Article Text

Abstract
Background Immune compromised mice are increasingly used for the preclinical development of monoclonal antibodies (mAb). Most common are non-obese diabetic (NOD) severe combined immunodeficient (SCID) and their derivatives such as NOD SCID interleukin-2 γ-/- (NSG), which are attractive hosts for patient-derived xenografts. Despite their widespread use, the relative biological performance of mAb in these strains has not been extensively studied.
Methods Clinically relevant mAb of various isotypes were administered to tumor and non-tumor-bearing SCID and NOD SCID mice and the mAb clearance monitored by ELISA. Expression analysis of surface proteins in both strains was carried out by flow cytometry and immunofluorescence microscopy. Further analysis was performed in vitro by surface plasmon resonance to assess mAb affinity for Fcγ receptors (FcγR) at pH 6 and pH 7.4. NOD SCID mice genetically deficient in different FcγR were used to delineate their involvement.
Results Here, we show that strains on the NOD SCID background have significantly faster antibody clearance than other strains leading to reduced antitumor efficacy of clinically relevant mAb. This rapid clearance is dependent on antibody isotype, the presence of Fc glycosylation (at N297) and expression of FcγRII. Comparable effects were not seen in the parental NOD or SCID strains, demonstrating the presence of a compound defect requiring both genotypes. The absence of endogenous IgG was the key parameter transferred from the SCID as reconstituting NOD SCID or NSG mice with exogenous IgG overcame the rapid clearance and recovered antitumor efficacy. In contrast, the NOD strain was associated with reduced expression of the neonatal Fc Receptor (FcRn). We propose a novel mechanism for the rapid clearance of certain mAb isotypes in NOD SCID mouse strains, based on their interaction with FcγRII in the context of reduced FcRn.
Conclusions This study highlights the importance of understanding the limitation of the mouse strain being used for preclinical evaluation, and demonstrates that NOD SCID strains of mice should be reconstituted with IgG prior to studies of mAb efficacy.
- immunotherapy
- antibodies, neoplasm
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
The growth in the numbers of monoclonal antibodies (mAb) being developed for the clinic, particularly for use in cancer, has led to the concurrent development of in vivo models enabling their preclinical evaluation.1 These models have increasingly made use of immune-compromised mice for growing patient-derived tumor xenografts and engrafting human immune or stem cells.2 3
Commonly used models include non-obese diabetic (NOD) severe combined immunodeficient (SCID) mice. The SCID mutation occurs in the Prkdc gene and impairs V(D)J recombination, leading to an absence of functional B and T cells and resulting in mice lacking endogenous IgG.4 5 The NOD phenotype results in reduced NK cell frequency and function and the absence of hemolytic complement activity.6 While these immune-deficient phenotypes make NOD SCID mice attractive recipients for cell transfers (such as human peripheral blood mononuclear cells (PBMC) and tumor xenografts), they may be further enhanced by additional genetic deletions such as the interleukin-2 γ-chain (NSG).7 8
While the effector function defects of NOD SCID mice and their related strains are often considered, one aspect regularly overlooked is mAb clearance, despite the fact that genetic alterations, as well as the lack of endogenous IgG in immune deficient strains, could readily impact on mAb pharmacokinetics, resulting in altered efficacy.9
The primary receptors responsible for mediating IgG mAb activity are the Fc gamma receptor (FcγR) family. It is composed of six receptors in humans and four in mice, which vary in expression pattern and affinity for IgG subclass.10 Another receptor capable of interacting with IgG in both humans and mice is FcRn, which is widely expressed throughout the body. The pH-dependent nature of FcRn-IgG interactions allows the receptor to scavenge IgG from lysosomes at an acidic pH, releasing it back into the circulation at neutral pH, providing the long in vivo half-life of antibodies.11–13
In addition to the potential issue of altered efficacy arising from the lack of endogenous IgG (and reduced competition for FcγR with therapeutic mAb) in NOD SCID mice, previous reports indicate that immune-compromised mice, such as NOD SCID and NSG, have reduced mAb half-life compared with related strains.14–16 More recently, it was reported that NOD SCID mice display an anomalous biodistribution of therapeutic antibodies, including reduced tumor targeting.17 This suggests further work is required to understand the limitations of these models and develop strategies to overcome their shortcomings to make more translationally relevant preclinical tumor models.
During a recent project examining the efficacy of a tumor targeting antibody in NOD SCID mice, we noted rapid mAb clearance of human (h) IgG1 and mouse (m) IgG2a isotypes. Using a Eμ-Tcl1 hCD20 +tumor model we found this rapid clearance resulted in reduced efficacy of clinically relevant mAb, such as rituximab. Employing genetically altered mice, we showed the rapid mAb clearance was dependent on the expression of the inhibitory FcγR, FcγRII. Additionally, we identified a reduced level of FcRn expression in NOD SCID mice, leading us to propose a novel hypothesis for how mAb half-life is regulated in these strains and means through which it can be overcome.
Materials and methods
In vivo experiments
Mice used in this study were bred and maintained in local facilities with experiments approved through local ethics committees and performed according to Home Office guidelines.
Generating bone marrow chimera
Recipient mice were provided with acid water, pH 2.5 on day −7 until 14 days after bone marrow receipt. Recipients received 1.1 Gy radiation on days −1 and 0 using a MultiRad 350 X-ray Irradiator (Faxitron). Bone marrow was harvested from donor mice and 3–8×106 cells injected intravenously (I.V.) into recipients. Systemic reconstitution was confirmed by flow cytometry 8–10 weeks after engraftment.
EµTCL-1 tumor model
This model has been described previously.18 19 Briefly, 1×107 cryopreserved Eµ-TCL-1 transgenic (Tg) or hCD20+ Eµ-TCL-1 Tg tumor splenocytes were injected intraperitoneally (I.P). into recipient mice. The presence of tumor was monitored in peripheral blood. Once tumor cells (CD19+CD5mid) were detectable by flow cytometry, mice were treated. The white cell count was determined using a Coulter Z1 particle counter with red blood cells (RBC) lysed using ZAP-OGLOBIN II (both Beckman Coulter) or by flow cytomery using Precision Count beads (Biolegend).
In vivo antibodies
All clinical antibodies were gifted from the Southampton General Hospital oncology pharmacy. Others were produced in-house. 18B12 and Rituximab isotype variants were cloned onto the appropriate IgG framework, produced in CHO cells and purified from culture supernatant with protein A. Purity was assessed by electrophoresis (Beckman EP; Beckman) and lack of aggregation confirmed by size exclusion (SEC) high performance liquid chromatography (HPLC). Unless otherwise stated, all antibodies were administered I.P. in 200 µL sterile PBS (Severn Biotech).
Flow cytometry
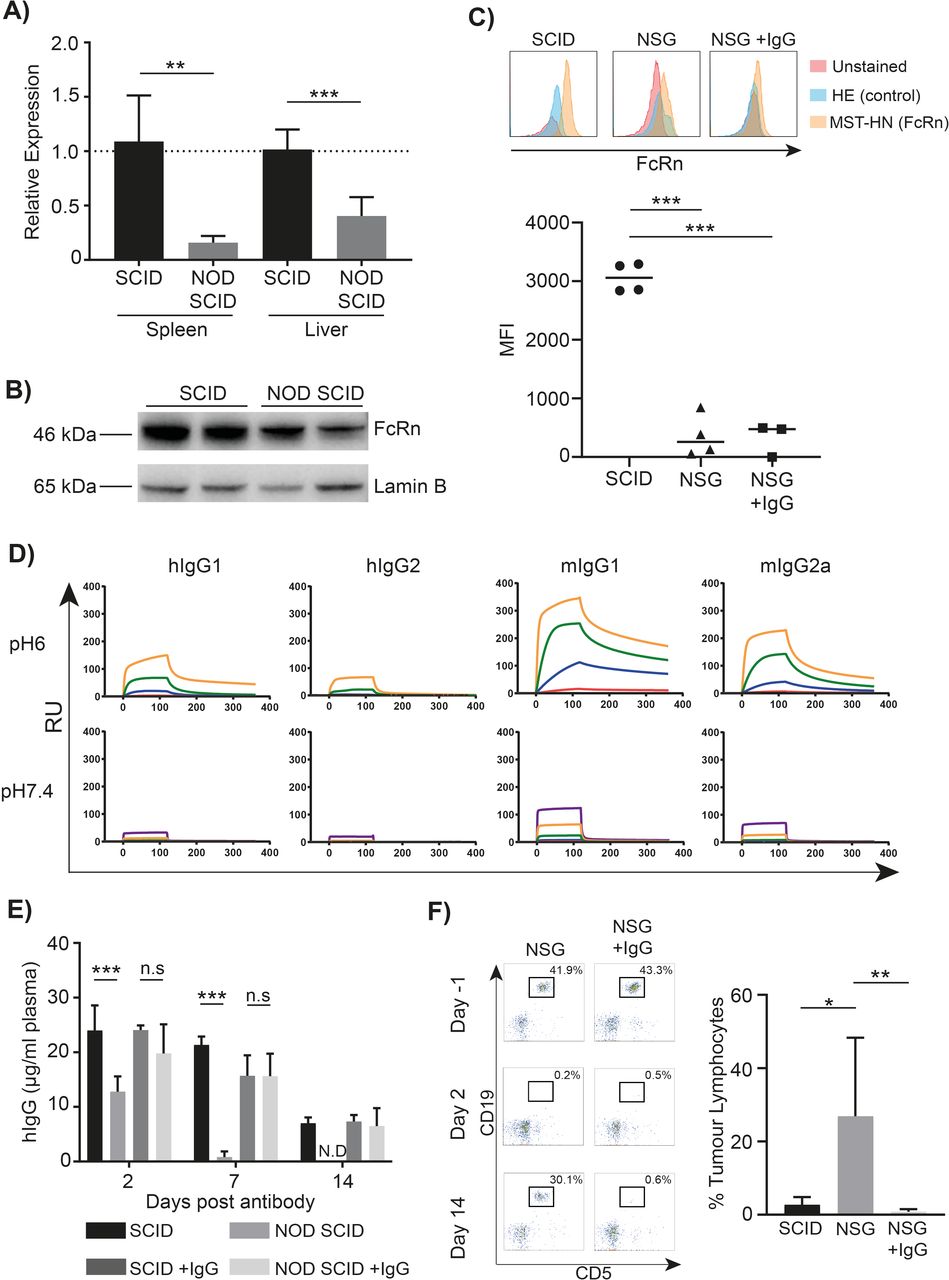
Flow cytometry was performed using the antibodies listed in online supplementary table 1. Anti-mFcγR has been reported previously.20 21 Following staining, RBC lysis buffer was added (AbD Serotec) and cells washed before analysis on a FACS Canto or FACS Calibur flow cytometer (BD Biosciences). Alexafluor 647 labeled MST-HN and H435A Abdegs for analyzing FcRn expression were a kind gift from Prof Sally Ward (University of Southampton) and used at 5 µg/mL with Fc block (2.4G2, 5 µg/mL) prior to extracellular staining.
Supplemental material
Generating bone marrow derived macrophages
The tibia and femur of mice were flushed with sterile complete RPMI (RPMI 1640 (Life Technologies), 2 mM L-glutamine, 1 mM sodium pyruvate, 100 U/mL penicillin, 100 µg/mL streptomycin (all Life Technologies), 10% fetal calf serum (Sigma-Aldrich). Cells were plated in 6-well plates at 0.8×106 cells/mL in complete RPMI +20% L929 conditioned media for 7–10 days.
Determining plasma IgG concentration
IgG concentration was determined by ELISA with reference to a standard curve of the same antibody as follows: for hIgG, maxisorp plates (Thermo Scientific) were coated with 5 µg/mL goat antihuman Fc-specific polyclonal antibody (Sigma-Aldrich) and blocked with PBS+1% BSA before addition of serum for 1 hour and washing. Detection was with horseradish peroxidase (HRP) conjugated F(ab’)2 goat anti-hFc specific antibody (Jackson Immunoresearch). Plates were incubated with o-phenylenediamine dihydrochloride (OPD) substrate and OD495 measured using an Epoch microplate spectrophotometer (Biotek). For quantification of mIgG, plates were coated with rabbit anti-mIgG and detected with HRP- rabbit anti-mIgG (both Jackson Immunoresearch).
Heat aggregation of IgG
Purified IgG was heated to 64°C for 30 min. The aggregated fraction was purified on a superdex S200 column (GE Healthcare). Aggregation was confirmed by HPLC using a Zorbax GF-250 column (Agilent).
Producing mFcγRII extracellular domain protein
RNA was isolated and cDNA generated from SCID or NOD SCID bone marrow derived macrophages (BMDM) and the mFcγRII gene amplified using gene specific primers. Subsequently, the extracellular domain (residues 1–207) of mFcγRII were cloned with the addition of a 6xHis tag. The construct was transfected into MEXi −293E cells (IBA lifesciences) and FcγRII-His expressed according to the manufacturer’s protocol and protein purified using a HisTrap HP column on an AKTA prime system (Both GE biosciences) and purity confirmed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE).
Surface plasmon resonance analysis
Surface plasmon resonance (SPR) was performed using a Biacore T100 system upgraded to a T200 (GE Life Sciences). For mFcγRII isoforms, anti-His capture antibody was immobilized on a CM5 chip (GE life Sciences). Purified FcγRII-His (10 µg/mL) was flowed over the chip at 30 µL/min for capture. IgG was injected at 30 µL/min. For all other analysis, IgG was immobilized via amine coupling with a target of 2000 RU. Recombinant mFcγRII or mFcRn (R&D systems) was flowed over the immobilized IgG in HBS-EP +buffer (GE Life Sciences) at pH7.4 or pH6.0. Affinity constants were determined by analysis with Biacore Bioevaluation software assuming 1:1 binding.
Quantitative PCR
mRNA was extracted from SCID or NOD SCID splenocytes and hepatocytes using an RNeasy Mini Kit (Qiagen) and cDNA generated using a Superscript III reverse transcription kit (Life Technologies). Quantitative PCR (qPCR) was performed using GoTaq qPCR master mix (Promega) using gene specific primers, with HPRT1 as a control. Ct values were normalized using HPRT1 values and the ΔΔCt method used to calculate fold change.
Western blotting
Lysates were produced from 5×106 SCID or NOD SCID splenocytes and hepatocytes using RIPA buffer (150 mM NaCl, 1% Triton X-100, 0.5% Deoxycholate, 0.1% SDS, 50 mM Tris, pH 8) and run on a 12% Novex Nupage BIS-TRIS gel (Thermo Fisher) before transfer to a methylcellulose membrane (GE Lifesciences). Primary antibodies were antimouse FcRn and Lamin B with detection using an HRP-conjugated donkey anti-goat antibody (online supplementary table 1).
Immunofluorescence
Liver tissue from BALB/c or NSG mice was embedded in OCT (CellPath) and frozen in isopentane on dry ice. 8 µm sections were cut and transferred to Superfrost plus slides (Thermo Scientific), air dried overnight and fixed in 100% acetone. Following blocking, primary antibodies against FcRn or FcγRII were added overnight before detection with Alexafluor 488-labeled secondary antibody (online supplementary table 1) for 45 min. Subsequently, primary antibodies against Clec4F or cytokeratin 8 were added for 2 hours before detection with AlexaFluor 549 or AlexaFluor 568 conjugated secondary antibodies (online supplementary table 1). Slides were mounted using Vectashield hardset with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories).
Images were collected using a CKX41 inverted microscope with a reflected fluorescence system equipped with a DP22 camera running CellSens software, using Plan Achromat 10×0.25 and 40×0.65 objective lenses (all from Olympus). Images were transferred to ImageJ (Fiji) or photoshop (Adobe) where background autofluorescence was removed, contrast stretched and brightness adjusted to maximize clarity, with all images treated equivalently.
Results
Antitumor mAb therapy is less effective in NOD SCID mice due to rapid antibody clearance
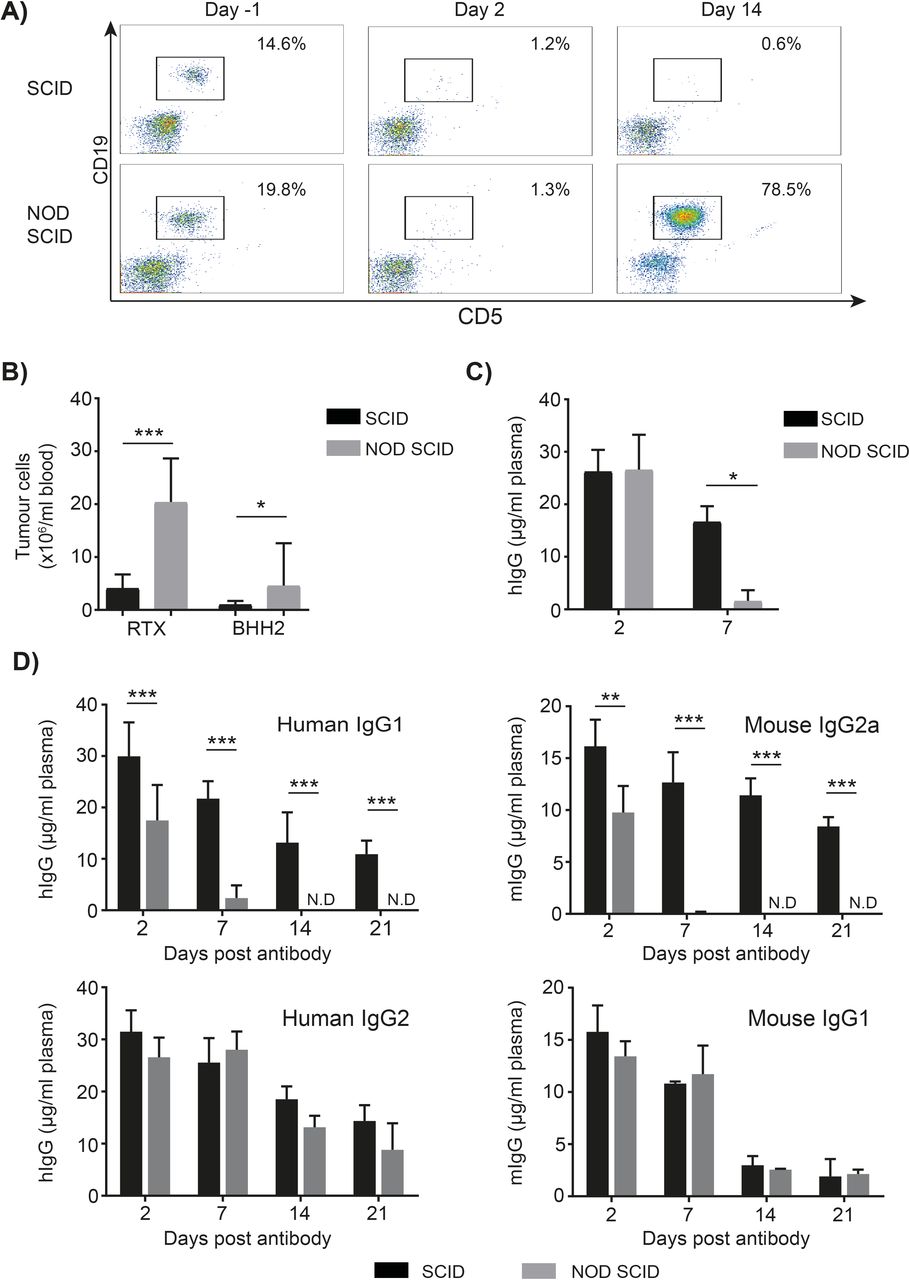
To explore potential differences of recipient mouse strains on immunotherapy efficacy, SCID and NOD SCID mice bearing established hCD20+ Eµ-TCL-1 tumors were treated with rituximab. Although initial tumor clearance was comparable between strains, 14 days after mAb treatment there were significantly more tumor cells in the peripheral blood of NOD SCID compared with SCID mice (figure 1A,B). To determine if this was associated with rituximab’s type I nature,22 23 we repeated the experiment with the type II anti-hCD20 mAb, BHH2,18 and observed the same reduced efficacy in NOD SCID compared with SCID mice (figure 1B).

Antibody mediated therapy is reduced in NOD SCID mice due to faster mAb clearance. (A–C) Eµ-Tcl1 x hCD20 Tg tumor cells were injected I.P. into SCID or NOD SCID mice. Once tumor was detectable in the peripheral blood animals were treated with 100 µg rituximab or BHH2 I.P. (A) 2 and 14 days after treatment, the percentage of tumor cells in the blood was assessed. (B) The number of tumor cells in the blood 14 days after treatment was determined (n=4–9). (C) The concentration of hIgG in the plasma was measured by ELISA with a significantly lower concentration of hIgG on day 7 in NOD SCID compared with SCID mice. hIgG was not detectable in the plasma of NOD SCID mice from day 14 onwards. (D) In the absence of tumor, 100 µg cetuximab or 100 µg rituximab hIgG2, mIgG2a or mIgG1 was administered I.P. to SCID or NOD SCID mice. The concentration of human or mouse IgG in the plasma was determined by ELISA. (hIgG1 n=6–7; combined data from two independent experiments, hIgG2, mIgG1 and mIgG2a n=3, representative of 2 independent experiments). ND=not detectable. statistics; two-way ANOVA with multiple comparisons *p<0.05, **p<0.01, ***p<0.001. No significant differences were observed between SCID and NOD SCID mice -receiving hIgG2 or mIgG1. ANOVA, analysis of variance; NOD, non-obese diabetic; SCID, severe combined immunodeficient.
To understand this difference in efficacy, the concentration of injected hIgG in the plasma of mice following treatment was determined (figure 1C). This revealed that 7 days after mAb treatment there was significantly less (~10-fold) hIgG in the plasma of NOD SCID compared with SCID mice (16.7 vs 1.6 µg/mL) suggesting that rapid hIgG1 clearance in NOD SCID mice was responsible for the less prolonged tumor deletion.
To investigate whether the rapid clearance was related to the mAb, strain and/or tumor, an alternative hIgG1 mAb, cetuximab, was administered to non-tumor bearing SCID and NOD SCID mice. Cetuximab was also more rapidly cleared from NOD SCID compared with SCID mice with hIgG being undetectable in the plasma of NOD SCID mice by day 7 post-administration (figure 1D). Similar results were also observed with other hIgG1 mAb including trastuzumab (online supplementary figure 1), showing that the rapid clearance is directly related to the NOD SCID strain, independent of tumor and a common feature of therapeutically-relevant hIgG1 mAb. Importantly, the hIgG1 clearance in SCID and NOD mice was comparable to that of immune-competent BALB/c mice (online supplementary figure 2 and previously shown24), confirming fast hIgG1 clearance in NOD SCID mice, rather than slow clearance in SCID or NOD mice. Furthermore, the lack of a difference in SCID mice demonstrates that rapid hIgG clearance does not result from the absence of endogenous IgG or immune deficiency per se.
Supplemental material
Supplemental material
Rapid mAb clearance in NOD SCID mice is isotype dependent and requires both SCID and NOD genotypes
To determine if rapid mAb clearance in NOD SCID mice extended beyond hIgG1, isotype switch variants of rituximab were generated and administered to SCID or NOD SCID mice. Similar to hIgG1, mIgG2a also had a significantly faster mAb clearance in NOD SCID mice (figure 1D), being no longer detectable in the plasma by day 14. In contrast, hIgG2 and mIgG1 had similar clearance rates in both strains. These results demonstrate that faster mAb clearance in NOD SCID mice is isotype dependent.
We next assessed whether the rapid IgG clearance occurred in NOD and NSG strains. NOD mice had a normal hIgG1 clearance rate, akin to that seen in SCID and BALB/c (figure 2A). However, NSG mice displayed rapid clearance, comparable to that in NOD SCID mice (figure 2B). These data demonstrate that both NOD and SCID phenotypes are necessary to confer rapid IgG clearance. Moreover, the differences between isotypes in NOD SCID mice also occurred in the NSG strain, with hIgG1 and mIgG2a but not mIgG1 exhibiting rapid clearance (figure 2B).

Rapid antibody half-life requires both the NOD and SCID phenotypes as well as functional Fc. (A) 100 µg cetuximab was injected I.P. into NOD or NOD SCID mice with the concentration of hIgG in the plasma determined by ELISA, over 14 days. (n=4–6). (B) 100 µg rituximab hIgG1, mIgG1 or mIgG2a was injected I.P. into NSG mice and the concentration of mouse or human IgG in the plasma determined by ELISA, (n=6–8), data combined from two independent experiments. (C) 100 µg rituximab (hIgG1) or rituximab hIgG1 N297Q (NQ) was injected I.P. into SCID or NOD SCID mice and the concentration of hIgG in the plasma determined by ELISA. (SCID n=3–4, NOD SCID n=7 combined from two independent experiments). Two-way ANOVA with multiple comparisons, *p<0.05, **p<0.01, ***p<0.001. ANOVA, analysis of variance; NOD, non-obese diabetic; SCID, severe combined immunodeficient.
Rapid hIgG1 clearance is dependent on FcγR binding
Given that mIgG2a and hIgG1 have similar FcγR binding profiles (binding to all mFcγR, with substantial affinity for several activatory FcγR), we hypothesized that the rapid mAb clearance of hIgG1 and mIgG2a isotypes in NOD SCID mice was mediated by FcγR.25 26 This was investigated using a N279Q (NQ)-mutant of rituximab which lacks glycosylation at N297 and does not robustly engage mFcγR (without compromising interaction with FcRn).27 The NQ-mutant remained present in the plasma of NOD SCID mice at significantly higher concentrations at all time-points, supporting mFcγR involvement in the rapid hIgG1 clearance in NOD SCID mice (figure 2C). Moreover, the concentration of rituximab-NQ was comparable between SCID and NOD SCID mice at all time-points suggesting that abrogation of mFcγR binding restored normal mAb clearance rate.
SCID and NOD SCID mice have comparable FcγR expression levels
Having established that the rapid hIgG1 clearance rate in NOD SCID mice was likely dependent on mFcγR, the relative expressions levels of these receptors in SCID and NOD SCID mice was investigated (figure 3A). While there were no statistically significant differences in expression levels (two-way analysis of variance p>0.05) trends toward differential expression were observed. mFcγRII expression was lower on both Ly6CHi and Ly6CLo monocytes in NOD SCID compared with SCID mice (figure 3B). Neutrophil and splenic macrophage FcγRIII expression was higher in SCID mice, with a similar expression profile for BMDM (online supplementary figure 3a). The expression of mFcγRI was not investigated as it is known to contain multiple polymorphisms in NOD SCID mice which prevent its detection using available antibodies.28 The subtle differences in activatory mFcγR expression detailed above appear to be compensatory with a similar overall expression of activatory mFcγR in each strain. In summary, only monocyte FcγRII was found to differ between SCID and NOD SCID mice; the relevance of this to mAb clearance rate remains to be determined.
Supplemental material

mFcγR expression profiling in SCID and NOD SCID mice. Splenocytes or peripheral blood from SCID and NOD SCID mice were stained with specific antibodies for Ly6C, Ly6G, CD11b or CD11c to identify monocytes, macrophages and neutrophils, concurrently with mAb to each mFcγR. n=3 combined data from two independent experiments, mean+range. No statistically significant differences were observed between strains (two-way ANOVA with Tukeys multiple comparison test, p>0.05). ANOVA, analysis of variance; FcγR, Fcγ receptors; mAb, monoclonal antibodies; NOD, non-obese diabetic; SCID, severe combined immunodeficient.
Rapid hIgG1 clearance in NOD SCID mice is dependent on FcγRII
To understand the contribution of specific mFcγR to rapid antibody clearance in NOD SCID mice, we made use of animals lacking different classes of mFcγR. In NOD SCID FcR γ-/- mice (which express no activatory FcγR at the cell surface29) there was no significant difference in the concentration of hIgG1 over time compared with NOD SCID mice (figure 4A), demonstrating that a lack of activatory mFcγR does not influence hIgG1 clearance. However, in NOD SCID mice deficient in the inhibitory mFcγRII, the concentrations of hIgG1 were significantly increased compared with wild-type NOD SCID mice retaining mFcγRII (figure 4B) and comparable with SCID mice. These results demonstrate that the rapid hIgG1 clearance in NOD SCID mice is dependent on mFcγRII. Moreover, this result suggests that the somewhat reduced FcγRII expression seen previously in NOD SCID mice is not responsible for the fast hIgG1 clearance rate.
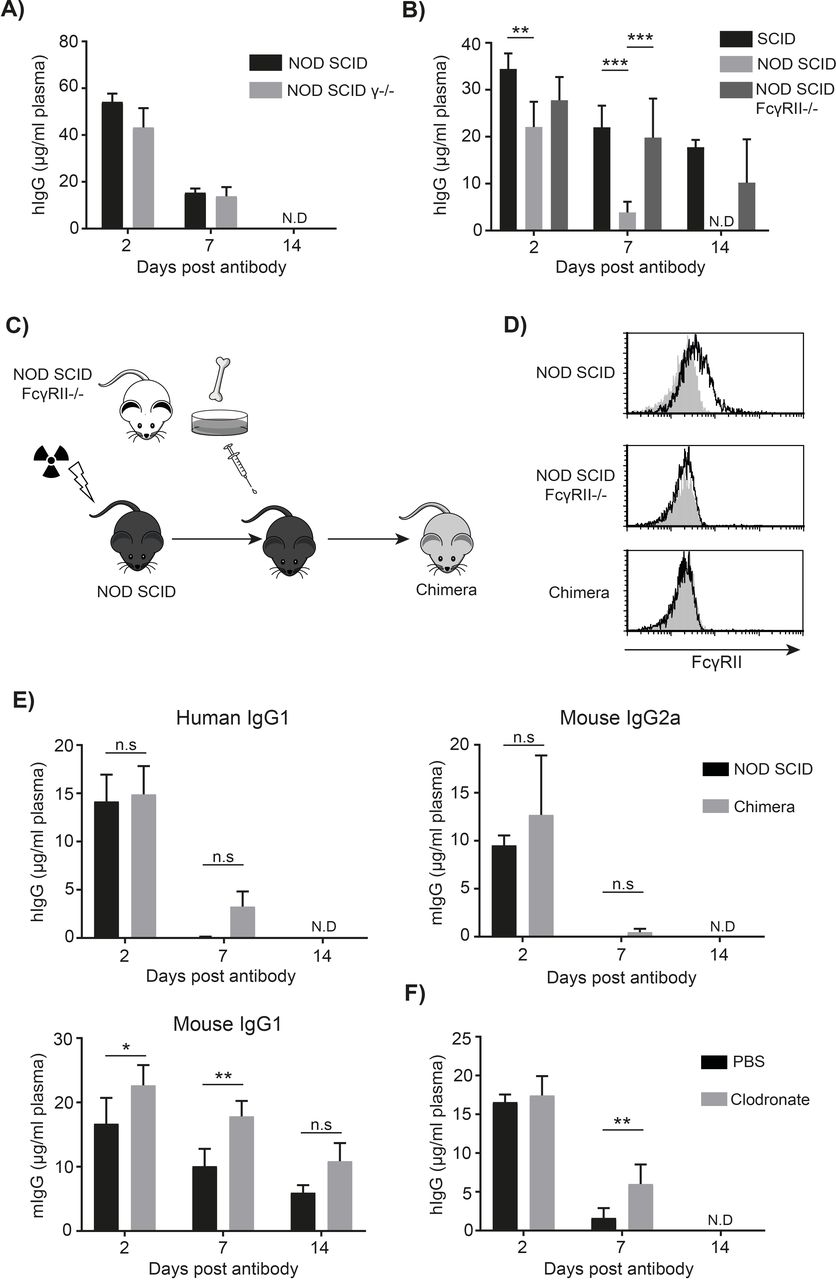
Rapid mAb clearance in NOD SCID mice is dependent on expression of mFcγRII but not activatory mFcγR. (A) NOD SCID and NOD SCID FcR γ-chain deficient mice (γ-/-) were injected with 100 µg trastuzumab hIgG1 I.V. Tail blood was collected and the concentration of hIgG in the plasma determined by ELISA. (n=3–4) mean+SD. By day 14 hIgG was not detectable in the plasma of both strains. (B) SCID, NOD SCID and NOD SCID mFcγRII-/- mice were injected with 100 µg cetuximab I.V. The concentration of hIgG in plasma was determined 2, 7 and 14 days later by ELISA. (n=3–4 per group), representative of two independent experiments. Two-way ANOVA with multiple comparisons *p<0.05 **p<0.01, ***p<0.001. (C) NOD SCID mice were irradiated and reconstituted with bone marrow cells harvested from NOD SCID FcγRII-/- mice. (D) Engraftment was confirmed by staining peripheral CD11b+ cells for mFcγRII expression. (E) 100 µg rituximab hIgG1 or mIgG2a was injected into these or control NOD SCID mice. The concentration of human or mouse IgG in the plasma was determined by ELISA. (n=3–4), mean+SD. (F) NOD SCID mice were injected I.V. with clodronate-containing or PBS-containing liposomes on days -3, –1, 6 and 13 to deplete macrophages. On day 0, 100 µg rituximab was administered I.P. and the concentration of hIgG in the plasma determined by ELISA. ANOVA, analysis of variance; FcγR, Fcγ receptors; mAb, monoclonal antibodies; NOD, non-obese diabetic; n.s, not significant; SCID, severe combined immunodeficient.
The polymorphic variants of mFcγRII have comparable affinity for hIgG1
A number of autoimmune strains including NOD express the ly17.1 form of mFcγRII while most other in-bred strains, including BALB/c, express the ly17.2 variant.30 These two polymorphic forms vary in four amino acids, three of which are located in the extracellular domain.30 The extracellular domain of FcγRII from SCID and NOD SCID mice was cloned and expressed; their relative affinity for IgG was then determined by SPR. Neither heat aggregated, pooled hIgG or individual isotypes of IgG displayed substantially different binding affinities to the ly17.1 and ly17.2 variants (table 1).
Affinity of mFcγRII variants for IgG subtypes
Absence of hematopoietic mFcγRII or phagocytes does not restore normal mAb clearance
mFcγRII is expressed on both hematopoietic and non-hematopoietic cells.30 31 We, therefore, sought to determine which mFcγRII-expressing cells were responsible for the rapid clearance of hIgG1. Accordingly, NOD SCID mice were irradiated and reconstituted with bone marrow from NOD SCID FcγRII-/- mice (figure 4C,D). These mice, reconstituted with hematopoietic cells lacking mFcγRII displayed rapid clearance of hIgG1 and mIgG2a, indicating that mFcγRII on cells of the hematopoietic system were not responsible for the rapid mAb clearance (figure 4E). We next considered whether phagocytes, particularly tissue resident macrophages, might be responsible and so deleted them with clodronate liposomes. This approach effectively removed macrophages (online supplementary figure 3b) but only resulted in a small increase in circulating hIgG1 7 days after mAb administration, with no hIgG1 detectable by day 14 (figure 4F). This suggests that phagocytes in NOD SCID mice are not primarily responsible for the rapid hIgG1 clearance and that a non-hematopoietic cell type is responsible. Given their high expression of mFcγRII, the liver sinusoidal endothelial cells (LSEC) seem the most likely candidate.31 We confirmed high expression of mFcγRII on these cells by immunofluorescence of livers from both BALB/c and NOD SCID mice (online supplementary figure 4a and b). Moreover, we found hIgG detectable at substantially higher levels within the liver of NSG than SCID mice following administration of hIgG1 mAb confirming a role for the liver as a site of hIgG1 accumulation (online supplementary figure 4c).
Supplemental material
NOD SCID mice have reduced FcRn expression
As mFcγRII is not known to directly regulate mAb clearance, we next considered whether FcRn might be involved in the process of controlling clearance rate in the NOD SCID mouse. Importantly, FcRn in NOD SCID mice does not to contain any sequence variations compared with other strains and has normal binding to both human and mouse IgG.14 However, qPCR revealed significantly lower FcRn transcription in both spleen and liver of NOD SCID versus SCID mice (figure 5A). This result was confirmed at the protein level by western blotting (figure 5B) and flow cytometry using the MST-HN protein which maintains FcRn binding at both acidic and neutral pH (figure 5C).32 This latter approach demonstrated a lower expression of FcRn in Ly6C+monocytes from NSG compared with SCID mice (MFI 354 vs 3061, mean of n=4). Combined, these results demonstrate that there is a lower expression of FcRn in the tissues known to be important for IgG recycling (spleen and liver) of NOD SCID mice compared with SCID.33 mFcγRII has a pH dependent affinity for IgG isotypes and is expressed on the same cell types as FcRn.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
NOD SCID mice have low expression of FcRn, associated with rapid mAb clearance, which can be overcome by IgG reconstitution. (A) cDNA was produced from SCID and NOD SCID spleen and liver lysates with FcRn transcript expression analyzed by qPCR using the ddCT method and expressed relative to that in SCID mice. (n=4–5), unpaired t-test **p<0.01, ***p<0.001. (B) Splenocytes from SCID and NOD SCID spleens were lysed and Western blot performed on the lysates for FcRn and Lamin B as a loading control. (C) Uptake of MST-HN Abdeg by splenocytes from SCID and NSG mice as well as mIgG reconstituted NSG mice, gated on CD11b+Ly6G-Ly6C+cells. No protein (red), H435A control (blue) and MST-HN (orange) are shown. N=3–4 with a representative example for each group shown. (D) Rituximab hIgG1, hIgG2, mIgG1 or mIgG2a was immobilized on Biacore CM5 chips and recombinant mFcγRII flowed over the chip at pH6 or pH7.4 with a highest receptor concentration of 1000 nM and 5-fold serial dilutions. (E) SCID or NOD SCID mice were reconstituted with 400 µg mIgG2a and 500 µg mIgG1 on day 0. An additional 200 µg mIgG2a was given on day 3, 6, 9, 12 and 15. 100 µg rituximab was then given I.P. on day 0 and the concentration of hIgG in the plasma determined by ELISA. (n=4–8), data combined from two independent experiments, mean+SD. Two-way ANOVA with multiple comparisons ***p<0.001, n.s.=not significant. (F) Eµ-Tcl1 tumor cells were injected I.P. into SCID or NSG mice. Once tumor was detectable in the peripheral blood a group of NSG mice were reconstituted with mIgG as described above, animals were then treated with 100 µg hIgG1 anti-mCD20 (18B12) I.P. representative flow cytometry plots are shown on the left. Fourteen days after treatment, the percentage of tumor cells in the blood was assessed and plotted. (n=5–6 per group), mean+SD. One-way ANOVA with multiple comparisons *p<0.05, **p<0.01. ANOVA, analysis of variance; FcγR, Fcγ receptors; NOD, non-obese diabetic; NSG, NOD SCID interleukin-2 γ-/-; SCID, severe combined immunodeficient.
While reduced FcRn could explain rapid IgG clearance, it does not provide an explanation for the isotype dependent nature of the effects seen, as all isotypes should be affected equally. In contrast, mFcγRII is known to display differential affinity for IgG isotypes (high for mIgG1, low for mIgG2a and hIgG1) and so we considered if mFcγRII specificity might be involved in regulating the clearance of the different isotypes. Using SPR and two different mAb of each isotype, we confirmed that at pH7.4 mFcγRII had ~10-fold higher affinity for mIgG1 (2.74×10–7M) vs mIgG2a (1.18×10–6M) and hIgG1 (3.02×10–6) while the affinity for hIgG2 was lower still (7.65×10–6M) (table 2 and figure 5D).
Affinity of mFcγRII and mFcRn for IgG subtypes
We next considered that mFcγRII might internalize cell-surface bound IgG and by virtue of its higher affinity, preferentially protect mIgG1 from degradation following internalization. To do this, it would need to remain bound to IgG in a low pH environment, akin to FcRn. We therefore repeated SPR analysis at pH6.0, and revealed that mFcγRII retained binding at low pH, with affinity for mIgG1, mIgG2a and hIgG1 ~100-fold higher than at pH7.4. Notably, the KD for mIgG1 binding mFcγRII was 2.77×10–9M,>10-fold higher than for hIgG1 and mIgG2a. This suggests that mFcγRII is capable of binding IgG at an acidic pH with the potential to protect IgG from degradation being greatest for mIgG1. Using previously published affinity data for IgG binding to mFcRn, we calculated the ratio of mFcγRII:FcRn binding for different isotypes at pH6.0 (table 2 and figure 5D).34 hIgG2 exhibited a high mFcγRII:FcRn ratio, suggesting preferential binding for FcRn at an acidic pH. In comparison, hIgG1 had a ratio around 1 (indicating no overall preference) whereas mIgG1 had a low ratio, preferentially binding with a higher affinity to mFcγRII than to FcRn.
Having hypothesized that the differential interaction with FcRn and FcγRII may play a role in the recycling of IgG, and with the knowledge that the liver expresses 75% of the FcγRII in the mouse, we sought to determine the distribution of these two receptors within the liver.31 The majority of mFcγRII was expressed by LSEC as determined by their morphology (online supplementary figure 4). We did not see mFcγRII onClec4F + Kupffer cells or Cytokeratin 8 + hepatocytes. This is consistent with a previous study reporting 90% of the liver mFcγRII as being expressed by LSEC when assessing immunofluorescence by pixel intensity.31 We found FcRn to be widely expressed throughout the liver including on Kupffer cells and hepatocytes (online supplementary figure 4). This is consistent with a previous report showing FcRn mRNA in various cell types, additionally identifying LSEC as having the highest expression level.31 Together, these results suggest that LSEC may be the predominant cell type coexpressing FcγRII and FcRn.
Overall, these data provided the possibility that antibody clearance in NOD SCID strains is controlled through differential engagement of the various isotypes by mFcγRII and FcRn. However, these effects have not been reported previously in standard in-bred strains, or the single NOD and SCID strains, indicating that the proposed pathway, which is at least in part mediated by isotype-dependent binding of mFcγRII, is only revealed in the absence of endogenous IgG.
Rapid hIgG1 clearance and reduced mAb efficacy can be overcome by IgG reconstitution
We, therefore, hypothesized that the addition of exogenous mIgG would restore normal mAb clearance rate in NOD SCID mice. Accordingly, SCID and NOD SCID mice were reconstituted with mIgG1 and mIgG2a to a level equivalent to that seen in the plasma of wild type BALB/c mice (online supplementary figure 5). Subsequently, the clearance of hIgG1 was investigated (figure 5E). Reconstitution with mIgG overcame the rapid clearance of hIgG1 in NOD SCID mice such that it became comparable to that observed in SCID mice. In contrast, mIgG addition did not significantly alter the clearance of hIgG in SCID mice. Additionally, reconstitution with mIgG substantially reduced the accumulation of hIgG in the liver (online supplementary figure 4c). Importantly, the expression of FcRn was not altered by reconstitution with mIgG (figure 5C). Finally, we sought to determine if overcoming rapid clearance of hIgG1 by mIgG reconstitution could improve therapy, using an Eμ-TCL1 tumor and a hIgG1 antibody targeting mouse CD20 (18B12). Using this second tumor model we found the differences in duration of therapy between SCID and NSG mice was maintained, with tumor growth recurring in NSG before SCID mice (figure 5F). We then compared the duration of tumor deletion in NSG mice versus NSG mice reconstituted with mIgG, using the same protocol as before. Reconstitution with mIgG was able to overcome the impaired therapy and restored comparable levels of tumor deletion and control to that observed in SCID mice both in terms of tumor percentage (figure 5F) and number of tumor cells (online supplementary figure 6). This demonstrates that reconstitution with mIgG is able to restore mAb efficacy in NSG mice.
Supplemental material
Supplemental material
Discussion
Our observations with rituximab showed reduced tumor control in NOD SCID compared with SCID mice. Given that equivalent initial tumor clearance was observed, and deletion is known to be dose dependent, this impairment was considered to be a direct result of reduced serum persistence and insufficient mAb at later time points.35 36 Rituximab is a type I mAb, known to be internalized through cis-binding to FcγRII following target binding, reducing its efficacy.18 37 Importantly, reduced tumor control was also seen with a type II anti-CD20 reagent, BHH2, indicating mAb internalization was not causal and that a separate phenomenon related to the mouse strain was responsible. In support of this, rapid clearance of wild-type and chimeric hIgG1 mAb was described recently in both NOD SCID and NSG mice.14 16 Furthermore, we saw the same rapid hIgG1 clearance for two additional hIgG1 mAb, lacking targets in the mouse, showing this phenomenon to be independent of the presence of tumor and unrelated to F(ab)-mediated antigen binding.
Separately, both SCID and NOD mice had normal clearance of hIgG1, comparable to that of immune competent BALB/c mice, as reported previously.24 This indicated that the genetic background of NOD mice, coupled with a lack of endogenous IgG (as a result of the SCID mutation) combined to elicit rapid hIgG1 clearance in NOD SCID mice. Importantly, our observations were replicated in NSG mice, indicating that rapid hIgG1 clearance is a feature of all NOD SCID derived strains. mIgG1 was found to have normal clearance in NOD SCID mice, whereas both hIgG1 and mIgG2a had short half-lives. Both humans and mice have multiple activatory FcγR, but a single inhibitory receptor, FcγRII (FcγRIIb in humans). These receptors interact differentially with the various mouse and human IgG isotypes: mIgG2a and hIgG1 bind preferentially to multiple activatory receptors and as such have high activatory:inhibitory (A:I) ratios.26 In contrast, mIgG1 exhibits binding to only a single activatory FcγR (FcγRIII) while retaining binding to FcγRII, yielding a corresponding low A:I ratio.38 These observations provided a potential clue toward the isotype-based effects observed. A role for FcγR in this process was subsequently confirmed by using a hIgG1 N297Q mAb which abrogates binding to FcγRs.27 Importantly, the N297Q mutation has been demonstrated not to alter hIgG1 clearance in immune compromised mice.27 The normal clearance rate of N297Q mAb in NOD SCID mice suggested that the rapid clearance of hIgG1 (and mIgG2a) in NOD SCID mice was dependent on Fc:FcγR interaction.
Despite establishing a likely role for mFcγR in rapid mAb clearance, no gross changes in activatory FcγR expression levels were observed in NOD SCID versus SCID mice. The subtle interstrain differences in mFcγRIII and mFcγRIV expression are likely to be compensatory and do not result in a large difference in the A:I ratio or amount of activatory FcγR on the cell surface. One caveat here was that the expression level of mFcγRI in NOD SCID mice could not be determined by flow cytometry as its sequence varies considerably from the canonical sequence seen in most other strains (by 17 residues), and so cannot be detected using our existing reagents.39 mFcγRII expression was found to be lower on monocytes from NOD SCID compared with SCID mice, likely due to the previously reported alterations upstream of the gene in NOD SCID mice associated with lower expression.40 However, subsequent results, particularly in mice lacking mFcγRII expression, demonstrate that the reduced expression of mFcγRII was unlikely responsible for the rapid mAb clearance.
We additionally investigated the expression levels of FcRn, the primary receptor responsible for IgG recycling and long half-life.41 While it has a broad tissue distribution, expression is particularly prominent in the spleen and liver.33 41 Analysis of these tissues demonstrated that there was lower expression of FcRn in NOD SCID compared with SCID mice. SCID mice have previously been reported to have comparable FcRn expression and tissue distribution to immune-competent mice; indicating that the reduction is a result of the NOD phenotype rather than elevated expression in SCID mice.33 However, given that the N279Q-mutated antibody which can interact with FcRn but not FcγR had a normal clearance rate in NOD SCID mice, the reduced FcRn expression in isolation cannot explain the differences in mAb clearance.
To determine the receptor responsible, we used mice deficient in either the activatory or inhibitory mFcγR by employing mFcR γ-chain -/- or mFcγRII-/- mice, respectively. Only the absence of mFcγRII restored normal hIgG1 clearance in NOD SCID mice. This observation supports the implications of the N297Q-mutant data, that failure to engage with mFcγRII is important and therefore phenocopies the effect in the mFcγRII-/- mice. Our observation that mFcγRII mediates this effect is in contrast to a previous study suggesting mFcγRIV is responsible.16 The previous study used an Fc-engineered antibody reported to have reduced binding to FcγRIV; such mutations often result in broader changes to FcγR binding profiles with causal effects of specific FcγR difficult to define. In contrast, in the present study we were able to specifically define the role of FcγRII by using mice genetically deficient in FcγRII.
Given the dependence on mFcγRII, we investigated this receptor in more detail. There are two polymorphic variants of mFcγRII; the ly17.1 haplotype expressed by NOD SCID mice, and the more common ly17.2 haplotype expressed by most other in-bred mouse strains, which differ by four amino acids, three of which are extracellular.42 43 We confirmed previous observations that these allotypes do not differ in their affinity for IgG. While this SPR analysis assesses the likely effects of the extracellular polymorphisms, further investigation is needed into the role of the I258S intracellular polymorphism to determine its influence on mAb internalization. This could be of importance given the role of the intracellular I232T polymorphism in hFcγRIIB, which alters the ability of the receptor to cluster into lipid rafts and deliver inhibitory signals.44
The data presented here indicate that no single factor can explain the isotype-dependent differences in mAb clearance in NOD SCID vs SCID mice. Instead, the data support a more complex model whereby multiple factors arising from the NOD and SCID backgrounds combine to deliver the observed defect. We propose a model whereby mFcγRII accelerates initial mAb internalization. The ability of FcγRII to mediate mAb internalization has been previously reported in DC-mediated antigen presentation and the internalization of rituximab.37 45 hIgG1 N297Q and hIgG2 do not bind appreciably to FcγRII, preventing receptor-mediated internalization, maintaining equivalent clearance in both SCID and NOD SCID mice. Similarly, in SCID mice with a normal level of FcRn, the IgG internalized via FcγRII can be efficiently recycled by FcRn to maintain serum persistence. However, In NOD SCID mice, this efficient FcRn mediated recycling does not occur due to the reduced FcRn expression levels, resulting in more rapid serum loss.
In trying to understand the isotype dependent nature of the rapid IgG clearance rate in NOD SCID mice, we unexpectedly observed an increased affinity of mFcγRII for all IgG at pH6.0. Crucially, however, the affinity of mFcγRII for mIgG1 at pH6.0 was retained and ~10-fold higher than for the other isotypes investigated. This raises the possibility that mIgG1 is protected from degradation under acidic conditions due to continued association with mFcγRII following internalization. In contrast, mIgG2a and hIgG1 would remain unprotected and become degraded following internalization, due to mFcγRII having lower affinity for these isotypes at pH6.0. We propose that this reduced degradation occurs only in the presence of reduced FcRn expression in NOD SCID mice, further work is, however, required to confirm this hypothesis. These data, therefore, suggest a complex role for FcγRII in mAb clearance; it is required for the internalization of IgG, preventing external catabolism, yet it also delivers IgG for internal lysosomal degradation unless it exhibits sufficient affinity for IgG binding at pH6.0.
Prior experiments in conditional knock-out mice suggested that both endothelial and hematopoietic cells regulate IgG levels in mice.46 47 The transfer of mFcγRII -/- NOD SCID bone marrow into NOD SCID mice resulted in mice deficient in hematopoietic mFcγRII but with mFcγRII expression on non-hematopoietic (predominantly endothelial) cells. hIgG1 clearance in these chimeras was unaltered compared with NOD SCID mice, demonstrating that hematopoietic cells were not responsible. The liver was previously found to be the main site of IgG clearance, accounting for 30% of all antibody degradation.48 The same organ has also been shown to contain 75% of the mFcγRII in the body, the receptor demonstrated here to be essential for rapid NOD SCID mAb clearance.31 Additionally, we were able to demonstrate greater accumulation of hIgG in the liver of NSG compared with SCID mice, an effect that was overcome by the addition of exogenous mIgG. Our immunofluorescence studies, combined with previous reports, suggest that LSEC are the predominant cell type in the liver expressing both FcγRII and FcRn, the receptors regulating the fast mAb clearance.31 49 LSEC have been described as having the highest rates of endocytic uptake in the body. In addition, mFcγRII is required for the efficient clearance of small immune complexes.31 50 This leads us to hypothesize that LSEC are the key cell type responsible for fast mAb clearance observed in the present study, with internalization of IgG mediated by mFcγRII.
Further investigation is required to determine if pH and isotype dependent affinity is restricted to mFcγRII or is common to the other FcγR. Moreover, it remains to be established if the same occurs with the inhibitory FcγRIIb in humans. This could have implications for mAb therapy as it is known that the A:I ratio of IgG binding to FcγR can determine the outcome of therapy, particularly where the expression of hFcγRIIb may increase, such as within the tumor microenvironment.51 Moreover, in this context, the acidic pH of the tumor microenvironment may further modify this ratio of A:I binding by altering the relative binding to individual receptors.
Given the importance of the rapid mAb clearance in NOD SCID mice on the therapeutic activity of direct targeting hIgG1 mAb, we sought a means of restoring normal pharmacokinetics. By reconstituting NOD SCID mice with physiological levels of mIgG, the rapid mAb clearance could be overcome, restoring persistence equivalent to that observed in SCID and BALB/c mice. Moreover, this increased persistence of therapeutic mAb was able to recover anti-tumor efficacy to the same level as seen in SCID mice. This result is in agreement with findings that the addition of human IVIg can restore the normal half-life of an antibody-drug conjugate.16 Moreover, it has been previously reported that the addition of exogenous IgG is able to overcome anomalous antibody biodistribution in NOD SCID mice, adding to the potential benefits of IgG reconstitution in tumor models.17 We suggest that in the presence of exogenous IgG, mFcγRII is occupied (most likely by the mIgG1 component, due to its higher affinity) and less internalization of hIgG1 can occur. Based on these observations, we propose that reconstitution with IgG should be a consideration when performing therapy experiments in NOD SCID mice in order to restore therapeutic antibody half-life.
The implications for the findings presented here are wide-reaching. With an increasing use of immune-compromised mice in preclinical investigation of mAb therapeutics, it is essential to understand how the choice of host strain can influence the outcome. The clearance rate of the most clinically relevant deleting isotypes are significantly shorter in NOD SCID and NSG mice than other immune-compromised strains such as SCID. This is likely to underplay the therapeutic efficacy of mAb used in these models and complicate comparisons between strains. Additionally, there are significant efforts ongoing to understand the isotype requirements for mAb directed against different targets, and with different Fc requirements.52 Our work suggests that NOD SCID mice may not be a suitable host strain for determining the optimal mAb isotype or therapeutic dose due to complications arising from different isotype-dependent clearance rates, unless exogenous mIgG reconstitution is also provided. Specifically, we suggest that caution should be exercised when interpreting results from immune compromised mice on the NOD SCID background with regard to differences in antibody activity that could be explained by mAb clearance rate.
Acknowledgments
The authors would like to thank all of the members of the Antibody and Vaccine Group (Cancer Sciences, Faculty of Medicine, University of Southampton, UK), Professor Falk Nimmerjahn and Professor Sally Ward for helpful discussions relating to the experiments reported herein. SW additionally provided the Abdeg reagents for quantifying FcRn expression.
References
Footnotes
Contributors RJO, CIM, SJ, KLC and VAP performed experiments. RJO analysed the data. RJO, MSC and MJG interpreted the results. PJD and HTCC generated critical reagents. RJO and MSC wrote the manuscript.
Funding Funding was provided through an iCASE studentship to RJO and MSC with Huntingdon Life Sciences from the MRC (1254288), Programme Grants from Bloodwise (12050) and Cancer Research UK (A24721) as well as CRUK center support C328/A25139.
Competing interests MSC is a retained consultant for Bioinvent and has performed educational and advisory roles for Boehringer Ingelheim, Merck KGaA, Baxalta and GLG. He has received research funding from Bioinvent, Roche, Gilead, Iteos, UCB and GSK.
Patient consent for publication Not required.
Ethics approval Animal experiments were cleared through local ethics committees and performed according to Home Office guidelines under project license PB24EEE31.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. All datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.