs, defined in the study protocol, online supplementary file 1) were assessed during the first 28 days of treatment. Regular safety assessments were performed, based on physical examination, ECOG performance status, laboratory parameters, and cardiac assessments. AEs, defined by the National Cancer Institute Common Terminology Criteria for Adverse Events V.4.03, were assessed at every visit.
Supplemental material
Response assessments
Efficacy was evaluated by local investigator’s assessment using RECIST v1.1 and irRC. Tumor assessments were performed at baseline and subsequent assessments were performed at 8 weeks, then every 8 weeks up to 40 weeks, then every 12 weeks until progression of disease per irRC, withdrawal of consent, or lost to follow-up.
PK assessments
Samples were collected for PK profiling on Days 1, 2, 3, 4, 8, 11, and 15 of Cycles 1 and 3, and Day 1 of Cycles 2, 4, 5, and 6, and at end of treatment. Serum concentrations were determined with liquid chromatography mass spectrometry.
PD analyses
An archival tumor sample and a newly obtained pretreatment tumor biopsy were collected at screening. There was no limit on the age of the archival tumor samples. An additional tumor biopsy was obtained during treatment, either in Cycle 2 or Cycle 3. PD-L1 levels in tumor cells were assessed centrally by immunohistochemistry (IHC) using a diagnostic developed for non-small cell lung cancer (NSCLC; Dako PD-L1 IHC 22C3 pharmaDx) and CD8+ levels were assessed by IHC, expressed as CD8+ staining as a percentage of the total sample area.
Results
Preclinical activity of spartalizumab
In vitro binding studies have shown that the mean dissociation constant (KD) values for spartalizumab binding to human and cynomolgus PD-1 were 0.83 and 0.93 nM, respectively; no binding was detected with mouse PD-1 (online supplementary table S1). In a cell-based assay, spartalizumab showed IC50 values of 0.9 and 1.3 nM for inhibiting the binding of PD-1 to PD-L1 and PD-L2, respectively (average of three experiments; online supplementary table S1). PD activity has also been demonstrated in vitro; spartalizumab induced a concentration-dependent increase in interferon gamma (IFNγ) release in a mixed lymphocyte reaction assay (eightfold to 13-fold increase relative to isotype control antibody), and enhanced interleukin-2 (IL-2) production in a Staphylococcal enterotoxin B ex vivo functional assay (twofold to threefold increase).
Supplemental material
Preclinical toxicity
In preclinical toxicology studies, repeated administration of spartalizumab was well tolerated in cynomolgus monkeys following once weekly intravenous administration at doses up to 100 mg/kg, for up to 14 weeks. Exposure in monkeys exceeded that seen in patients at 400 mg intravenous Q4W. Microscopic changes comprised macrophage infiltrates in the spleen, perivascular mononuclear cell infiltrates and/or fibrosis at the intravenous injection site, and vascular/perivascular mononuclear cell infiltrates in multiple tissues, which were generally of minimal to mild severity. These findings are consistent with the known biology associated with PD-1 blockade, which leads to the disinhibition of endogenous immune responses.
Clinical patient population, treatment, and disposition
Patient demographics and baseline characteristics are shown in table 1. The median age was 54.5 years (range 23–82) and 98% of patients had an ECOG performance status of 0 or 1. Patients had a wide range of primary tumor types, most commonly sarcoma (16 patients, 28%) and metastatic renal cell carcinoma (6 patients, 10%); other tumor types were reported in three or fewer patients each. The majority of patients had received prior antineoplastic therapy (93%), including neoadjuvant and/or adjuvant therapies, with a median of three prior regimens (range 0–11). Five patients had received prior immunotherapy, none of which were checkpoint inhibitors. At the data cut-off date (October 5, 2018), 58 patients had been treated in the phase 1 part of the study. Patients received spartalizumab, intravenously, at doses of 1, 3, or 10 mg/kg, Q2W (n=42), or 3 or 5 mg/kg, Q4W (n=16). All patients discontinued from the study due to progressive disease (81%), patient or physician decision (10%), death (7%; due to study indication (n=2), progressive disease (n=1), and respiratory failure (n=1)), or AEs (2%; grade 3 dyspnea, not suspected to be related to treatment).
Baseline patient demographics and characteristics, by treatment group
DLTs, PK, and RP2D
No patient experienced a DLT, assessed in the first 28 days of treatment. PK parameters are shown in table 2 and concentration–time profiles by dose/regimen are shown in figure 1. Exposure was approximately dose-proportional from 1 to 10 mg/kg Q2W and from 3 to 5 mg/kg Q4W based on Cycle 1 PK parameters. The observed mean half-life for spartalizumab ranged from 11 to 41 days, but this must be interpreted with caution due to the short observation window. The targeted Cycle 3 AUC0–336h levels of over 1000 day*µg/mL were reached with doses of 3 mg/kg Q2W and 5 mg/kg Q4W, comparable to levels achieved with other PD-1 inhibitors. The PK data were used to build a population PK model which examined the effect of bodyweight on spartalizumab PK. Based on the safety profiles observed at 3 mg/kg Q2W and 5 mg/kg Q4W and the population PK modeling, the corresponding flat dose of 400 mg Q4W was selected as the RP2D.11 An every 3 week dosing regimen with the equivalent weekly dose, 300 mg Q3W, was selected as an alternative regimen.11 These two doses achieved similar exposure; alternate dosing regimens may permit flexibility for combination with other therapies.

Median concentration–time profiles for spartalizumab, by treatment group. (A) Cycle 1. (B) Cycle 3. Q2W, once every 2 weeks; Q4W, once every 4 weeks.
Pharmacokinetic parameters for spartalizumab, by treatment group
Safety and tolerability
All patients experienced at least one AE and 36 patients (62%) experienced grade 3/4 AEs, regardless of relationship to study treatment, as detailed in online supplementary table S2. No fatal AEs were observed. AEs suspected to be related to study treatment were experienced by 34 patients (59%; table 3); the most frequent (≥10% of patients) were fatigue (13 patients; 22%), diarrhea (10 patients; 17%), pruritus (8 patients; 14%), hypothyroidism, and nausea (each 6 patients; 10%). Grade 3/4 AEs suspected to be related to study treatment were reported in two patients (3.4%); autoimmune colitis (biopsy confirmed) in one patient and decreased weight, hyperglycemia, and hypocalcemia in one patient. Both patients continued treatment; the patient with autoimmune colitis had an interruption to treatment. Grade 3 autoimmune colitis was the only on-treatment AE classified as a serious AE suspected to be related to treatment; this patient also experienced serious grade 2 pneumonitis over 2 months after receiving the last dose of treatment. The most commonly observed AEs of special interest (including immune-related AEs) suspected to be related to treatment were diarrhea (10 patients; 17%), pruritus (8 patients; 14%), and hypothyroidism (6 patients; 10%; online supplementary table S3).
Adverse events (any grade, occurring in ≥5% of patients, suspected to be related to study drug), by treatment group
Preliminary efficacy
The median duration of exposure to treatment was 13 weeks (range 2–110). Thirty-six per cent of patients received treatment for ≥20 weeks, and four patients received treatment for longer than 12 months (figure 2B). Forty-four patients out of 58 were evaluable for best percentage change in sum of target lesion diameters from baseline (figure 2A); a subset of patients was not evaluable due to unavailable post-baseline assessments (n=9: discontinued due to progressive disease, n=5; death, n=2; subject/guardian decision, n=1; AE not suspected to be related to treatment, n=1); non-measurable disease (n=4), or post-baseline assessment method different from baseline assessment (n=1). Partial responses were observed in two patients and stable disease was experienced by 22 patients, including one patient with undifferentiated liposarcoma who had stable disease persisting for over 24 months (table 4; figure 2C).
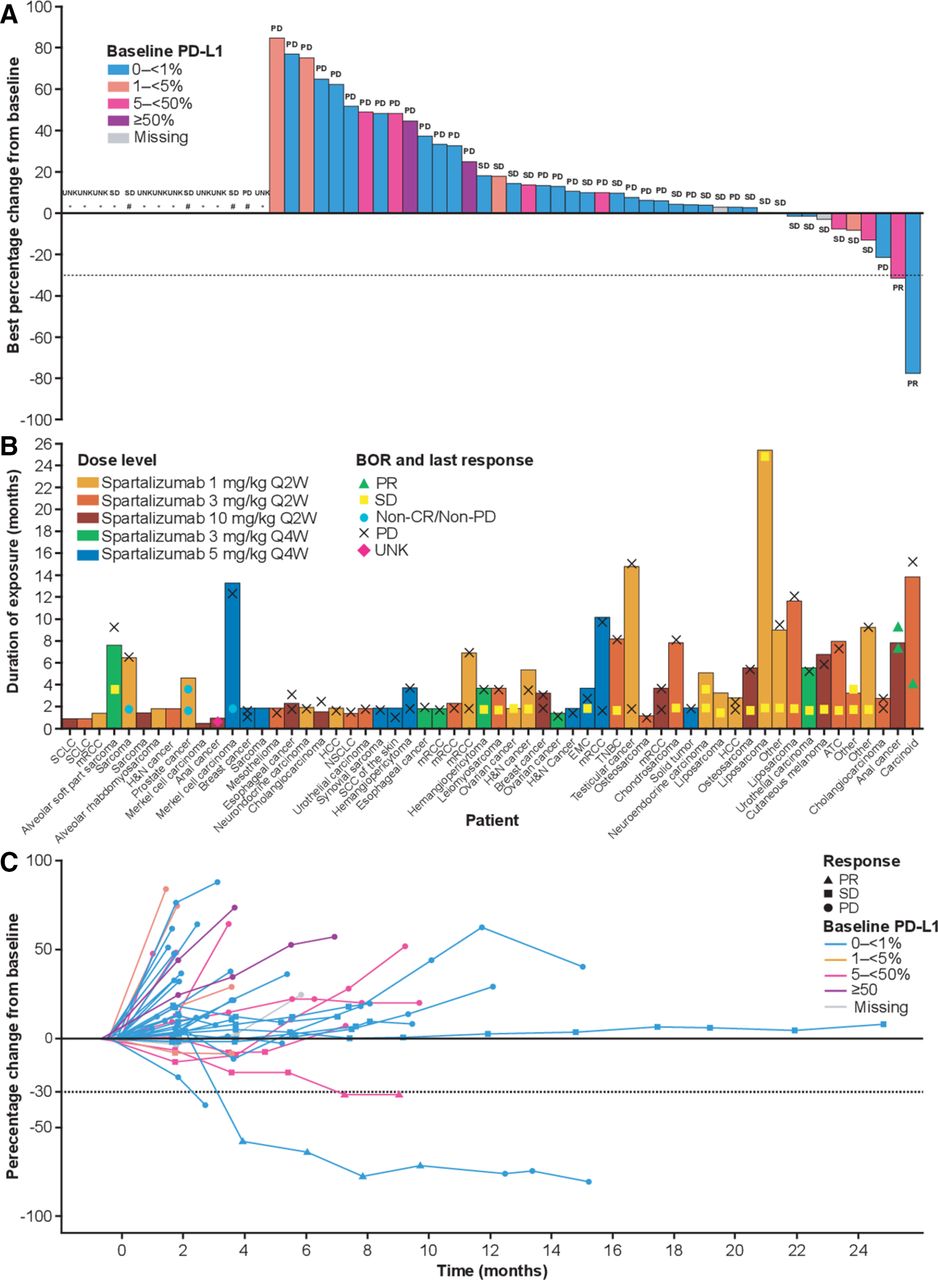
Percentage change from baseline in target lesions and duration of exposure to spartalizumab. (A) Best percentage change from baseline in target lesions, by PD-L1 expression at baseline. Best overall response is shown for each patient according to RECIST v1.1. (B) Duration of exposure to spartalizumab, by treatment group. Best overall response and response at last assessment are shown. (C) Percentage change from baseline in target lesions over time, by PD-L1 expression at baseline. Best overall response is shown for each patient according to RECIST v1.1. * Unavailable; # Non-measurable disease. ATC, anaplastic thyroid cancer; BOR, best overall response; EMC, extraskeletal myxoid chondrosarcoma; H&N, head and neck; HCC, hepatocellular carcinoma; mRCC, metastatic renal cell carcinoma; non-CR/non-PD, non-complete response/non-progressive disease; NSCLC, non-small cell lung cancer; PD, progressive disease; PD-L1, programmed death ligand 1; PR, partial response; Q2W, once every 2 weeks; Q4W, once every 4 weeks; RECIST, Response Evaluation Criteria In Solid Tumors; SCC, squamous cell carcinoma; SCLC, small cell lung cancer; SD, stable disease; TNBC, triple-negative breast cancer; UNK, unknown.
Best overall response (investigator assessed according to RECIST v1.1)
Of the two responding patients, one patient (10 mg/kg Q2W) had a diagnosis of anal cancer, for whom the duration of response was approximately 1.8 months at the last assessment, and the best percentage decrease from baseline in sum of target lesion diameters was 31%. The response was ongoing when this patient discontinued from the study. This patient experienced serious autoimmune colitis and pneumonitis and discontinued due to patient decision; the patient subsequently died due to disease progression, over a year after discontinuation from the study. The other responding patient (3 mg/kg Q2W) had a diagnosis of atypical carcinoid tumor of the lung (neuroendocrine). The duration of response was approximately 8.5 months, after which the patient developed progressive disease. The best percentage decrease from baseline in sum of target lesion diameters was 77%, and responses were seen in multiple lesions, in the liver, pleura, and lymph nodes (figure 3A,B).

{kind=link}
{kind=link}
{kind=link}
Tumor response assessments and immunohistochemistry of CD8+ lymphocyte infiltration in patient with atypical carcinoid tumor of the lung with a partial response to treatment. (A) Reduction in overall tumor burden (upper) and individual lesions (lower). (B) CT scans of the liver: (i) liver metastasis pretreatment; (ii) pseudo-progression at first restaging; (iii) response at second restaging. (c) High levels of CD8 detected by immunohistochemistry during Cycle 2. PD, progressive disease; PR, partial response; Q2W, every 2 weeks; Q4W, every 4 weeks; SD, stable disease.
Biomarker analyses
Tumor tissue samples obtained at baseline and on treatment were analyzed by IHC for PD-L1 expression (Dako PD-L1 IHC 22C3 pharmaDx) and CD8+ T cell infiltration (positive IHC staining as a percentage of total sample area; online supplementary table S4). The majority of patients (n=39; 67%) were negative for baseline PD-L1 expression (<1%), 12 patients (21%) had baseline PD-L1 expression ≥5%, and four patients (7%) had baseline PD-L1 expression ≥50%. Approximately 50% of patients had both baseline and on-treatment (Cycle 2 or 3) biopsy samples. A correlation was seen between an on-treatment increase in biomarker evidence of inflammation and a decrease in sum of target lesion diameters, both for PD-L1 expression (n=28; Spearman coefficient=−0.49) and CD8+ T cell infiltration (n=32; Spearman coefficient=−0.42; online supplementary figure S1).
For the patient with anal cancer who experienced a partial response, baseline PD-L1 expression was 10% with an increase to 50% observed at the Cycle 2 Day 1 (C2D1) biopsy; CD8+ T cell infiltration was 0.5% in the tumor sample obtained at baseline and 2.7% in the sample obtained on C2D1. For the patient with atypical carcinoid tumor of the lung who experienced a partial response, baseline PD-L1 expression was 0%; however, CD8+ lymphocyte infiltration was observed in a fresh tissue biopsy obtained from this patient at baseline (CD8=5.6%), with an increase to 37.6% observed at the C2D1 biopsy (figure 3C).
For the patient with liposarcoma who experienced stable disease for >2 years, both PD-L1 expression (baseline PD-L1=0%) and CD8 staining were low (baseline CD8=0.2%; C2D1 CD8=0.2%). A second patient with liposarcoma (well-differentiated) received treatment for >1 year (stable disease >9 months) and on-treatment increases were seen for both PD-L1 expression (baseline PD-L1=0%; C3D1 PD-L1=80%) and CD8 staining (baseline CD8=0.2%; C3D1 CD8=18.8%). A number of other patients with stable disease were found to have on-treatment increases in one or both markers, including a patient with triple-negative breast cancer (TNBC) and stable disease lasting ~8 months (baseline PD-L1=0%; C2D1 PD-L1=10%; baseline CD8=1.6%; C2D1 CD8=2.5%) and a patient with ovarian cancer and stable disease lasting ~2 months (baseline PD-L1=0%; C2D1 PD-L1=5%; baseline CD8=0.7%; C2D1 CD8=2.8%).
Discussion
Treatment with spartalizumab was generally well tolerated in this phase 1 study. The incidence of grade 3/4 AEs suspected to be related to treatment was low, and toxicities were consistent with what has been observed with other anti–PD-1 antibodies.12 13 The PK parameters of spartalizumab were characterized from 1 to 10 mg/kg Q2W and from 3 to 5 mg/kg Q4W. The data generated were used to build a population PK model which examined the effect of bodyweight on spartalizumab PK. At 3 mg/kg Q2W and 5 mg/kg Q4W, PK exposure exceeded 1000 day*μg/mL, which is comparable with marketed PD-1 inhibitors.14 Based on the population PK model, a flat dose of 400 mg Q4W was selected as the RP2D, and the predicted steady-state concentration of spartalizumab at this dose was sufficient for PD-1 blockade.11 15 This dose was therefore selected as the RP2D, with an alternative dosing schedule of 300 mg Q3W expected to achieve similar exposure. The availability of two dosing schedules should facilitate combination with other antineoplastic regimens.
The clinical activity observed in this phase 1 study was limited; however, this may be explained in part by the fact that the patient population was heavily pretreated and relatively depleted of patients with tumor types now known to be responsive to anti–PD-1 antibodies, such as NSCLC and melanoma8 16–18; only one patient with NSCLC and one with melanoma were included in this phase 1 study. Furthermore, less than a third of patients in the phase 1 part had tumor biopsies positive for PD-L1 expression, which is associated with response to anti–PD-1 therapies.19 Response rates of 7%–28% have been observed in the preliminary reports from the phase 2 part of this study, in patients with NSCLC, melanoma, and anaplastic thyroid cancer (ATC).20 21
Partial responses were observed in two patients in the dose escalation part of the study reported here; one response in a patient with anal cancer and one durable response in a patient with atypical carcinoid. Both patients had evidence of an on-treatment immune response, and the patient with anal cancer had PD-L1+ tumor cells at baseline. A number of other patients had stable disease, and although this alone is not evidence of clinical activity, paired tumor biopsies from some of these patients showed an increase in CD8+ lymphocyte infiltration on-treatment, consistent with the mechanism of action for spartalizumab. Evidence of immune activation in tumors, including an increase in intratumoral CD8+ lymphocytes and plasma IFNγ, has also been seen in patients with melanoma receiving a combination of spartalizumab with BRAF and MEK inhibitors.22 It may be that patients with a PD response to spartalizumab treatment harbor additional resistance mechanisms that prevent a tumor response. If so, these patients may benefit from combination treatment with spartalizumab and other therapies.
The phase 2 part of this study is ongoing and is investigating the safety and efficacy of spartalizumab in patients with ATC, TNBC, NSCLC, and melanoma; exploration of potential predictive biomarkers is also ongoing.20 21 Spartalizumab is also being investigated in combination with other immunotherapies and targeted therapies.
Conclusions
Spartalizumab was well tolerated in patients with advanced solid tumors. The RP2D was selected as 400 mg Q4W or 300 mg Q3W. Limited clinical activity was seen in this heavily pretreated patient population, which enrolled relatively few patients with tumor types known to be responsive to PD-1 blockade or with positive tumor PD-L1 expression. Treatment with spartalizumab resulted in immune activation in tumors, consistent with the mechanism of action for spartalizumab.
Acknowledgments
The authors would like to thank the patients who participated in the trial and their families. The authors would also like to thank the physicians, nurses, research coordinators, and other staff at each site who assisted with the study. The authors would also like to thank Geraldine Bostel, Victor Antona, and Dominique Pinet for their important contributions to this study. The study was sponsored by Novartis Pharmaceuticals Corporation. Editorial assistance was provided by Laura Hilditch, PhD, and was funded by Novartis Pharmaceuticals Corporation.
References
Footnotes
Twitter @AnaingMD
Contributors SC, JF, JM, APS, HS, and HW designed the study. Acquisition of data was done by AN, JG, HG, PMF, MOB, C-CL, SS, MOdO, AV, MT, JHMS, APS, and TMB. Analysis and interpretation of data was done by AN, JFG, HG, PMF, MOB, C-CL, SS, MOdO, AV, MT, JHMS, HW, HS, JF, JM, SC, and TMB. Statistical analysis was done by HW. Writing, review, and/or revision of the manuscript was done by all authors.
Funding This study was funded by Novartis Pharmaceuticals Corporation.
Competing interests AN received research funding from NCI, EMD Serono, MedImmune, Healios Oncology Nutrition, Atterocor, Amplimmune, ARMO BioSciences, Eli Lilly, Karyopharm Therapeutics, Incyte, Regeneron, Merck, BMS, Pfizer, Neon Therapeutics, Calithera Biosciences, TopAlliance Biosciences, Kymab, PsiOxus, and Immune Deficiency Foundation (spouse); travel/accommodation expense from ARMO BioSciences; and consultancy fees and research funding from CytomX Therapeutics and Novartis. JG received grant funding from Array, Tesaro, Moderna, Adaptimmune, and Alexo; personal fees from Oncorus, Regeneron, Pfizer, Incyte, Agios, Amgen, and Ironwood Pharmaceuticals; and grant funding and personal fees from Bristol-Myers Squibb, Genentech/Roche, Takeda, Blueprint, Loxo, Novartis, and Merck. PMF received research funding from Corvus and Kyowa; consultancy fees from AbbVie, Boehringer, EMD Serono, Inivata, Janssen, Lilly, and Merck; and research funding and consultancy fees from AstraZeneca, BMS, and Novartis. MOB received funding from Novartis, personal fees from BMS, Adaptimmune, GSK, Novartis, EMD Serono, Sanofi, Immunocore, and Turnstone; grant funding from Takara Bio; and personal fees and grant funding from Merck. C-CL received personal fees from BeiGene, Daiichi Sankyo, Roche, and Novartis. SS received research funding from GSK, Millennium, MedImmune, Johnson & Johnson, Gilead, Plexxikon, Onyx, Bayer, Blueprint, XuanZhu, Incyte, Toray, Celgene, Hengrui, OncoMed, Tesaro, AADi, Merck, Inhibrx, AMAL, and Syndax; personal fees and research funding from Novartis; equity from Iterion Therapeutics, Proterus Therapeutics, ConverGene, and Stingray Therapeutics; honoraria from Exelixis, Loxo Oncology, Natera, Hengrui Therapeutics, Tarveda Therapeutics, Dracen Pharmaceuticals, and Barricade Therapeutics; and owns stock with LSK BioPharma and Salarius Pharmaceuticals. MT received consultancy fees from Eisai, Bristol-Myers Squibb, Array Biopharma, Blueprint Medicines, Arqule, Loxo Oncology, Bayer, Novartis, and Genentech. JHMS received personal fees from Modra Pharmaceuticals. TMB received grant funding from Daiichi Sankyo, Medpacto, Incyte, Mirati Therapeutics, MedImmune, AbbVie, AstraZeneca, MabVax, Stemline Therapeutics, Merck, Lilly, GlaxoSmithKline, Novartis, Genentech, Deciphera, Merrimack, Immunogen, Millennium, Phosplatin Therapeutics, Calithera Biosciences, Kolltan Pharmaceuticals, Principa Biopharma, Peleton, Immunocore, Roche, Aileron Therapeutics, Bristol-Myers Squibb, Amgen, Onyx, Sanofi, Boehringer-Ingelheim, Astellas Pharma, Five Prime Therapeutics, Jacobio, Top Alliance BioScience, Janssen, Clovis Oncology, Takeda, Karyopharm Therapeutics, Foundation Medicine, and ARMO Biosciences; grant funding and consultancy fees from Leap Therapeutics; grant funding, consultancy fees, and non-financial support from Ignyta and Moderna Therapeutics; grant funding, personal fees, and consultancy fees from Pfizer; grant funding, personal fees, and non-financial support from Loxo and Bayer; personal fees and non-financial support from Guardant Health; and personal fees from Exelesis. HW, HS, and JM are employees of Novartis. APS and JF are employees of Novartis and own stock with Novartis. SC is an employee of Novartis and has patents with Novartis.
Patient consent for publication Not required.
Ethics approval The study protocol was approved by an independent ethics committee or institutional review board for each center. The study was conducted according to the principles of the Declaration of Helsinki and was performed in compliance with Good Clinical Practice guidelines. Written informed consent was obtained from each patient.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.