Article Text

Abstract
Background Tumor ablation techniques, like cryoablation, are successfully used in the clinic to treat tumors. The tumor debris remaining in situ after ablation is a major antigen depot, including neoantigens, which are presented by dendritic cells (DCs) in the draining lymph nodes to induce tumor-specific CD8+ T cells. We have previously shown that co-administration of adjuvants is essential to evoke strong in vivo antitumor immunity and the induction of long-term memory. However, which adjuvants most effectively combine with in situ tumor ablation remains unclear.
Methods and results Here, we show that simultaneous administration of cytidyl guanosyl (CpG) with saponin-based adjuvants following cryoablation affects multifunctional T-cell numbers and interleukin (IL)-1 induced polymorphonuclear neutrophil recruitment in the tumor draining lymph nodes, relative to either adjuvant alone. The combination of CpG and saponin-based adjuvants induces potent DC maturation (mainly CpG-mediated), antigen cross-presentation (mainly saponin-based adjuvant mediated), while excretion of IL-1β by DCs in vitro depends on the presence of both adjuvants. Most strikingly, CpG/saponin-based adjuvant exposed DCs potentiate antigen-specific T-cell proliferation resulting in multipotent T cells with increased capacity to produce interferon (IFN)γ, IL-2 and tumor necrosis factor-α in vitro. Also in vivo the CpG/saponin-based adjuvant combination plus cryoablation increased the numbers of tumor-specific CD8+ T cells showing enhanced IFNγ production as compared with single adjuvant treatments.
Conclusions Collectively, these data indicate that co-injection of CpG with saponin-based adjuvants after cryoablation induces an increased amount of tumor-specific multifunctional T cells. The combination of saponin-based adjuvants with toll-like receptor 9 adjuvant CpG in a cryoablative setting therefore represents a promising in situ vaccination strategy.
- adjuvants, immunologic
- adaptive immunity
- CD8-positive T-lymphocytes
- dendritic cells
- immunomodulation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
In interventional oncology, a large variety of in situ tumor destruction techniques, like cryoablation and radiotherapy, are successfully applied. Extreme cold temperatures are applied in cryoablation to destroy tissue,1 resulting in direct cell death, via lethal hypothermia-induced necrosis, as well as through delayed apoptotic cell death in the outer ablation zones via injury of mitochondria.2 Cryoablation results in the release of unique antigens from a heterogeneous population of tumor cells, thereby creating in situ availability of ablated tumor material.3 Besides a reduction in tumor size as result of freezing and the released tumor antigens, cryoablation releases multiple bioactive molecules such as damage-associated molecular patterns (DAMPs), which all result in different innate and adaptive immune effects.1 Antigen-presenting cells (APCs), including dendritic cells (DCs) are able to phagocytose tumor debris4 and subsequently maturate and migrate to the lymph nodes where they present antigens to naïve T cells. Alternatively, tumor antigens may passively enter the circulation and be transported to lymph nodes where these antigens can be taken up by lymph node-resident DCs. DCs can present peptides derived from such extracellular antigens to MHC class I restricted CD8+ cytotoxic T cell response via a process termed antigen cross-presentation.5 As opposed to in vitro generated DC-based vaccines, the in vivo loading of DC with tumor-associated antigens released following in situ tumor ablation is also referred to as in vivo DC vaccination.3 4 Since the production of DC vaccines is time consuming and an expensive process, in situ cancer vaccination is an attractive way to generate an unbiased T cell response against tumor antigens, including neoantigens. Indeed, we and others have shown that in situ cancer vaccination can be highly effective to trigger systemic antitumor immunity provided that immune adjuvants are co-provided to further induce and amplify the immune responses.
The majority of adjuvants used in the clinic induce high antibody production but poor cellular responses. For anticancer vaccines, however, adjuvants ideally induce strong Th1-skewed cellular immunity. Microbial-derived adjuvants like toll-like receptor (TLR)9-agonist cytidyl guanosyl (CpG) adjuvants6–8 and R8489 skew immunity toward Th1 responses. CpG stands out as a cancer vaccine adjuvant, because of its preferential induction of Th1 responses and tumor-specific cytotoxic CD8+ T lymphocytes (CTLs10–13). On binding to TLR9, predominantly expressed on DCs and B cells, CpG is internalized and results in immune activation. The co-localization of antigen and CpG within DCs, and thus the timing of CpG administration relative to the release of antigens, plays an essential role in failure or success of CpG as a vaccine adjuvant.6 13 Saponin-based adjuvants like immune-stimulating complexes (ISCOMs) matrix adjuvants are another type of adjuvant that can induce cellular immunity. ISCOMs are 40 nm-sized particles consisting of saponin, cholesterol and phospholipids.14–16 They are also known for their Th1-skewing properties and induction of solid CD4+ and CD8+ T cell responses. Most importantly, ISCOM Matrix facilitates antigen presentation by DCs17 18 without the need of additional pattern recognition receptor triggering components.19 20 ISCOM Matrix adjuvants induced strong humoral and T cell-mediated immune responses and reduced relapse rates in patients with fully resected melanoma. However, no clinical benefit in patients with advanced melanoma was observed, possibly due to immunosuppressive mechanisms.21 This shows the safety and feasibility of clinical application, but also underlines that combinatorial approaches, for instance, together with other adjuvants like CpG, should be further explored.
The question remains what adjuvant combinations are best to boost the ablation-induced immune effects and induce powerful antitumor immunity. Here, we report that in our mouse model of in situ tumor ablation, co-injection of CpG plus ISCOMs resulted in higher production of interleukin (IL)-1β by DCs in vitro, which was associated by an increase in polymorphonuclear neutrophil (PMN) influx in the lymph nodes in vivo when combined with tumor ablation. Moreover, the adjuvant combination plus tumor ablation increased the amount and quality of T cells, producing multiple pro-inflammatory cytokines, relative to either adjuvant alone. In conclusion, this study provides a rational for clinical testing of these adjuvants in in situ tumor vaccination strategies.
Materials and methods
Mice and cells lines
Female C57BL/6 N and J mice (aged 6–8 weeks) were purchased from Charles River Wiga (Sulzfeld, Germany). IL-1ɑβ−/− mice (aged 25 weeks) on a C57BL/6 background22 were a kind gift of Dr Y. Iwakura (University of Tokyo, Center of Experimental Medicine, Tokyo, Japan) and were bred in house (confirmed at gene level and IL-1β excretion level (online supplementary figure 1a,b). OT-I mice (aged 8–14 weeks) that produce CD90.1+ CD8+ T cells with a transgenic T-cell receptor specific for the chicken ovalbumin (OVA) epitope SIINFEKL (OVA257-264) presented on MHC class I H-2kb were bred and held in the Nijmegen animal facility. Drinking water and standard laboratory food pellets were provided ad libitum and mice were allowed to settle for at least 1 week before random assignment into specific treatment groups. All mice were maintained under specific pathogen-free barrier conditions at the Central Animal Laboratory (Nijmegen, The Netherlands).
Supplemental material
The murine melanoma cell line B16F10 (ATCC) was cultured in complete medium (Minimum Essential Medium (MEM), 5% fetal bovine serum (FBS, Greiner Bio-One), 100 U/mL penicillin G sodium and 100 µg/mL streptomycin (Pen/Strep), MEM sodium pyruvate (1 mM), 2% NaHCO3, 1,5% MEM vitamins, 1% MEM non-essential amino acids (all from Gibco) and 50 µM beta-mercaptoethanol). OVA-transfected B16F10 (B16OVA, clone MO5) was cultured in complete B16F10 medium supplemented with 60 µg/mL hygromycin (Merck) and 1 mg/mL G418 (Gibco). B3Z cells, a T-cell hybridoma specific for the immunodominant OVA Kb peptide in H-2Kb, which carries a β-galactosidase construct driven by NF-AT elements from the IL-2 promotor,23 were cultured in IMDM supplemented with 5% FBS, Pen/Strep, 50 µM beta-mercaptoethanol, 0.5 mg/mL hygromycin and 2 mM L-glutamine.
Adjuvants, reagents and antibodies
CpG-ODN 1668 (‘5-TCCATGACGTTCCTGATGCT-3’) with total phosphorothioate-modified backbone was purchased from Sigma Genosys (Haverhill, UK). As saponin-based adjuvants, the Quillaja Saponaria-derived adjuvants Matrix C ISCOMs and Quil A saponin (kindly provided by MSD Animal Health) were used as indicated.
For flow cytometry experiments, the following antibodies (clone name in brackets, followed by supplier and dilution) conjugated to various fluorophores were used: extracellular; MHC class II-BV510 (M5/114.15.2, Antibodychain, 1:500), CD80-A488 (16-10A1, Antibodychain, 1:1000), Ly6G-FITC (1A8, Biolegend, 1:300), CD8 TCRβ-FITC (53–5.8, BD Biosciences, 1:100), CD40-PE (23/3, Antibodychain, 1:200), CD11b-PerCP (M1/70, Biolegend, 1:600), CD115-PeCy7 (AFS98, eBioscience, 1:200), CD11c-APC (HL3, BD Biosciences, 1:400), CD86-APCCy7 (GL-1, Biolegend, 1:800), Ly6C-APCCy7 (AL-21, BD Biosciences, 1:400), CD90.1-biotin (HIS51, eBioscience, 1:1000), CD62L-PerCP (MEL-14, Biolegend, 1:400) and intracellular; tumor necrosis factor (TNF)-α-A488 (XMG1.2, Biolegend, 1:100), IL-2-PE (JES6-5H4, Biolegend, 1:100), interferon (IFN)γ-APC (XMG1.2, Antibodychain, 1:300), streptavidin-PE (BD Biosciences, 1:250) and streptavidin-BV510 (BD Biosciences, 1:250).
After incubation, medium was removed from samples, and cells were washed with phosphate buffered saline (PBS) supplemented with 0,5% bovine serum albumin and 0,05% sodium azide (PBA). Samples were resuspended with Fc-block (CD16/32, BD Biosciences, 1:800) and centrifuged. Incubation with fluorescently labeled antibodies in PBA was performed for 20 min on ice. If applicable, samples were incubated with streptavidin antibodies in PBA after wash for 10–20 min on ice. Subsequently, cells were washed and diluted in PBA and measured on FACS Verse or stated otherwise, or additional intracellular staining was performed according to the protocol of the manufacturer (Fixation/Permeabilization kit, BD Biosciences).
Cryoabation and in vivo and ex vivo readouts
Tumors were induced and when established, treated with cryoablation as previously described.12 24 In short, tumor cells were suspended in a mixture of PBS and Matrigel (2:1, BD Biosciences, GFR, PR&LDEV free), and 0.5×106 cells in a total volume of 50 µL were injected subcutaneously at the right femur. When tumor diameters measured between 6 and 8 mm (generally at day 9–10), mice were randomly assigned to treatment groups. Cryoablation (Cryo) was performed under isoflurane/O2/N2O anesthesia using a liquid nitrogen cryoablation system (CS76, Frigitronics, Shelton, CT or liquid nitrogen-cooled CryoPro Maxi device, Cortex Technologies, Hadsund, Denmark) of which the tip is cooled by a continuous flow of circulating liquid nitrogen. During two to three treatment cycles of freezing and thawing, the tumor was macroscopically frozen, while leaving surrounding healthy tissue intact. Adjuvants were injected peritumorally (40 µL divided over two injections in PBS), concentrations as indicated. All injections were performed within 30 min after ablation.
For the rechallenge model, mice were carefully examined for recurrent tumors at the site of ablation. When recurrences appeared, mice were excluded from further analysis. To monitor the induction of long-lasting tumor protection, mice were re-challenged with 15×103 B16F10 cells 40 days after cryoablation. This number of cells was chosen after careful titration and ensures a solid 100% tumor take. Rechallenges were injected in 100 µL PBS subcutaneously on the right flank. Mice were sacrificed when tumor volume (=(A×B2)×0.4) exceeded 1000 mm3 or when occasionally tumors brake through the skin barrier.
To identify PMN mobilization, different organs were processed to single cell suspensions. Bone marrow was flushed out of tibia and femurs from female C57BL/6 n mice. Bone marrow and spleen were mashed over a nylon mesh with pore size 100 µm. Erythrocytes in splenocytes were lysed using ACK treatment (150 nM NH4CL, 10 mM KHCO3 and 0.1 mM Na2-EDTA, pH 7.2). Blood was harvested retro-orbital, under isoflurane/O2/N2O anesthesia, and collected in BD Biosciences Vacutainer blood collection tubes. Inguinal lymph nodes were excised, torn apart using needles and digested in collagenase type III (Worthington) and DNase I (Merck) for 30 min at 37°C. After addition of EDTA and re-suspension, cells were filtered through a nylon mesh to remove large debris. In vivo depletion of PMNs was achieved using anti-Gr-1 (RB6-8C5, 500 µg intraperitoneally, BIOXCell) and anti-Ly6G (1A8, 500 µg, intraperitoneally) antibodies.
To analyze DC biology, DCs were isolated from the inguinal tumor draining lymph nodes, as stated above, 2 days after cryoablation. DC were enriched by positive selection using anti-CD11c-labeled magnetic micro beads (clone N418, Miltenyi Biotec, Bergisch Gladback, Germany). To study the fate of antigen and DC maturation after cryoablation, tumors were injected with fluorescently labeled OVA protein (OVA-Alexa488, Molecular Probes, 20 µg/20 µL) intratumorally just prior to ablation. CD11c-enriched cells were stained with anti-CD11c APC, anti-CD80 biotin (1G10/B7, BD Biosciences) and strep-PE. Expression of CD80 was analyzed in CD11c+ cells by flow cytometry (FACS Calibur, Becton Dickinson).
To assess the amount of antigen-specific T cells, 7–10 days after ablation, draining lymph node cells were stained with allophycocyanin-labeled or phycoerythrin-labeled iTag OVA-Kb tetramers (Beckman Coulter) and CD8 TCRβ. Cytokine profiles were assessed after re-stimulation with 0.1 µg/mL OVA Kb peptide and 10 µg/mL Brefeldin A (Merck Millipore, 203729) for 5 hours. Subsequently, cells were stained for CD8 TCRβ, fixed and permeabilized according to Cytofix/Cytoperm kit instructions (BD Pharmingen), and intracellular cytokine staining was carried out for IFNγ.
To determine the role of cross-priming and CTL induction, in vivo cytotoxicity assays were performed. Wild-type C57BL/6 splenocytes were differentially labeled with the dye carboxyfluorescein succinimidyl ester (CFSE) (1 µM vs 10 µM) according to manufacturer’s protocol (Thermo Scientific). CFSElow cells were pulsed with irrelevant HPV peptide (RAHYNIVTF, Kb), while the CFSEhigh cells were pulsed with OVA Kb peptide. These two fractions were mixed 1:1 (3×106 cells per fraction) and injected intravenously 10 days after cryoablation into recipient mice that received the indicated ablation treatments with B16OVA tumors. All recipient mice received intratumoral injection of 20 µg OVA just prior to ablation to boost precursor frequencies. Twenty hours later, recipient spleens and lymph nodes were isolated and analyzed for remaining target cells. Injection into an OT-I mouse served as a positive control.
BMDC culture and in vitro cross-presentation and OT-I assays
Bone marrow flushed out of tibia and femurs was mashed over a nylon mesh with pore size 100 µm. Cells (4×106 in 10 cm dish) were cultured for 8 days in RPMI medium, supplemented with 10% FBS, 2 mM glutamine, 1% Pen/Strep and 50 µM β-mercaptoethanol (BMDC medium) in the presence of 20 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF) (PeproTech), at 37°C with 5% CO2 (GM-CSF DCs). Protocol was adapted from previously described protocols.18 25 Expression of maturation markers were assessed using flow cytometry 16–24 hours after incubation with adjuvants. Supernatant was collected 16–24 hours after incubation with adjuvants, and cytokines (IL-6, IL-12, TNF-α and IL-1β) were measured using ELISA kits according to manufacturer’s protocols. Dilutions of supernatant used were 1:40, 1:4, 1:4, and 1:2, respectively.
For the in vitro cross-presentation assays, 80×103 DCs (B3Z assays) or 10×103 DCs (OT-I assays) were pulsed with indicated amounts of endotoxin-free chicken egg OVA (Endograde, Hyglos GmbH, Germany) in the presence of the indicated adjuvants. After 5 hours, cells were washed and cultured overnight with 80×103 B3Z cells or for 72 hours with 50×103 CFSE-labeled (3 µM) CD8+ OT-I cells (isolated with EasySep Mouse CD8+ T cell isolation kit, StemCell Technologies, 19 853A). As a control for cell viability and/or MHC class I expression levels, DCs were washed 30 min before adding the B3Z cells and pulsed with 5 ng/mL OVA Kb peptide (257–264, AS-60193, Tebu-bio). The presentation of OVA Kb peptide in H-2Kb results in production of β-galactosidase (LacZ) by B3Z cells, which can be detected by adding 0.15 mM chlorophenolred-h-D-galactopyranoside (Calbiochem), 9 mM MgCl2, 0.125% NP40 and 7.5 mM dithiothreitol in PBS. Plates were incubated for 3–5 hours at 37°C and absorbance values were measured at 595 nm using a photo spectrometer (Bio-Rad). For the readout of OT-I proliferation, T cells were stained extracellularly for CD90.1 and CD25. CFSE dilution, CD90.1 and CD25 expression were measured by flow cytometry.
To analyze cytokine production in T cells, 50×103 GM-CSF DCs were pulsed with OVA protein or OVA Kb peptide in round bottom 96-well plates for 5 hours at 37°C, 5% CO2. Plates were washed twice with BMDC medium and incubated overnight with 120×103 CD8+ OT-I T cells and 5 µg/mL brefeldin A at 37°C, 5% CO2. Next, T cells were stained extracellularly for CD90.1, followed by intracellular staining for TNF-α, IL-2 and IFNγ, according to manufacturer’s protocol (BD Bioscience).
RNA isolation and qPCR
In a 48-well plate, 0.4×106 GM-CSF DCs were plated and stimulated for 6 hours with indicated adjuvants. Total RNA was isolated from cells using a RNA isolation kit (Zymo Research), and treated with DNAse I on column. RNA quantity and purity were determined on a NanoDrop spectrophotometer. Equal amounts of RNA per sample was reverse transcribed into cDNA by using random hexamers and Moloney murine leukemia virus reverse transcriptase (Invitrogen). mRNA levels for IL-1ɑ (FW: CGTGTTGCTGAAGGAGTTGCC RV: TTCCAGAAGAAAATGAGGTCGGT) and IL-1β (FW: GGACAGAATATCAACCAACAAGTGATA RV: GTGTGCCGTCTTTCATTACACAG) were determined with a CFX96 sequence detection system (Bio-Rad) using the Faststart SYBR green mastermix (Roche) with SYBR Green as the fluorophore and gene-specific oligonucleotide primers. Reaction mixtures and program conditions were used that were recommended by the manufacturer (Bio-Rad). Quantitative PCR data were analyzed with the CFX Manager V1.6.541.1028 software (Bio-Rad) and checked for correct amplification and dissociation of the products. mRNA levels of the genes of interest were normalized to mRNA levels of the reference gene Rer1 (FW: GCCTTGGGAATTTACCACCT RV: CTTCGAATGAAGGGACGAAA) and were calculated according to the cycle threshold method.
Statistical analyses
Depending on the experimental layout, data were analyzed using a one-way analysis of variance, or with post hoc Tukey test, as indicated in the figure legends. Kaplan-Meier survival curves were analyzed using a log rank test, followed by a post hoc Bonferroni test. Differences were considered statistically significant when p values were <0.05.
Results
Co-administration of CpG and saponin-based adjuvants induces solid antitumor immunity following tumor ablation
We previously demonstrated that peritumoral injection of either CpG or saponin-based adjuvants directly following ablation of established B16 tumors resulted in enhanced CD8+ T cell-mediated antitumor immunity, via improved DC functioning.13 24 As recent studies have shown that combinations of adjuvants can improve vaccination outcome,26–28 we set out to explore the combination of the TLR9 adjuvant CpG and ISCOMs in a tumor ablation setting. To compare the induction of tumor-specific memory responses following administration of the various adjuvants in a stringent setting, wild-type B16F10 tumor-bearing mice were treated with cryoablation when the tumors measured 6–8 mm in diameter (figure 1A). Peritumoral injections of either CpG-ODN 1668, Quil A Saponin, ISCOMs or combinations thereof, were performed within 30 min following ablation. Forty days after ablation, tumor-free mice were rechallenged with B16F10 cells (see detailed time schedule in figure 1B). As we have shown before,4 cryoablation alone results in a small delay in the outgrowth of rechallenge tumors and partially induces protection (24%), compared with naïve controls (figure 1C). As expected, co-injection of CpG and either ISCOMs or saponin resulted in a clear enhanced protection (38%, 71% and 79%, respectively) of the mice.24 Importantly, simultaneous administration of both saponin-based adjuvants with CpG resulted in a memory response (82%–91%) protecting almost all mice from outgrowth of the rechallenge tumor, indicating the induction of a strong antigen-specific memory response (figure 1C). Even though statistical significance is not reached at day 80, a clear positive trend (11%–12% increase in survival by the combination with CpG over saponin-based adjuvants alone) is observed in this stringent model reaching its limits. In a setting where the peritumoral area was injected with CpG and/or ISCOMs, but the primary tumors were left untreated (no ablation), neither CpG or ISCOMs nor their combinations were able to prevent outgrowth of the primary tumor24 (data not shown). These results demonstrate that both cryoablation as well as the adjuvants are necessary for the observed tumor protection.

Potent antitumor memory response following ablation combined with saponin-based adjuvants and CpG-ODN. (A) Visual representation of cryoablation on 6–8 mm tumors. (B) Established B16F10 tumors subcutaneously on the right femur were treated with cryoablation alone, or in combination with peritumoral injection of the indicated adjuvants (CpG-ODN and ISCOMs 30 µg, Quil A saponin 20 µg), within 30 min after ablation. Forty days later, naïve and tumor-free mice received a re-challenge with tumor cells subcutaneously on the flank. (C) Growth of this re-challenge is depicted as a Kaplan-Meier survival curve demonstrating superior protection from tumor outgrowth after combination of ablation with saponin-based adjuvants Quil A saponin or ISCOMs and CpG-ODN. Data pooled from three to four independent experiments, with 19–34 mice per group in total. Statistical significance was calculated using a log-rank test with post hoc Bonferroni correction. *P<0.05 for Cryo/CpG vs Cryo/CpG/ISCOMs and Cryo/CpG/saponin at day 80. Cryo, cryoablation; CpG, cytidyl guanosyl; ISCOMs, immune-stimulating complexes.
In conclusion, these data show that the weak immune responses developing after in situ tumor destruction can be effectively improved by combining cryoablation with injection of adjuvants. The solid antitumor immunity observed after cryoablation with co-administration of CpG and saponin-based adjuvants (10% increase relative to single adjuvant treatment) led us to further investigate the immune effects generated by this adjuvant combination.
Similar co-stimulatory molecule expression and cross-presentation but enhanced cytokine production for CpG/ISCOMs stimulated DCs in vitro
Previously, we reported that post-ablation immune responses critically depended on antigen uptake and antigen cross-presentation by DCs.6 We therefore analyzed these parameters for various combinations of the adjuvants using GM-CSF-induced bone marrow-derived DCs. As reported before, ISCOM administration alone did not result in altered expression of the maturation markers CD80 and CD86, whereas stimulation with CpG does.18 24 Combining CpG with ISCOMs did not lead to a further increment of CD80 or CD86 (figure 2A). Next, we assessed the levels of antigen cross-presentation on CpG/ISCOMs exposure, using OVA as a model antigen and B3Z cells as a readout system. B3Z cells detect OVA peptide presented in H2-Kb independent of co-stimulatory molecules. Although cross-presentation is greatly increased due to the addition of ISCOMs, the combination with CpG led to a lower rather than higher level of cross-presentation, while stable presentation of exogenous OVA peptide was maintained (figure 2B). We next investigated cytokine secretion by GM-CSF DCs following overnight CpG/ISCOMs exposure. The production of the cytokines IL-12 and IL-6 was induced by CpG, but no further increase in production was observed when ISCOMs were added (figure 2C). However, the combination of ISCOMs with CpG released some additional TNF-α and especially a significantly higher amount of IL-1β as compared with either adjuvant alone (figure 2C). Interestingly, Wilson et al previously showed the ability of saponin-based adjuvants to release IL-1β by DCs on combination with TLR4-ligand lipopolysaccharide (LPS).29 Collectively, these in vitro data suggest that the increased memory response following ablation in combination with ISCOMs and CpG cannot be simply explained by a direct effect of the adjuvants on DC maturation or cross-presentation, but could possibly involve the increased production of cytokines, like IL-1β.

Combination of CpG and ISCOMs does not increase maturation, nor cross-presentation of antigens, but does affect cytokine production in vitro. Bone marrow DC were cultured with GM-CSF, and 2×105 (A), 0.8×105 (B) or 1–2×105 (C) cells were exposed to 400 ng/mL ISCOMs and/or 1 µg/mL CpG-ODN and (B) 80 µg/mL endotoxin-free ovalbumin (OVA) or OVA Kb peptide. (A) After 16–24 hours, CD80 and CD86 expression was determined in CD11c+ population by flow cytometry (n=5). (B) After 5 hours stimulation, cells were washed and cultured overnight with 0.8×105 B3Z cells, which produce LacZ on TCR recognition of OVA peptide in the context of H-2Kb. Next, LacZ production by the B3Z cells, as a measure of cross-presentation, was evaluated using a colorimetric assay (n=3). (C) After 16–24 hours, supernatant from GM-CSF DCs was harvested and the cytokines IL-6 (n=3), IL-12 (n=3), TNF-α (n=3) and IL-1β (n=21) were measured by ELISA. Results are shown as means with SEM. Statistical significance was calculated using a one-way analysis of variance with Tukey multiple comparison correction. *P<0.05; **p<0.01; ***p<0.001. CpG, cytidyl guanosyl; DC, dendritic cell; GM-CSF, granulocyte-macrophage colony-stimulating factor; IL, interleukin; ISCOMs, immune-stimulating complexes; ns, not significant; TNF, tumor necrosis factor.
Tumor ablation with CpG/ISCOMs releases IL-1 and induces PMN mobilization into lymph nodes
To study the in vivo release of IL-1 following ablation with adjuvant co-administration conditions, we investigated the influx of PMNs in various lymphocytic compartments in time, as mobilization of these cells is a well-documented effect after IL-1 release.30–32 Ablation alone leads to a rapid depletion of PMN from the bone marrow compared with naïve mice (figure 3A), as they are massively recruited to the site of tissue damage via positive feedback mechanisms33 to defend against infection, aid in wound debridement and produce mediators to activate other cells important for the repair process.34 35 The percentage of PMNs in the bone marrow is restored over the course of a week. An early increase of these cells in the blood at day 1 post-treatment suggests that these bone marrow cells get mobilized to the blood, before reaching the site of action. The enhanced PMN levels in the blood are more apparent when adjuvants were added, and the duration of these enhanced levels are longer, especially when (a combination with) ISCOMs were injected. Strikingly, only the cryoablation with CpG/ISCOMs adjuvant combination allowed PMN to enter the draining lymph nodes, as illustrated by a dramatic influx at day 1 and 2 (figure 3A). Indeed, performing cryoablation in IL-1ɑβ-/- mice confirmed that a large part of the PMN influx in draining lymph nodes observed following ablation plus CpG/ISCOMs injection is dependent on IL-1 release (figure 3B and C).

CpG/ISCOMs causes IL-1 release, as reflected by PMN influx. (A–C) Established B16F10 tumors subcutaneously on the right femur of WT (A–C) and IL-1αβ-/- (B, C) mice were left untreated or cryoablated alone, or in combination with peritumoral injection of indicated adjuvants, within 30 min after ablation. After indicated time points (A, n>4) and 2 days postablation (B, C, n=2–3), bone marrow, blood, spleens (B) and draining lymph nodes (A–C) were collected and percentage of PMNs (Ly6C+Ly6G+CD11b+) were detected by flow cytometry. Results are shown as means with SEM. Statistical significance was calculated using a one-way analysis of variance with Tukey multiple comparison correction. **P<0.01. CpG, cytidyl guanosyl; IL, interleukin; ISCOMs, immune-stimulating complexes; PMN, polymorphonuclear neutrophil; WT, wild-type.
Summarizing, our data indicate in vivo release of IL-1 and related PMN mobilization following cryoablation, and specific PMN recruitment into draining lymph nodes after CpG/ISCOMs administration.
CpG/ISCOMs exposed DCs induce qualitatively better T-cell responses
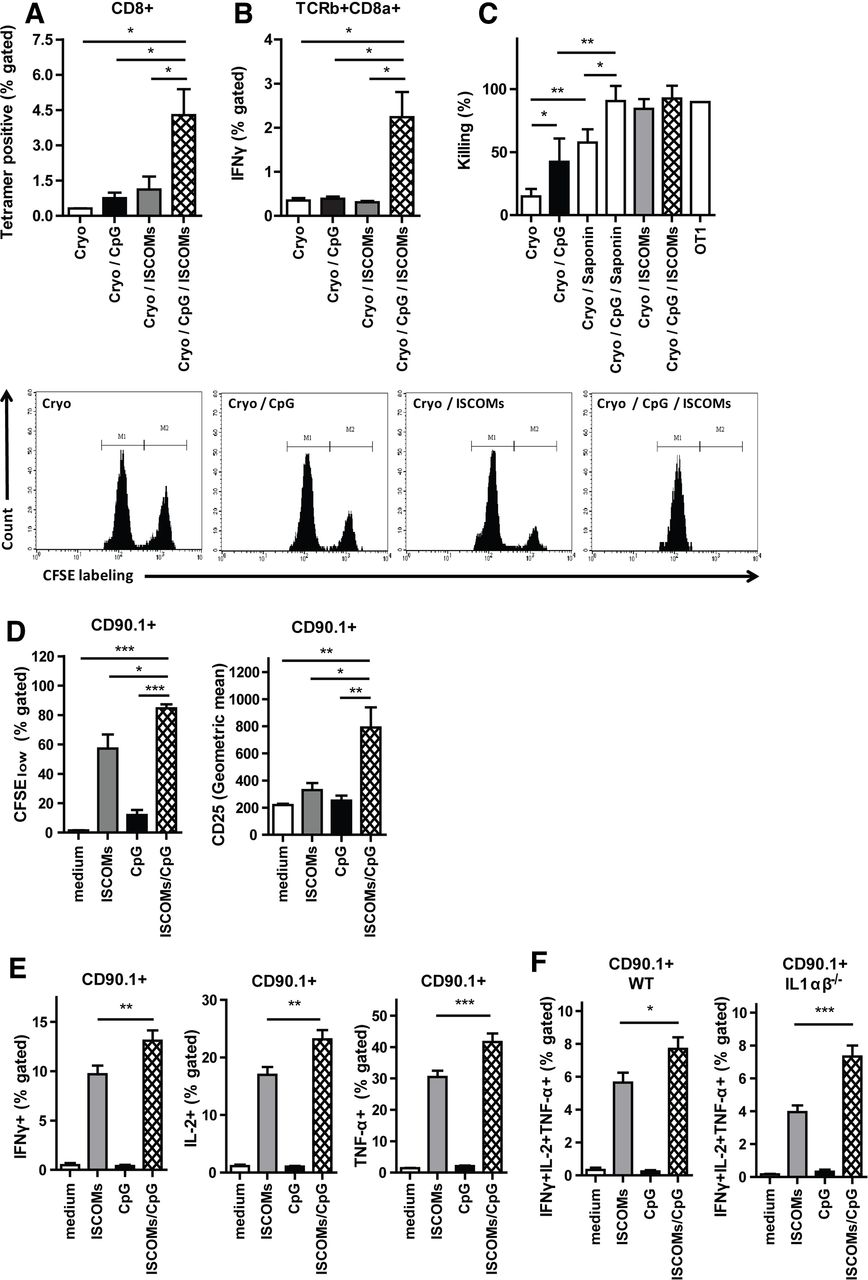
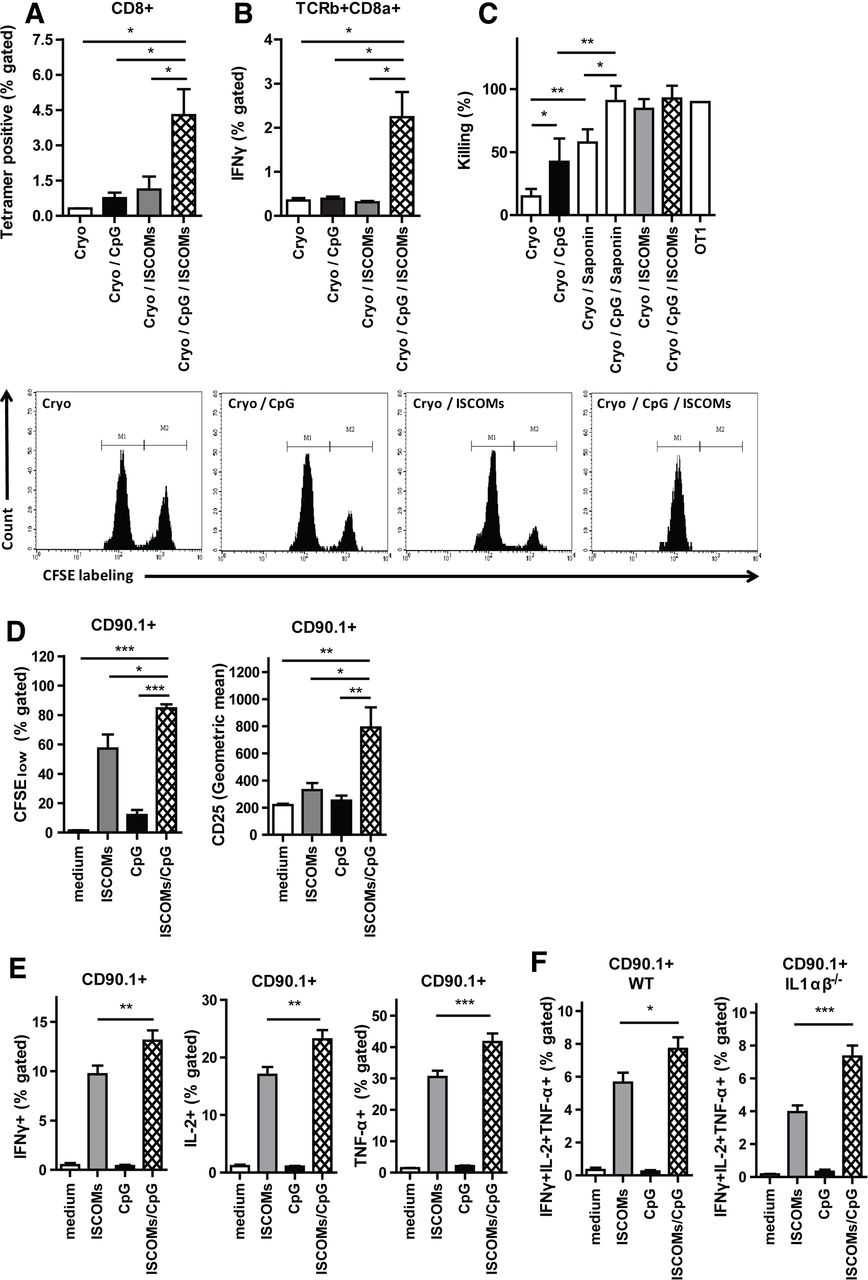
Although their exact functions within lymph nodes are not well understood, PMN and IL-1 have both been implicated in modulating innate and adaptive immunity.32 36 37 Recently, T-cell-intrinsic IL-1R signaling has been shown to license production of effector cytokines in T cells.38 This prompted us to analyze CD8+ T-cell responses in vivo in OVA-tumor bearing mice treated with ablation with or without the different adjuvants. Quantification of OVA antigen-specific CD8+ T cells isolated from draining lymph nodes showed that mice treated with cryoablation and injected with ISCOMs and CpG had a strong increase in antigen-specific T cells as compared with either adjuvant alone (figure 4A). Moreover, intracellular staining of total lymph node CD8+ T cells restimulated with OVA Kb peptide also showed superior IFNγ production by these T cells (figure 4B). IFNγ derived from cytotoxic T lymphocytes directly enhances their motility and cytotoxicity.39 To confirm the enhanced functional capacity of these T cells, an in vivo cytotoxicity assay was performed. Killing of OVA-peptide loaded target cells (high CFSE labeling) versus irrelevant peptide-loaded target cells (low CFSE labeling) was highly dependent on the co-administration of adjuvants following ablation, with ISCOMs being more efficient than CpG co-administration, which is in line with our previous findings.40 Strikingly, co-administration of saponin-based adjuvants together with CpG showed unprecedented killing levels almost reaching maximum levels of target killing in our model (figure 4C).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CpG/ISCOMs adjuvant treatment to DCs results in multifunctional T cells. (A–C) Established B16OVA tumors were treated with cryoablation alone, or in combination with the indicated adjuvants. (A) Seven days after ablation, lymph node cells were stained for OVA-Kb tetramers. (B) Seven days after ablation, cells from draining lymph nodes were restimulated with OVA Kb peptide and brefeldin A for 5 hours and TCR-β+CD8α+ cells were analyzed for the expression of IFNγ. (A, B) Results are shown as means with SEM (n=2). (C) After 10 days, the cryo-ablated and adjuvant injected mice, or one OT-I mouse, received CFSEhigh splenocytes pulsed with the target peptide (OVA Kb peptide), along with CFSElow splenocytes pulsed with the control peptide (HPV Kb peptide), in a ratio of 1:1. Inguinal lymph nodes of these mice were harvested 20 hours later and the relative numbers of CFSEhigh and CFSElow target cells were determined by flow cytometry. Data depicted are mean percentages of target cell killing (n=6 mice) with SD, corrected for background cell-death in naïve mice, and representative plots. Bone marrow DCs were cultured with GM-CSF, and 0.1×105 (D) or 0.5×105 (E, F) cells were exposed to 400 ng/mL ISCOMs and/or 1 µg/mL CpG-ODN and 80 µg/mL endotoxin-free ovalbumin (OVA protein). After 5 hours, cells were washed and cultured with 50×103 CFSE-labeled CD8+ T cells for 72 hours (D) or 1.2×105 CD8+ OT-I T cells with brefeldin A overnight (E, F). (D) Cells were stained for CD90.1 and CD25. The expression of CD25 and the CFSE labeling in CD90.1+ population as a measure of proliferation were assessed by flow cytometry (n=3). (E, F) Intracellular build-up of IFNγ, IL-2 and TNF-α was measured using flow cytometry (E, WT, n=9 and F, IL-1ɑβ-/-, n=4). (D–F) Results are shown as means with SEM. (A–F) Statistical significance was calculated using a one-way analysis of variance with Tukey multiple comparison correction. *P<0.05; **p<0.01; ***p<0.001. CpG, cytidyl guanosyl; DC, dendritic cell; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN, interferon; IL, interleukin; ISCOMs, immune-stimulating complexes; TNF, tumor necrosis factor; WT, wild-type.
As DCs respond to both adjuvants by producing IL-1, which is believed to be crucial for T cell priming,38 41 42 we analyzed the capacity of adjuvant-activated DCs to activate antigen-specific T cells and induce effector functions in vitro. OVA-specific OT-I cells incubated with CpG/ISCOMs-stimulated DCs proliferated more and expressed higher levels of CD25 after 72 hours of co-culture relative to DCs matured by either stimulus alone (figure 4D). Intracellular cytokine staining demonstrated increased pro-inflammatory cytokine production (IFNγ, IL-2 and TNF-α) in OT-I cells activated by DCs treated with CpG/ISCOMs, as compared with DCs treated with the single adjuvants (figure 4E). Also, when gated for triple cytokine positive T cells, we observed an increase in the percentage of multifunctional T cells when exposed to CpG/ISCOMs incubated DCs (figure 4F). Next, we used IL-1αβ-/- DCs to determine the role of IL-1 produced by ISCOM/CpG-activated DC in the observed effects. Absence of IL-1α and -β production by DCs alone is not sufficient to affect the induction of multifunctional OT-I T cells in vitro (figure 4F). Collectively, these data show that CpG/ISCOM stimulation of DCs result in a higher quantity and quality of antigen-specific CD8+ T cells.
Altogether, our data suggest that in situ tumor destruction by cryoablation together with a combination of saponin-based adjuvants plus the TLR9-ligand CpG leads to a superior systemic antitumor response, as compared with either adjuvant modality alone. The combination of saponin-based adjuvants with CpG induces multifunctional T cells, able to produce high amounts of pro-inflammatory cytokines.
Discussion
Tumor ablation regimens allow for direct in situ (neo)antigen loading of DCs without delivery of predefined tumor antigens or peptides as in conventional DC vaccination. The challenge within the field of interventional oncology is to create an in situ vaccine that drives systemic immune responses following tumor ablation that is powerful enough to cause a broad and effective systemic antitumor immune response. Previously, we have shown that cryoablation can induce effective antitumor memory responses in multiple animal models provided that adjuvants are co-administered at the time of ablation.4 6 12 13 24 40 In this study, we report that cryoablation plus co-injection of both saponin-based adjuvants and TLR9-ligand CpG unleashes a more potent antitumor immune response than either adjuvant alone. Mechanistically, this adjuvant combination results in 1) induction of IL-1 production by DCs in vitro and PMN influx in lymph nodes in vivo, 2) the generation of more multifunctional T cells able to produce higher amounts of pro-inflammatory cytokines.
TLR agonist-type adjuvants, like CpG, ensure essential DC maturation and upregulation of co-stimulatory molecules in vitro and in vivo after cryoablation,6 12 13 24 while saponin-based adjuvants excel in stimulation of antigen cross-presentation by DC.18 24 In line with previously published data, addition of ISCOMs to in vitro cultured inflammatory DCs alone did not upregulate maturation marker expression.43 In contrast to the in vitro data, we and others have shown that ISCOMs induce a level of maturation of DCs in vivo, alone or when combined with in situ tumor destruction (online supplementary figure 2a).24 44 This in vivo-in vitro discrepancy is most likely due to endogenous mediators able to mature the DCs that are released by other (immune) cells not present in vitro that respond to the saponin adjuvant in vivo and indirectly enhance DC maturation and/or the tumor ablation treatment. Indeed, cryoablation in itself is known to induce cytokine release, DAMPs and mature especially the antigen-positive DCs.24
Supplemental material
Our current data show that the combination of adjuvants decreases rather than increases cross-presentation by DC in vitro. In vivo, somewhat less antigen-specific DCs numbers are present in the draining LNs (online supplementary figure 2b) after ablation with dual adjuvant treatment compared with single ISCOMs treatment. Besides DC maturation and antigen presentation also cytokines are key for immune response induction, T-cell expansion and effector T-cell differentiation. ISCOMs are known to initiate localized inflammatory responses at the subcutaneous site of injection in a MyD88-independent way,44 while CpG signals via MyD88 axis to activate immune cells, including cytokine production. These different pathways could crosstalk and thereby enhance or counteract each other. Interestingly, in vitro cultured DCs secreted increased amounts of TNF-ɑ when CpG and ISCOMs were combined. Most strikingly, only the combination of the two adjuvants resulted in high IL-1β production in vitro, indicating inflammasome activation in DCs. These data are in line with other reports showing synergy between NALP3 inflammasome activation and TLR stimulation. In peritoneal macrophages, an ISCOMs-based adjuvant OVA formulation with a different TLR-ligand, LPS, resulted in IL-1β and IL-18 production in vitro.29
Our data show that saponin-based adjuvants injected after cryoablation induced PMN mobilization from the bone marrow to the blood. Likewise, ISCOMs containing adjuvants cause influx of monocytes and neutrophils in subcutaneous air pouches in 4–24 hours.44 Strikingly, we show that only the combination of CpG and ISCOMs result in PMN recruitment into the tumor-draining lymph nodes after cryoablation, which is in line with the current knowledge that IL-1β secretion results in neutrophil recruitment.30–32 The enhanced production of TNF-ɑ by the DCs after combination treatment might also play a role in this neutrophil recruitment.31 45 The functions exerted by neutrophils in lymph nodes are diverse and include transport of antigens and attracting other leukocytes into the lymph node.32 Crosstalk between neutrophils and DCs, via contact-dependent and independent ways, has been suggested to contribute to DC maturation.46 Preliminary data in total IL-1αβ-/- mice or partial PMN depletion using anti-Ly6G and anti-Gr1 antibodies (online supplementary figure 3a) did not yield clear effects on either DC maturation (online supplementary figure 3b) or T cells (online supplementary figure 3c).
Supplemental material
T-cell intrinsic IL-1R signaling has recently been shown to permit effector cytokine production by pre-committed T-cell lineages.38 Ben-Sasson et al have shown that IL-1 enhances CD4+ T cell, but also CD8+ T cell responses.41 42 Following cryoablation and the adjuvant combination, an increase in amount of IFNγ production by antigen-specific CD8+ T cells was observed. In vitro, enhanced T cell proliferation was induced on co-culture with DCs treated with ISCOMs and CpG. Interestingly, these CD8+ T cells also showed enhanced production of different immune-stimulating cytokines. Thus, combination treatment results in a change in quantity and in multifunctionality of T cells. As the co-stimulation-independent B3Z assay showed lesser cross-presentation at these conditions, co-stimulatory pathways must play an important role. Analysis of IL-1ɑβ-/- DCs revealed they still are able to induce increased numbers of multifunctional T cells in vitro, indicating IL-1ɑ/β is not essential. Interestingly, multifunctional T cells after ISCOM treatment have also been described in CD4+ T cell population.47 In conclusion, the adjuvant combination alters DCs in their ability to induce multifunctional CD8+ T cells that are superior in producing immune-stimulating cytokines. Further research is needed to investigate the precise mechanism by which the combination of ISCOMs and TLR-ligands result in superior DC activation and anti-tumor immunity induction.
Saponin-based adjuvants are promising as they can induce robust cell-mediated immune responses.26 48 The only study up to now using ISCOMs in a human cancer setting is using full-length NY-ESO-1 protein formulated in ISCOMs adjuvant. Although Cebon et al demonstrate strong humoral and T cell-mediated immune responses, which reduced relapse rates in patients with fully resected melanoma, patients with advanced melanoma showed no clinical responses to vaccination.21 Unfortunately, patients with advanced melanoma had a significantly higher proportion of circulating regulatory T cells compared with those with minimal residual disease. CpG has been tested as vaccine adjuvant in clinical trials for several diseases,49 50 and is considered to be safe.51 Interestingly, a vaccine formulation combining the saponin-based adjuvant QS21 and CpG also containing monophosphoryl lipid A called AS15, is well tolerated52–54 and showed enhanced survival in patients with melanoma in a phase II trial.52
The in situ vaccination strategy ensures that the immune system is well instructed and the adaptive arm is properly initiated. However, immune suppression that antigen-specific CD8+ T cells encounter when arriving at the tumor site could still be a potential bottleneck.21 Therefore, the combination of activating adjuvants (already approved for clinical use) at the time of tumor ablation with agents that create an immune permissive TME, like anti-PDL1, are currently pursued. For cancer-vaccine development aiming to induce strong T-cell responses, both antigen cross-presentation and DC maturation are required.26 These two prerequisites can be achieved by a combination of saponin-based adjuvants, and TLR-ligands like CpG, respectively. Indeed, as regular vaccine adjuvants the combination of the saponin-based adjuvants and TLR-ligands have been explored in mouse models and result in enhanced antibody responses as well as improved IFNγ-producing T cell responses.55 56 We have now shown that the combination of these two adjuvants in a tumor-ablative setting results in CD8+ T cells with enhanced capacity to produce pro-inflammatory cytokines as well.
Conclusions
Our data demonstrate that antitumor immunity following cryoablation can be enhanced by co-injection of saponin-based adjuvants and CpG. We showed that these adjuvants promote adaptive immunity via an improved functioning of DCs and increased quality of CTLs. The fact that saponin-based adjuvants like ISCOMs as well as CpG are currently evaluated for clinical application makes these adjuvants outstanding tools to combine with in situ tumor destruction.
Acknowledgments
The authors would like to thank C. Schrier, E. Rijke, W. Degen, E. Mombarg, J. Sigmans, M. Minderman, C. Büll, N. Balneger and L. Cornelissen for technical assistance and/or helpful discussions; and F. van der Loo for kindly providing the IL1αβ-/-mice.
References
Footnotes
RJEvdB, MHdB, MA and GJS contributed equally.
Contributors TR, RvdB, MdB, MW, AdG, JW and SN performed the animal and analytical experiments. MdB, GJS and GA conceived the project. TR, MdB, SN and GA designed the experiments. TR, RvdB, MdB, SN, MA and GA interpreted the data. TR, RvdB, MdB and GA wrote the paper. All authors read and approved the final manuscript.
Funding This work was supported by the Dutch Cancer Society (KUN2013-6111) to MdB and GA.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The experiments were approved by the Animal Experiments Committee of the Radboud University Medical Center, and were performed in accordance with institutional, national and European guidelines.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request.