Article Text

Abstract
Background CD40 is a compelling target for cancer immunotherapy, however, attempts to successfully target this pathway have consistently been hampered by dose-limiting toxicity issues in the clinic that prevents the administration of efficacious doses.
Methods Here, using cytokine and cytokine receptor depletion strategies in conjunction with a potent CD40 agonist, we investigated mechanisms underlying the two primary sources of CD40 agonist-associated toxicity, hepatotoxicity and cytokine release syndrome (CRS).
Results We demonstrate that CD40 agonist -induced hepatotoxicity and CRS are mechanistically independent. Historical data have supported a role for interleukin-6 (IL-6) in CRS-associated wasting, however, our findings instead show that an inflammatory cytokine network involving TNF, IL-12p40, and IFNγ underlie this process. Deficiency of TNF or IFNγ did not influence CD40-induced hepatitis however loss of IL-12p40 significantly decreased circulating concentrations of liver enzymes and reduced the frequency of activated CD14+MHCII+ myeloid cells in the liver, indicating a role for IL-12p40 in liver pathology.
Conclusions As clinical research programs aim to circumnavigate toxicity concerns while maintaining antitumor efficacy it will be essential to understand which features of CD40 biology mediate antitumor function to develop both safe and efficacious agonists.
- immunotherapy
- drug evaluation, preclinical
- cytotoxicity, immunologic
- cytokines
- costimulatory and inhibitory t-cell receptors
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- immunotherapy
- drug evaluation, preclinical
- cytotoxicity, immunologic
- cytokines
- costimulatory and inhibitory t-cell receptors
Background
Immune checkpoint therapies have provided entirely new modalities to treat cancer by leveraging the patient’s immune system. Immunotherapeutics, exemplified by PD-1/PD-L1 and CTLA-4 antagonists, have shown the potential to induce durable remissions in numerous tumor types.1 However, the benefits of these approaches are restricted to a small subset of patients and new modalities are needed to expand the opportunities promised by immunotherapy.
The TNF family receptor CD40 (Tfnsf5) is expressed on dendritic cells, monocytes, macrophages, B cells and stromal populations.2 CD40–CD40L interactions license dendritic cells to promote CD8 T cell responses,3 prime macrophages for cytotoxic functions4 and elicit B cell activation.5 These unique features position CD40 as a critical effector molecule in antitumor immunity and there has been considerable interest in developing CD40 agonists for cancer immunotherapy.6 In preclinical models, CD40 agonists result in tumor regression and CD8 T cell memory responses.7 8 Clinical trials of CD40 agonists (eg, CP-870,893, dacetuzumab, ADC-103, Chi Lob 7/4 and APX005M) have shown signs of clinical activity in multiple indications,9–11. However, these studies are frequently associated with adverse events leading to dose-limiting toxicity. The most common adverse events include cytokine release syndrome (CRS), characterized by increased circulating IL-6 and TNF,12 and hepatotoxicity associated with increased concentrations of circulating liver enzymes alanine aminotransferase (ALT), aspartate aminotransferase (AST) and glutamate dehydrogenase (GLDH).12 In a preclinical cancer model, TNF contributes to CD40 agonist-induced hepatotoxicity,13 whereas IL-12 mediates wasting and CRS by CD40 agonists in colitis.14 Preclinical tumor models have also implicated myeloid-derived suppressor cells (MDSCs) in CD40-induced hepatotoxicity via their production of inflammatory Nox2.15
Multiple mechanisms are implicated in CD40-mediated pathology. It is still unclear how, or if, CD40-induced CRS is implicated in hepatotoxicity, and if so, what molecular mechanisms couple these processes. Here we investigate these relationships using a potent CD40 agonist while ablating cytokine signaling in MC38 tumor-bearing mice. Our findings demonstrate that IL-12p40, IFNγ, and TNFR mediate CRS-induced bodyweight loss. However, IL-6, IFNγ, and TNFR do not influence hepatotoxicity. Instead, CD40 agonist-mediated hepatotoxicity is reduced in IL-12p40−/− mice, and this reduction in toxicity correlated with decreased macrophage activation in the absence of IL-12p40.
Methods
Mice
Six-week to 8-week-old female C57BL/6, IFNγ−/−, TNFR−/−, IL12p40−/− and PepBoy CD45.1 mice were purchased from The Jackson Laboratory.
Cell lines
The MC38 cell line was provided by Dr Antoni Ribas at UCLA (Los Angeles, California, USA) in 2011. IDEXX tested (IDEXX Laboratories, ME), pathogen-free cells cultured in Roswell Park Memorial institute-1640 (RPMI-1640), 10% Fetal bovine serum (FBS) and 2 mmol/L l-glutamine were harvested for implantation while growing in the exponential phase.
In vivo tumor efficacy studies
0.5×106 MC38 cells were implanted subcutaneousl in 100 µL phosphate-buffered saline (PBS). Tumors were treated at 70–100 mm3 with anti-mouse CD40 antibodies dosed subcutaneously twice, 3 days apart. Bodyweight and tumor volumes were measured twice a week. Tumors were measured using calipers, and volume calculated as V=0.5 L × W2, where L and W are the longest and shortest diameter of the tumor, respectively.
Antibodies for immunotherapy
Anti-mouse CD40 clone 8F2 was generated by immunizing American hamsters with mCD40-6xHis recombinant protein three times at triweekly intervals. Splenocytes were harvested and fused with P3-X63Ag8.653 cells to generate hybridomas. Hybridoma supernatants were screened and clone 8F2 selected based on functional properties. Antibody variable regions were sequenced and cloned into the pARC vector for mIgG1 recombinant expression. Variable sequences from Rat FGK45 were obtained from Genbank (accession numbers: heavy chain AEI27236.1, light chain AEI27235.1) and engrafted into mouse IgG1 constant region vectors. Rat IgG1 anti-mouse IL-6 (clone MP5-20F3) was purchased from BioXcell.
Mouse toxicology studies
Female C57BL/6 (n=5/group) mice were given a single subcutaneous administration of test articles or control (PBS) and clinical and anatomic pathology was analyzed 7 days later. At necropsy, the liver was fixed in 10% neutral-buffered formalin for histology. Livers were processed, paraffin-embedded, sectioned, stained with H&E, and analyzed by a board-certified veterinary pathologist. Blood samples were collected for clinical chemistry analysis using Advia 1800 (Siemens). Alternatively, for figures, 1F and 3I GLDH was analyzed by ELISA (Cusabio) in accordance with the manufacturer’s instructions. Serum samples were analyzed for liver enzymes by the Pfizer Groton Clinical Pathology Laboratory and Pfizer Cancer Immunology Discovery in San Francisco. Cytokine analysis was performed by Pfizer Cancer Immunology Discovery in San Francisco.
CD40-bone marrow (BM) chimeras
BM chimeras were established as previously described.16 Recipients were lethally irradiated and reconstituted with 1×106 BM cells and 1×106 splenocytes. Engraftment was confirmed to be >90% by flow cytometry in peripheral blood 7 weeks postirradiation by monitoring donor CD45.1+ cells in CD45.2+ recipients, or vice versa.
Liver immune cell FACS analysis
Mouse livers were isolated for FACS staining as previously described.17 Hepatic immune cells were resuspended in PBS stain using the LIVE/DEAD Fixable Blue Dead Cell Stain Kit (Invitrogen). Mouse Fc Block (10 µg/mL, BD Biosciences) was added for 10 min, and cells were then stained directly with cell surface staining cocktail diluted in brilliant stain buffer (BD Biosciences) for 45 min at 4°C. Anti-mouse phenotyping antibodies purchased from BD Biosciences or Biolegend were CD45 (30F11), CD90.2 (53–2.1), B220 (RA3-6B2), CD4 (GK1.5), CD8a (53–6.7), CD11b (M1/70), CD11c (N418), Ly6C (HK1.4), Ly6G (1A8), CD14 (Sa14.2), F4/80 (BM8) and I-A/I-E (M5/114.15.2). Flow cytometry data were acquired using LSR Fortessa (BD Biosciences) and analyzed using FlowJo (TreeStar Inc.).
Statistical analysis
Results were expressed as mean±SEM. Statistical analyzes were performed using GraphPad Prism V.6.0 one-way or two-way analysis of variance to compare the differences among multiple groups. A p value of <0.05 was considered a significant difference.
Results
Antitumor efficacy of CD40 agonist treatment is associated with CRS and hepatotoxicity
We began by characterizing the efficacy and safety profiles of two agonistic anti-CD40 antibodies. Anti-CD40 IgG1 clone 8F2 was generated in-house, possesses strong agonistic activity, and a KD of 98 nM, whereas the commercially available FGK45 clone binds with a KD of ~2000 nM and possesses weaker antitumor efficacy. For these studies, we grafted the agonistic anti-CD40 clone FGK45, a rat IgG2 isotype,18 onto a mouse IgG1 backbone to facilitate the comparison with 8F2. The antitumor efficacy of these two antibodies was evaluated in the MC38 colorectal carcinoma model (figure 1). Implanted groups were randomized and treated at 80 mm3 every 3 days for a total of two injections. Antibodies were dosed at 10 mg/kg, which was the maximum tolerated dose for 8F2 based on bodyweight loss. Relative to isotype, 8F2 impaired tumor growth by 76.5% 24 days postimplantation (figure 1A) and was associated with severe systemic toxicity characterized by bodyweight loss of 15%–20%, 2–3 days following antibody treatment (figure 1B). FGK45 controlled tumor growth to a lesser extent (50.1% at day 25, figure 1C); however, bodyweight loss was markedly lower in the 7%–10% range (figure 1D). In parallel, we measured circulating inflammatory cytokines and liver enzymes as markers of CRS and hepatotoxicity, respectively. An increase in the circulating concentrations of TNF, IL-6, IFNγ, and IL-12p40 was observed on CD40 agonist treatment (figure 1E). Consistent with the efficacy data, concentrations of circulating inflammatory cytokines were higher on treatment with 8F2 compared with FGK45, except for IFNγ, which was initially higher on FGK45 treatment but lower than 8F2 at later timepoints. Analysis of liver enzymes showed little increase in circulating GLDH concentrations 7 days after treatment with FGK45. However, a substantial increase in GLDH was associated with 8F2 treatment (figure 1F) as well as, in a different study, increased concentrations of AST and ALT (online supplementary figure S1). Histopathological assessment of liver samples following 8F2 treatment demonstrated dense infiltrations of mononuclear cells around the central veins, portal tracts and within the tissue parenchyma compared with isotype control-treated samples (figure 1G). In summary, CD40 agonist treatment induces systemic inflammatory cytokine secretion and hepatotoxicity, the extent of which correlates with tumor growth inhibition.
Supplemental material
Supplemental material

Anti-CD40 agonistic antibody eradicates tumor and induces transient bodyweight loss, cytokine release and liver injury. (A–D) MC38 tumor-bearing mice (n=10) were injected with the indicated anti-CD40 Ab at 10 mg/kg subcutaneously on day 7, 10, and 13 post-tumor inoculation (A–D) and tumor volume and bodyweight monitored. Data are presented as mean±SEM. Two-way ANOVA, Fisher’s LSD test, **p<0.01, *** p<0.001, **** p<0.0001. Serum were collected on day 0 (prior to treatment in B and D) and at days 1, 4, and 7 post-treatment and analyzed for either cytokines release. (E–G) MC38 tumor-bearing mice were injected with the indicated anti-CD40 Ab at 10 mg/kg subcutaneously once on day 7 post-tumor inoculation and circulating cytokines (E) or GLDH concentrations assessed (F). Data were presented as mean±SEM. Unpaired t test, * p<0.05, **p<0.01, *** p<0.001, **** p<0.0001. All data represent one of at least three repeated studies. Liver tissues obtained from day 7 post-treatment were processed, embedded in paraffin, sectioned, and stained with H&E (G). ANOVA, analysis of variance; PBS, GLDH, glutamate dehydrogenase; PBS, phosphate-buffered saline.
Hematopoietic CD40 is responsible for CD40 agonist-induced toxicity
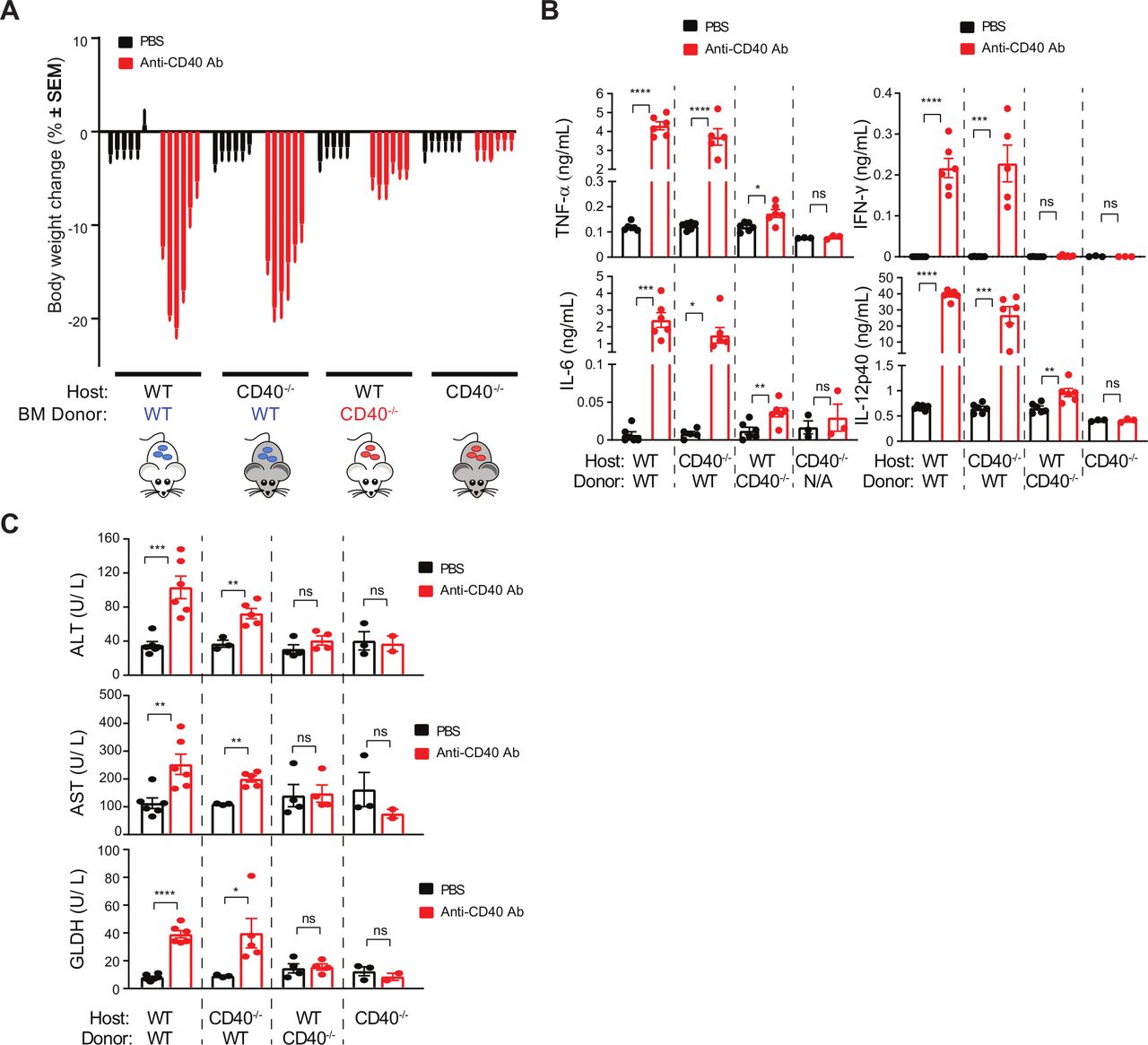
We sought to understand the sources of toxicity arising from anti-CD40 agonist treatment. To determine the relative contribution of hematopoietic cells we established BM chimeras using wild-type (WT) and CD40−/− mice. Clone 8F2 was used for subsequent studies owing to its more potent toxicity profile. Seven weeks following reconstitution, non-tumor bearing mice were challenged with anti-CD40 and toxicity parameters measured (figure 2). CD40 agonist significantly decreased bodyweight in both WT and CD40−/− mice reconstituted with WT BM (figure 2A). In contrast, we observed vastly reduced bodyweight loss in both WT and CD40−/− mice reconstituted with CD40−/− BM. Mirroring these findings, CD40 agonist increased serum concentrations of TNF, IFNγ, IL-6 and IL-12p40 in mice that received WT BM irrespective of host genotype (figure 2B). Concentrations of these cytokines were unaffected by 8F2 in mice that were completely deficient of CD40. However, IL-6, TNF and IL-12p40 were marginally higher on 8F2 treatment in WT mice that received CD40−/− BM (figure 2B). Analysis of liver enzymes further revealed a hematopoietic contribution to hepatotoxicity (figure 2C). 8F2 treatment increased ALT, AST and GLDH concentrations in WT BM recipients (figure 2C) and was also associated with cellular infiltrate in the liver (data not shown). In contrast, WT mice receiving CD40−/− BM did not increase liver enzymes and, although cellular infiltrates were still present in the liver, they were less severe than in mice receiving WT BM. These findings show that BM-derived cells are the primary cellular source of CD40-mediated CRS and bodyweight loss and contribute significantly to liver toxicity.
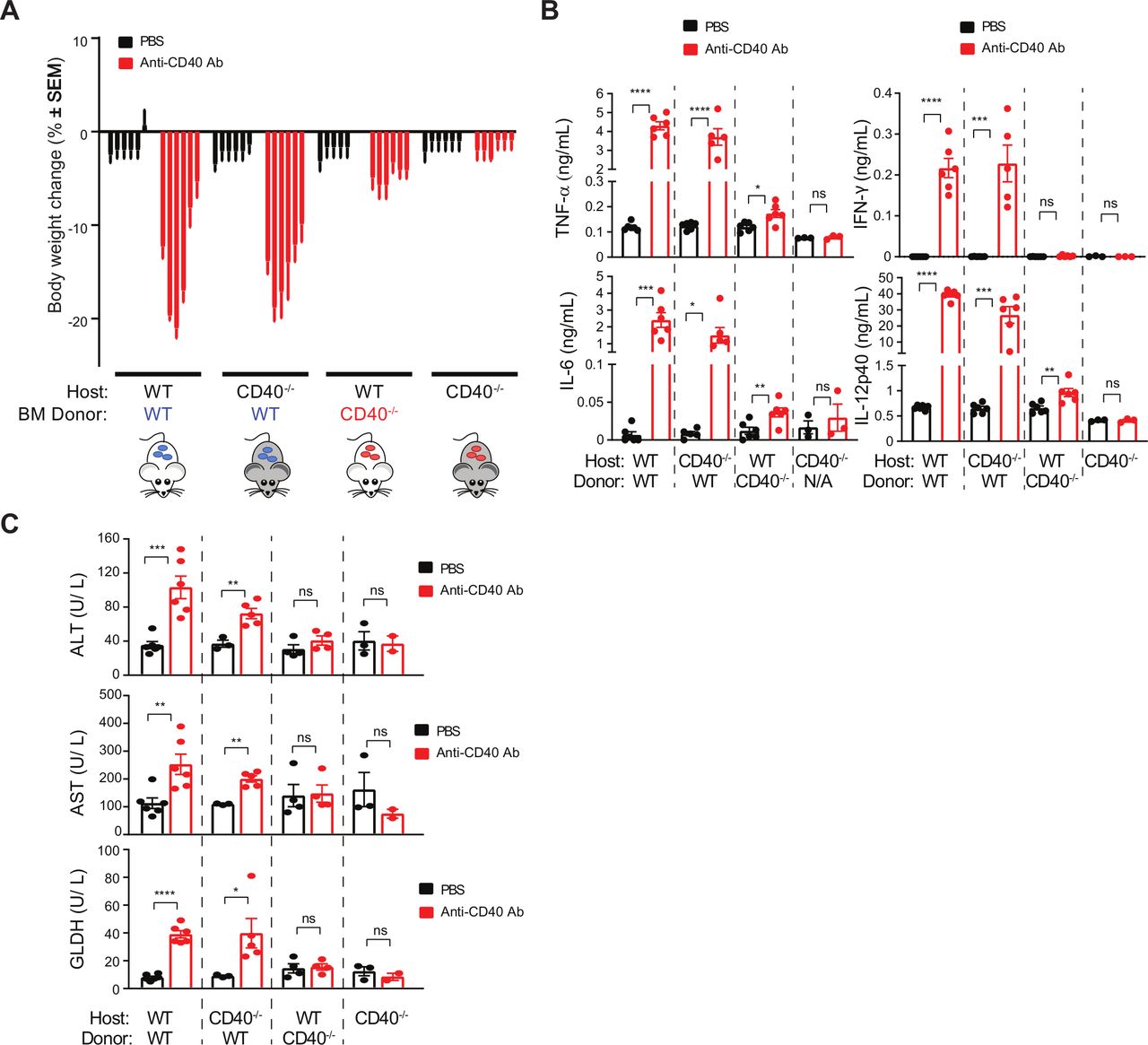
CD40 agonist-induced toxicity stems from hematopoietic cells, rather than hepatocytes. A–C. Three groups of BM chimeras were generated by transplanting (a) WT BM into WT recipients (n=12), (b) WT BM into CD40 −/− recipients (n=12), (c) CD40 −/− BM into WT recipients (n=12), and (d) CD40 −/− mice (n=6). Animals were injected once with either 8F2 (n=6 mice per group) at 10 mg/kg, or PBS (n=6 mice per group). (A) Bodyweight change at 1, 2, 3, 4, 7 and 8 days post-treatment, each bar represents one timepoint. (B) Serum samples obtained at 24 hours post-treatment were analyzed for cytokines, and (C), serum was collected 7 days post-treatment for liver enzyme analysis. Data were presented as mean±SEM Unpaired t test, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. BM, bone marrow; PBS, phosphate-buffered saline; WT, wild-type.
Inflammatory cytokine production underlies bodyweight loss but not hepatotoxicity
As CD40 expressed on immune cells is responsible for the CRS and hepatotoxicity associated with CD40 agonist treatment, we sought to understand the molecular mechanisms underpinning this toxicity. Clinical studies using CP-870,893 determined that CRS is associated with a rapid increase in IL-6 and TNF concentrations.12 Thus, we assessed the influence of these cytokines on anti-CD40-induced toxicity in MC38 tumor-bearing mice. IL-6 neutralization reduced IL-6, and increased circulating concentrations of IFNγ on 8F2 treatment but did not affect TNF or IL-12p40 concentrations (online supplementary figure S2A). Additionally, IL-6 blockade did not influence GLDH concentrations (figure 3A); however, we did observe a slight reduction in bodyweight loss induced by 8F2 on IL-6 blockade (figure 3B). These findings suggest that IL-6 is not responsible for the systemic or liver toxicity associated with CD40 agonist treatment.
Supplemental material
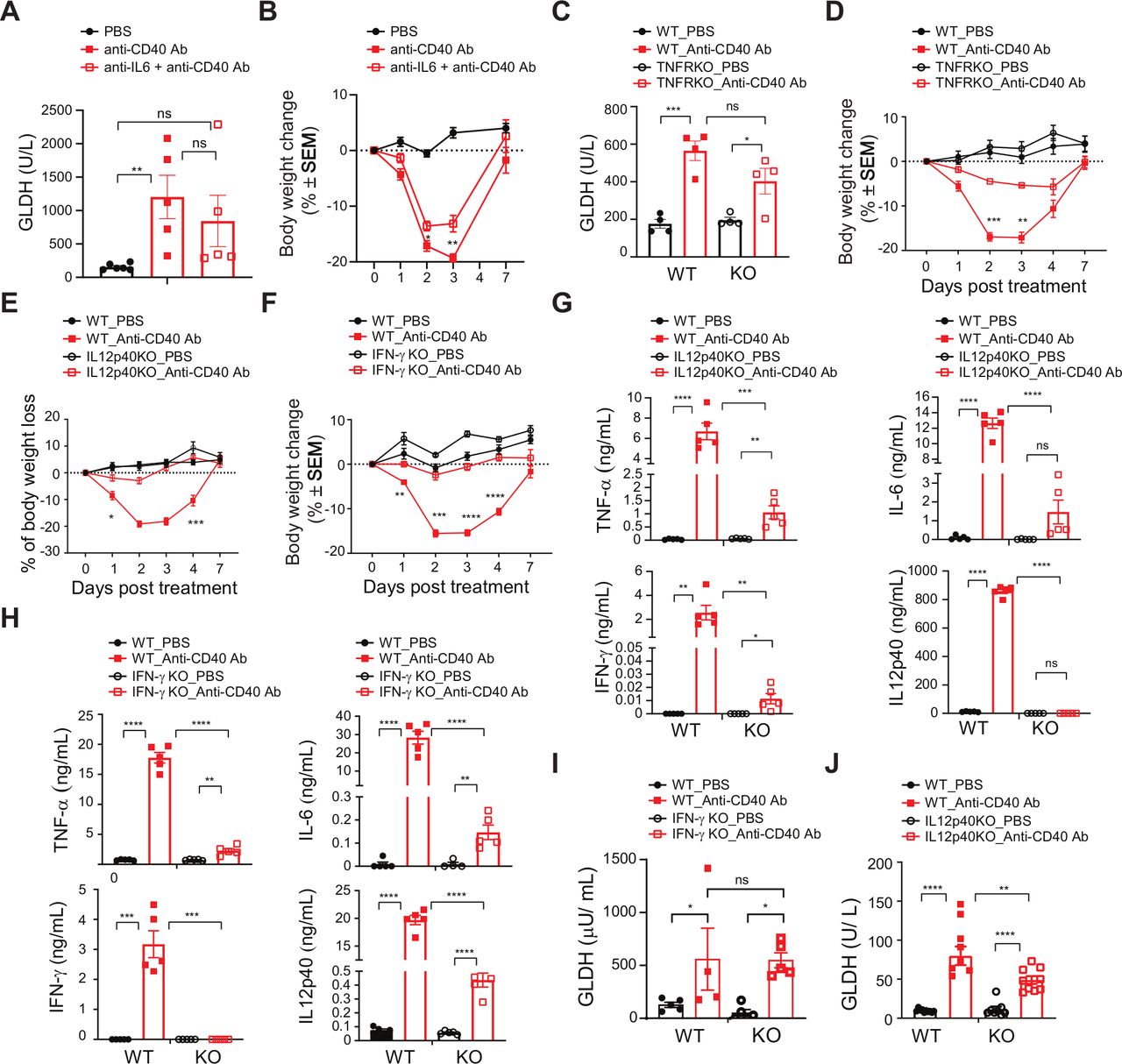
CD40 agonist-induced liver injury is independent of TNF-α, IL-6 and IFNγ. (A–B) MC38 tumor-bearing WT mice were injected with 10 mg/kg 8F2 in the presence or absence of a 20 mg/kg pretreatment of anti-IL-6 blockade antibody administered Intraperitoneal 15 min prior to CD40 antibody dosing (n=5 mice per group). (A) Circulating liver enzymes were assayed 7 days post-treatment, and (B) bodyweight change in response to 8F2 treatment. (C–D) MC38 tumor-bearing TNFR−/− (and WT control) animals (n=5 mice per group) were injected with 10 mg/kg 8F2 Ab once or PBS. (C) Circulating liver enzymes were assayed 7 days post-treatment, and (D) bodyweight change in response to 8F2 treatment. (E–J) MC38 tumor-bearing IL-12p40−/−, IFN-γ−/− and WT control mice (n=10 mice per group) were injected with 10 mg/kg anti-CD40 Ab or PBS once. (E) WT versus IL12p40−/− bodyweight change, (F) WT versus IFN-γ−/− bodyweight change. (G) Serum cytokine concentrations in IL-12p40−/− and WT mice 24 hours post-treatment. (H) Serum cytokine concentrations in IFN-γ−/− and WT mice 24 hours post-treatment. (I–J) GLDH concentrations 7 days post-treatment in (I) IFN-γ−/− and WT mice, and (J) IL-12p40. Data are presented as mean±SEM. Unpaired t test, * p<0.05, **p<0.01, *** p<0.001, **** p<0.0001. GLDH, glutamate dehydrogenase; PBS, phosphate-buffered saline; WT, wild-type.
Next, we evaluated the role of TNF in toxicity using TNFR−/− mice. 8F2 increased circulating TNF concentrations (online supplementary figure S2B), presumably due to a decreased uptake in the absence of TNFR expression. Global TNFR deficiency curtailed IL-6, IFNγ and IL-12p40 concentrations (online supplementary figure S2B) yet did not influence hepatotoxicity (figure 3C). Importantly, bodyweight loss was markedly reduced in TNFR−/− mice on 8F2 treatment (figure 3D). On average, TNFR−/− mice lost 6.8% of their bodyweight as compared with 17.1% for WT mice on day 3, demonstrating that TNF rather than IL-6 is primarily associated with CD40 agonist-induced bodyweight loss.
Because IFNγ and IL-12p40 were decreased in 8F2 treated TNFR−/− mice, we asked whether either of these contribute to CRS-associated bodyweight loss. Strikingly, both IL-12p40−/− and IFNγ−/− mice showed complete protection from CD40 agonist-induced body weight loss (figure 3E,F), accompanied by decreases in all measured inflammatory cytokines (figure 3G,H). IFNγ deficiency did not impact GLDH concentrations (figure 3I). hHowever, IL-12p40−/− mice demonstrate a significant reduction in circulating GLDH concentrations (figure 3J). Thus, a TNF-IL-12p40-IFNγ cytokine axis regulates CD40-dependent body weight loss. Furthermore, our findings establish a role for IL-12p40 in liver toxicity elicited in response to CD40 agonist treatment.
CD40-mediated hepatotoxicity is associated with an accumulation of MHCII-positive macrophages
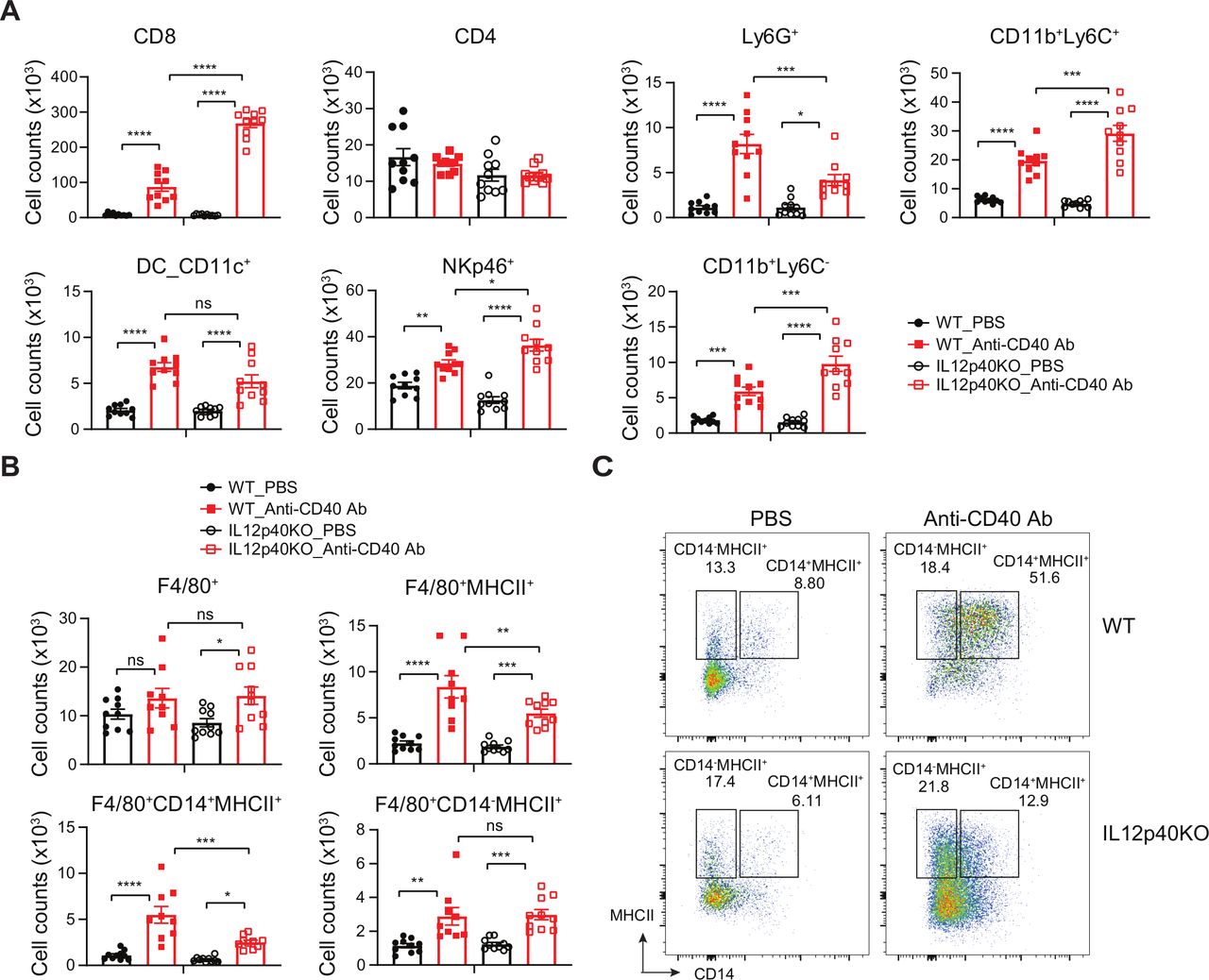
As IL-12p40 deficiency provided partial protection from liver toxicity we sought to understand the cellular basis of this phenotype. WT and IL12p40−/− mice were injected with a single dose of 8F2 and liver immune cell phenotypes were assessed by FACS 7 days postinjection (figure 4 and online supplementary figure S3A). Following 8F2 treatment, we observed increased CD45+ cell frequencies in IL12p40−/− compared with WT mice (figure 4A). This was defined by increased CD8+ T cells, NK cells, and CD11b+Ly6C+ and Ly6C− monocytes. CD40 agonists have been reported to mediate hepatotoxicity via activation of monocyte-derived macrophages.15 Thus, we determined the influence of 8F2 on liver macrophage activation in WT and IL-12p40−/− deficient mice. 8F2 did not affect total macrophage numbers 24 hours postinjection (figure 4B). However, the frequency of MHCII+ macrophages were increased by 8F2 (figure 4B,C), and this was somewhat diminished in IL12p40−/− mice. Further assessment revealed that the accumulating IL12p40-dependent MHCII+ macrophages express CD14 and CD11b (figure 4B,C and online supplementary figure S3B), and are thus likely monocyte-derived rather than CD11b negative Kupffer cells.19 These findings support the notion that monocytes contribute to liver toxicity on CD40 agonist treatment, and that an IL-12p40-dependent process contributes to this axis.
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
CD40 ligation-induced liver CD14+MHCII+F4/80+ immune cell increase is correlated with hepatotoxicity and driven by IL-12. (A) Enumeration of liver-infiltrating CD45+ cell subsets as defined by FACS gating in online supplementary figure S3A in WT and IL12p40−/− mice injected once with 8F2 (10 mg/kg), liver infiltrate was analyzed 7 days post-treatment. (B) Numbers of liver macrophages post 8F2 treatment as defined by gating in (C) and online supplementary figure S3A. (C) Representative flow cytometry plots showing CD14−MHCII+ and CD14+MHCII+ cells were gated on F4/80+ populations. Bar represents mean±SEM. Unpaired t test, * p<0.05, **p<0.01, *** p<0.001, **** p<0.0001. WT, wild-type.
Discussion
Here we investigate the mechanisms underpinning CD40 agonist-induced bodyweight loss and hepatotoxicity using a potent CD40 agonistic antibody. IL-6 and TNF were previously associated with anti-CD40-induced CRS in patients.12 Here we show that TNF rather than IL-6 mediates this pathology as TNFR deficiency, rather than IL-6 blockade, prevented CD40-agonist-dependent bodyweight loss. Notably, protection from CD40 agonist-induced bodyweight loss was observed in IL12p40−/−, IFNγ−/− and TNFR−/− mice and loss of any one of these cytokines substantially impaired the production of the others. This supports the concept that inflammatory cytokine networks underpinning disease-induced cachexia20 may also be relevant CRS-associated bodyweight loss observed with immunotherapeutics such as CD40 agonists.
Despite the amelioration of wasting in inflammatory cytokine-deficient models, loss of these factors did not substantially mitigate hepatotoxicity. Our BM chimeras demonstrated a role for hematopoietic CD40 in hepatotoxicity. A much less-severe, CD40 agonist-dependent, inflammation was, however, still observed in livers of WT mice reconstituted with CD40-deficient BM. This may be explained by anti-CD40 activity on liver stromal, or radio-resistant immune populations that persist in WT mice following CD40−/− BM reconstitution,21 and that immune-mediated damage is exacerbated by CD40 expressing BM-derived cells.
Deficiency in IL-12p40 reduced liver toxicity associated with CD40 agonist treatment; however, there was a trend towards lower GLDH in both IFNγ and TNFR-deficient mice. This is consistent with a recent study showing that multiple cytokines produced in response to liver injury regulate monocytic infiltration.22 Absence of IL-12p40 did not influence 8F2-dependent macrophage accumulation but did reduce activation, defined by decreased Cd11b+CD14+F4/80+MHCII+ macrophages. CD14+ monocytic MDSCs contribute to CD40-induced hepatotoxicity in EL4 lymphoma.15 Here, the expansion of monocytic subsets led to hepatotoxicity in tumor-bearing mice at doses that were safe in non-tumor-bearing mice. Current strategies for CD40 agonist immunotherapies aim to maximize the therapeutic index to establish efficacy while mitigating toxicity.6 Such approaches will need to consider variations in circulating MDSC frequencies and mechanism of agonist activity either by drug design or patient selection. Some approaches, including tumor-targeted CD40 agonists, are showing promise in preclinical models,23 yet their relative safety versus efficacy profiles are yet to be determined.24
Acknowledgments
The authors thank German Vergara and Teresa Radcliffe for excellent technical assistance with the animal work; Christopher Kimberlin and Sherman Michael Chin for generous support with anti-mouse CD40 antibody generation and purification and Qing Zong for support with clinical chemistry.
References
Footnotes
CB and GT are joint first authors.
Contributors CB, JH, YQ, and SSA performed the conception and design. CB, WH, BJ, JH, and BB helped in development of methodology. Data acquisition was done by CB, WH, BJ, JH, BB, ML, and SP. Analysis and interpretation of data was done by CB, WH, BJ, YQ, JH, BB, GT, and SSA. Writing, review, and/or revision of the manuscript was performed by CB, YQ, GT, and SSA. Administrative, technical, or material support was done by WC, JCR, and DM.
Funding This study was funded by Pfizer, Inc.
Competing interests Pfizer employees may hold stock/stock options in the company.
Patient consent for publication Not required.
Ethics approval Mice were maintained and all animal experiments were conducted according to the protocols approved by the Institutional Animal Care and Use Committee of CID Pfizer (South San Francisco) and Worldwide Research and Development (La Jolla), Pfizer Inc. No human samples were used in this work.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.