Article Text

Abstract
Background Immune checkpoint inhibitors (ICIs) have produced significant survival benefit across many tumor types. However, immune-related adverse events are common including autoimmune responses against different endocrine organs. Here, a case of ICI-mediated hypoparathyroidism focusing on long-term follow-up and insights into its etiology is presented.
Case and methods A 73-year-old man developed severe symptomatic hypocalcemia after the initiation of ipilimumab and nivolumab for the treatment of metastatic melanoma. Hypoparathyroidism was diagnosed with undetectable intact parathyroid hormone (PTH). Immunoprecipitation assays, ELISAs, and cell-based functional assays were used to test the patient for antibodies against the calcium-sensing receptor (CaSR). NACHT leucine-rich repeat protein 5 (NALP5) and cytokine antibodies were measured in radioligand binding assays and ELISAs, respectively.
Results The patient’s symptoms improved with aggressive calcium and vitamin D supplementation. At 3 years and 3 months since the diagnosis of hypoparathyroidism, PTH was still inappropriately low at 7.6 pg/mL, and attempted discontinuation of calcium and calcitriol resulted in recurrent symptomatic hypocalcemia. Analysis for an autoimmune etiology of the patient’s hypoparathyroidism indicated that CaSR antibodies were negative before treatment and detected at multiple time points afterwards, and corresponded to the patient’s clinical course of hypoparathyroidism. CaSR antibodies purified from the patient’s serum activated the human CaSR. The patient was seronegative for NALP5 and cytokine antibodies, indicating that their hypoparathyroidism was not a manifestation of autoimmune polyendocrine syndrome type 1.
Conclusion The etiology of hypocalcemia is likely autoimmune hypoparathyroidism caused by the development of CaSR-activating antibodies that might prevent PTH release from the parathyroid.
- antigens
- immunity, humoral
- immunotherapy
- melanoma
- epitope mapping
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Monoclonal antibodies that inhibit immune checkpoint proteins such as the cytotoxic T lymphocyte antigen-4 (CTLA-4) and programmed cell death 1 receptor (PD-1) have revolutionized the treatment approach to many solid tumors.1 2 Furthermore, the combination of ipilimumab, an anti-CTLA-4 antibody, and nivolumab, a PD-1 inhibitor, has demonstrated synergistic antitumor activities, significantly improving objective response rates and overall survival in patients with advanced melanoma or renal cell carcinoma.3 4 Given these results, we expect the number of emerging indications for immune checkpoint inhibitors (ICIs) to continue to grow. However, these novel agents can cause life-threatening immune-related adverse events (irAEs) since checkpoint blockade can lead to unrestrained immune attack on healthy tissues.5 In addition to the skin and the gastrointestinal tract, the endocrine system is one of the most commonly affected organs.5 6
Thyroid dysfunction and hypophysitis have the highest incidence among the endocrine irAEs of ICIs.5 6 Thyroid disorders are more common with the anti-PD-1/programmed cell death receptor ligand 1 antibodies, either as monotherapy or in combination with an anti-CTLA-4 antibody.7 8 Hypophysitis is more frequently observed with ipilimumab as single agent or in combination with other ICIs.5 Rare cases of type 1 diabetes, Graves’ ophthalmopathy, and autoimmune adrenalitis have been observed.5 6 Hypoparathyroidism is exceedingly rare.9–11
Here, we describe the long-term follow-up of a patient who developed severe symptomatic hypocalcemia after the initiation of ipilimumab and nivolumab combination for the treatment of metastatic melanoma. A possible autoimmune etiology of the patient’s hypoparathyroidism was investigated. The patient’s serum samples were tested for: (1) Calcium-sensing receptor (CaSR) antibodies, as they can be present in patients with hypoparathyroidism.12–14 (2) NACHT leucine-rich repeat protein 5 (NALP5) antibodies, as they can be indicative of hypoparathyroidism in the context of autoimmune polyendocrine syndrome type 1 (APS1).15 (3) Cytokine antibodies to confirm or deny a diagnosis of APS1 of which hypoparathyroidism is a key manifestation.16 The effect of the patient’s CaSR antibodies on CaSR activity was also tested.
Case presentation
A 73-year-old man with widely metastatic BRAF V600K mutated melanoma was treated in the frontline setting with ipilimumab 3 mg/kg plus nivolumab 1 mg/kg combination therapy intravenously every 3 weeks. One week after his second cycle, he presented to the emergency department with increased fatigue, dizziness, difficulty maintaining balance, and abdominal bloating and discomfort. His wife also noticed slowing of speech and change in facial expression. Review of systems was positive for perioral numbness, paresthesia in both hands and feet, and numbness over his left abdomen. Physical exam was remarkable for marked ataxia when walking.
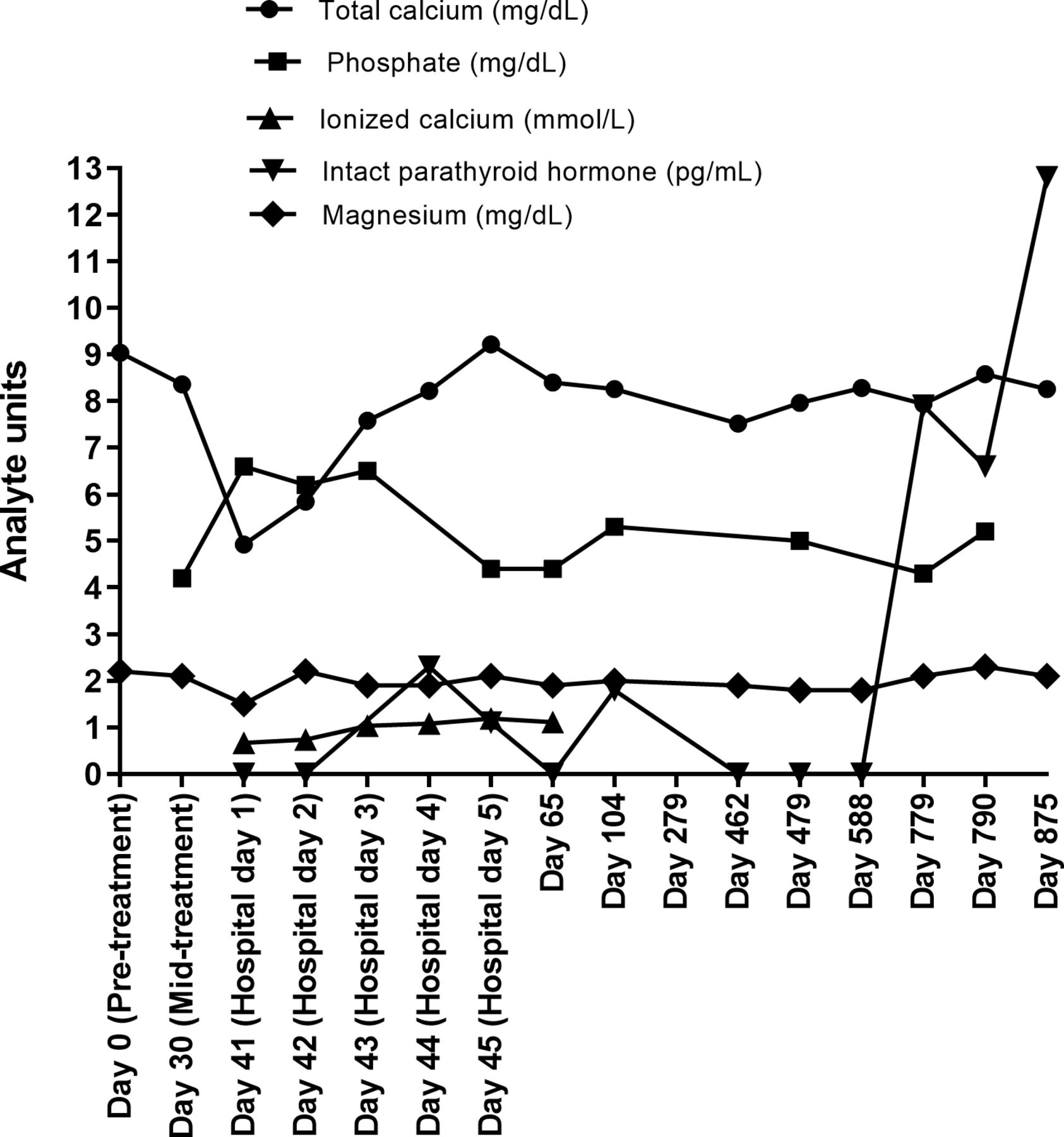
Laboratory tests at presentation revealed a low total calcium of 5 mg/dL (normal range: 8.4–10.2 mg/dL), low ionized calcium of 0.67 mmol/L (normal range: 1.13–1.32 mmol/L), high phosphate of 6.6 mg/dL (normal range: 2.5–4.5 mg/dL), low magnesium of 1.5 mg/dL (normal range: 1.8–2.9 mg/dL), and undetectable intact parathyroid hormone (PTH) at <1 pg/mL (normal range: 15–65 pg/mL) (figure 1). In addition, the patient had normal albumin of 4.1 g/dL (normal range: 3.5–5.2 g/dL), normal creatinine of 0.9 mg/dL (normal range: 0.67–1.17 mg/dL), but a high urinary calcium of 266.6 mg/24 hours. The patient also had low 25-hydroxyvitamin D of 18 ng/mL (normal range: 30–100 ng/mL) and normal 1,25-dihydroxyvitamin D of 29 pg/mL (normal range: 18–64 pg/mL). The 1,25-dihydroxyvitamin D level would be expected to be low because of severely reduced PTH. It is likely that the hypoparathyroidism event was acute such that 1,25 dihydroxyvitamin D was still normal, although low, at the point of presentation. The laboratory analyses led to a diagnosis of severe symptomatic hypocalcemia due to primary hypoparathyroidism.

Time course of biochemical tests. The results of biochemical tests are shown for corrected calcium (normal range: 8.4–10.2 mg/dL), phosphate (normal range: 2.5–4.5 mg/dL), ionized calcium (normal range: 1.13–1.32 mmol/L), magnesium (normal range: 1.8–2.9 mg/dL), and intact parathyroid hormone (PTH) (normal range: 15–65 pg/mL) from the start of acute hypocalcemia to the most recent follow-up visit.
The patient was initiated on a calcium gluconate drip (5000 mg/500 mL calcium gluconate with 5% dextrose at an infusion rate of 50 mL/hour), oral calcium carbonate supplement (equivalent to 1500 mg of elemental calcium three times daily), calcitriol (0.5 µg twice daily), and ergocalciferol (50 000 IU daily for 5 days and weekly, thereafter). Mild hypomagnesemia was corrected (figure 1). Despite normalization of calcium and magnesium levels, PTH remained undetectable (figure 1). Nevertheless, the patient’s symptoms improved in parallel with serum calcium normalization. He was discharged 4 days later on calcium carbonate (equivalent of 1000 mg elemental calcium twice daily), calcitriol (0.25 µg twice daily) and ergocalciferol (50 000 IU weekly).
The patient received cycle 3 of ipilimumab and nivolumab therapy 3 weeks later. Additional irAEs emerged including thyroiditis with a thyrotoxicosis phase (free T4 of 4.52 ng/mL, normal range: 0.93–1.70 ng/mL; thyroid-stimulating hormone (TSH) of <0.02 µU/mL, normal range: 0.27–4.20 µU/mL; total T3 of 226 ng/dL, normal range 80–200 ng/dL; thyroid peroxidase antibody-negative, and TSH receptor antibody-negative), and hepatitis (alanine aminotransferase of 706 U/L, normal range: 7–56 U/L; aspartate aminotransferase of 447 U/L, normal range: 14–46 U/L) requiring high doses of steroids and permanent discontinuation of ICI therapy 4 weeks after the hypoparathyroidism diagnosis.
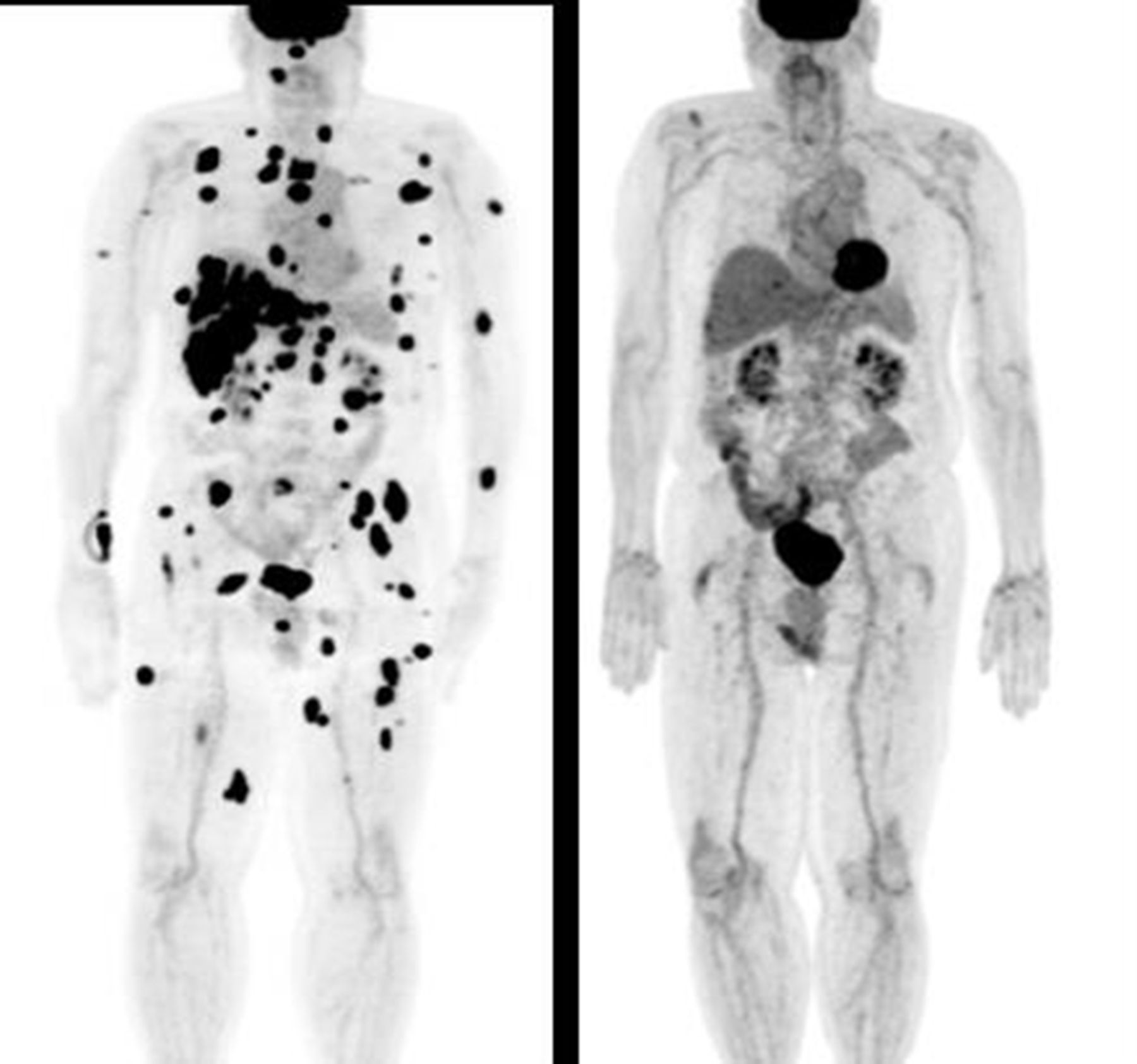
Despite his toxicities, the patient achieved a complete response to treatment with disappearance of all his metastatic tumors and remains in remission and asymptomatic from his previous irAEs almost 2 years later (figure 2). His calcium level continued to be stable, between 8.0 mg/dL and 8.5 mg/dL (figure 1) with low doses of calcium (equivalent of 1000 mg elemental calcium per day), calcitriol (0.25 µg daily) and ergocalciferol (50 000 IU weekly). PTH remained undetectable (<0.1 pg/mL) for over 2 years (figure 1). However, 2 years and 3 months since this sentinel event, his PTH level was 12.8 pg/mL with a corrected calcium of 8.2 mg/dL (figure 1). A trial of calcium and calcitriol discontinuation was attempted, however, the patient developed recurrent symptoms of hypocalcemia and, therefore, was given calcium and calcitriol. At his last follow-up at 3 years and 3 months, his PTH was again inappropriately low at 7.6 pg/mL.

Response of the patient to ipilimumab plus nivolumab treatment. Positron emission tomography (PET) scans showing the patient prior to treatment (left-hand panel) and the complete response to treatment (right-hand panel) with disappearance of all the patient’s cancer lesions 2 years after the start of immune checkpoint inhibitor (ICI) therapy.
With respect to the etiology of the patient’s hypoparathyroidism, he denied a history of past radiation to the neck or prior surgery to thyroid and parathyroid glands which could have damaged the organs prior to ICI treatment. No familial hypocalcemic disorders were present. The patient had no history of autoimmune disease or parathyroid inflammation. In addition, the patient’s total calcium level prior to immunotherapy initiation was normal at 9.2 mg/dL. This information excluded the possibility that he had a primary parathyroid dysfunction prior to treatment with ICIs. The patient’s hypoparathyroidism was therefore considered to be related to his treatment with ipilimumab and nivolumab and that it maybe autoimmune in etiology as irAEs against endocrine organs often are.
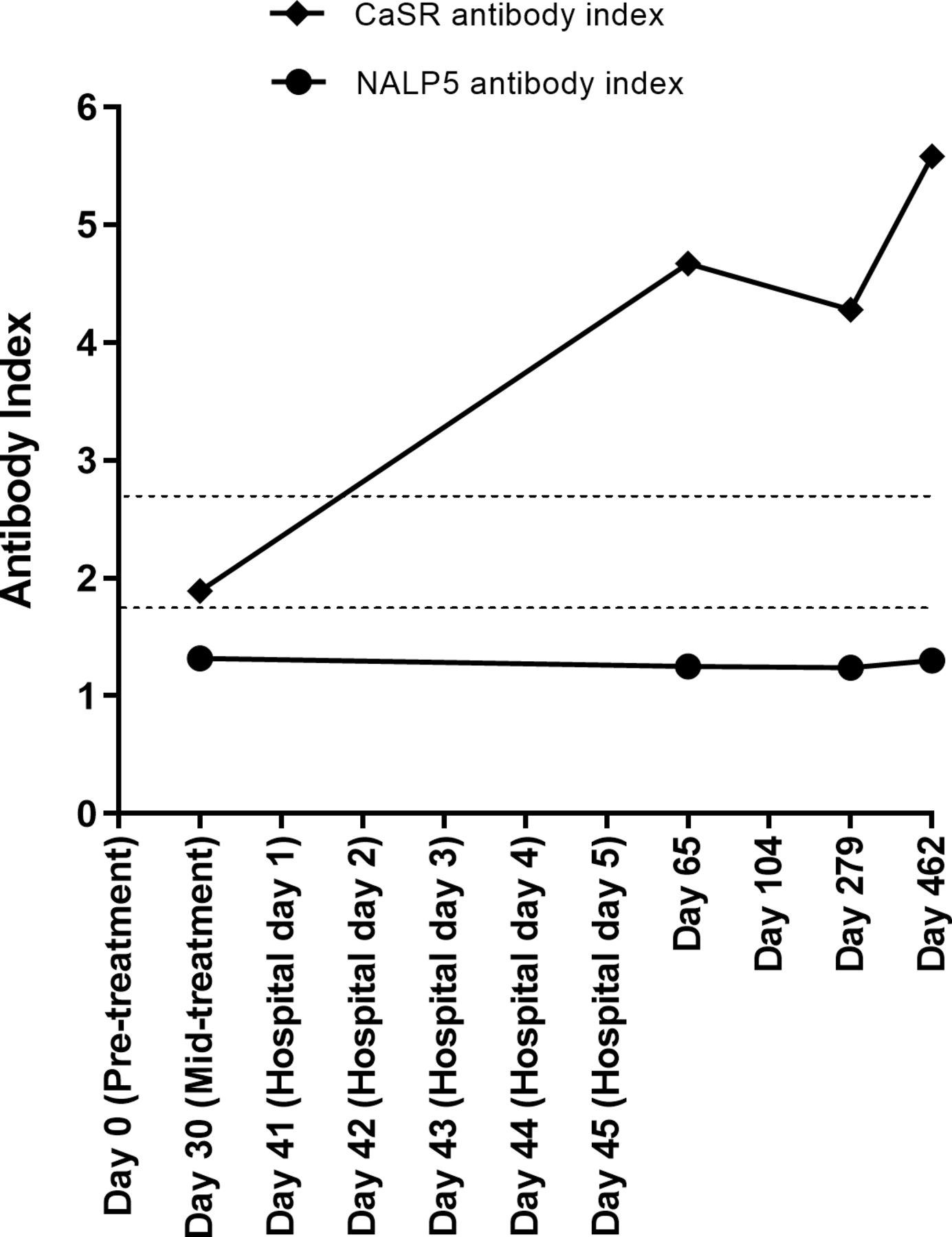
Autoimmune hypoparathyroidism can occur in isolation or as part of Autoimmune Polyglandular Syndrome Type 1 (APS1).12 14 Initially, to eliminate APS1 as the disease context for the patient’s hypoparathyroidism, serial archived serum samples from the patient were evaluated in ELISAs for cytokine antibodies against interleukin (IL) 22, IL-17F, IL-17A, interferon (IFN)-ω, IFN-α2A, and IFN-λ1.16 These are disease markers for APS1. The patient was negative for all cytokine antibodies tested, indicating that the patient was unlikely to have APS1. In addition, serum samples were investigated using radioligand binding assays for the APS1 hypoparathyroidism markers, NALP5 antibodies.15 However, NALP5 antibody serology was negative at all time points (figure 3).

Calcium-sensing receptor (Casr) and NACHT leucine-rich repeat protein 5 (NALP5) antibody tests. The upper limit of healthy control sera in the CaSR antibody assay was a CaSR antibody index of 2.69. The upper limit of healthy control sera in the NALP5 antibody assay was an NALP5 antibody index of 1.73. The upper limit for each assay is shown by a dotted line.
Antibodies against the CaSR have been detected in patients with autoimmune hypoparathyroidism with and without the context of APS1.12–14 17 In the present case, CaSR antibodies were detected at multiple time points, with time at positivity correlating with the patient’s clinical course of hypoparathyroidism (figure 3). His serum samples were negative for CaSR antibodies at two time points prior to hypoparathyroidism diagnosis, and increased at the time of hypoparathyroidism diagnosis and remained high during follow-up.
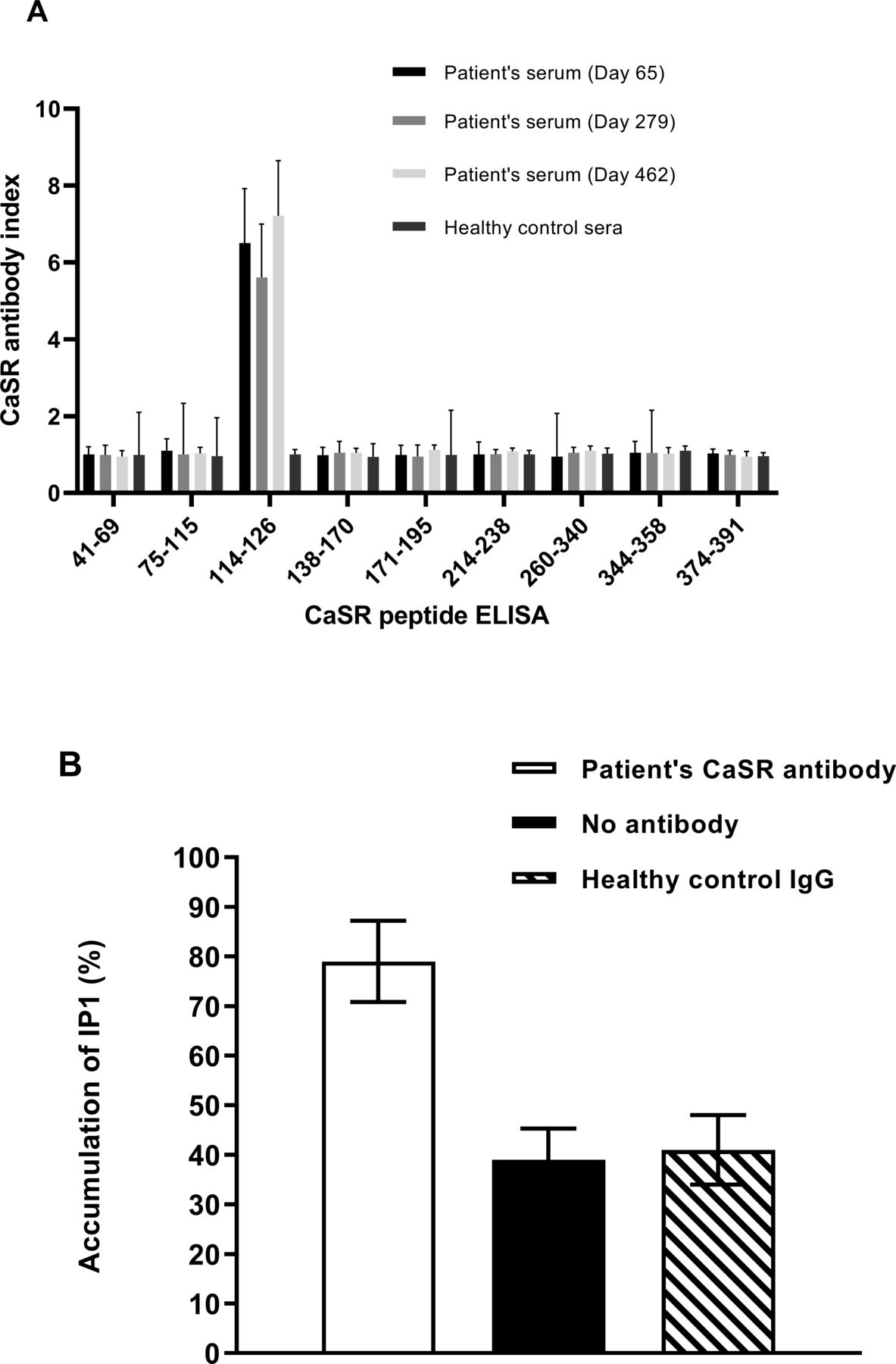
Patient serum samples post-ICI therapy were analyzed in ELISAs using CaSR peptides spanning amino acids 41–69, 75–115, 114–126, 138–170, 171–195, 214–238, 260–340, 344–358, and 374–391 as the target antigens.13 The results indicated that the patient’s CaSR antibodies were against the CaSR peptide 114–126, an epitope previously identified.13 No other CaSR antibody-binding specificities were detected (figure 4A). Subsequently, peptide affinity chromatography13 was used to purify CaSR antibodies against CaSR peptide 114–126 from the patient’s serum.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Characterization of the patient’s calcium-sensing receptor (CaSR) antibodies. (A) Epitope identification. Patient post-treatment serum samples and sera from 10 healthy controls were evaluated at a dilution of 1:100 in ELISAs for antibodies against CaSR peptides 41–69, 75–115, 114–126, 138–170, 171–195, 214–238, 260–340, 344–358, and 374–391. The CaSR antibody index (mean±SD) is shown for three experiments. (B) Effect of the patient’s CaSR antibodies on CaSR activity. Changes in inositol-1-phosphate (IP1) accumulation were measured in response to Ca2+ in human embryonic kidney 293 cells expressing the receptor (HEK293-CaSR) cells preincubated with the patient’s purified CaSR antibody against CaSR peptide 114–126 at a 1:100 dilution prior to stimulation with Ca2+ at a final concentration of 1.5 mM. HEK293-CaSR cells without preincubation with antibody and preincubated with IgG from a healthy individual were included as controls. Intracellular IP1 accumulation was measured using a IP-One ELISA. The mean (±SD) IP1 accumulation is shown for three experiments.
In some cases, CaSR antibodies have been shown to modulate the receptor’s function such that its response to calcium is adversely affected.14 17 To determine the effects of the patient’s CaSR antibodies on CaSR function, human embryonic kidney 293 cells expressing the receptor (HEK293-CaSR) were preincubated with the patient’s purified CaSR antibodies prior to measurement of Ca2+-induced inositol-1-phosphate (IP1) accumulation in an IP-One ELISA.17 The results showed that preincubation of HEK293-CaSR cells with the patient’s CaSR antibody significantly increased the levels of IP1 accumulation when compared with Ca2+-stimulation of HEK293-CaSR cells that were not preincubated with CaSR antibody (figure 4B). The results indicated that the patient’s CaSR antibodies were able to stimulate the receptor so that even at lower than optimum calcium levels PTH secretion from the parathyroids would be reduced leading to hypocalcemia.
Discussion
Chronic hypoparathyroidism is a rare endocrinopathy.18 Surgical intervention to the neck is responsible for 75% of cases with the remainder occurring secondary to autoimmune, genetic, or infiltrative etiologies.18 19 Autoimmune hypoparathyroidism is more commonly seen as part of APS1, but can arise as an isolated disease.19 20 It can be caused by immune-mediated damage to the parathyroid gland or by functional hypoparathyroidism due to antibody-induced activation of parathyroid function.14 17 20 Such activating antibodies are directed towards the CaSR.14 By decreasing the receptor’s set point, PTH is not released at lower than optimum blood calcium concentrations resulting in hypocalcemia.14 20 Although rare, autoimmune hypoparathyroidism can also manifest following the use of ICIs for the treatment of cancer.9–11
Here, we report a patient with metastatic melanoma who developed hypocalcemia resulting from autoimmune hypoparathyroidism following ICI treatment. The case described demonstrates a cause of hypoparathyroidism that is likely immune-mediated secondary to ICIs, given the temporal proximity of hypoparathyroidism to the initiation of treatment without other mechanism for acute injury to the parathyroid glands. The detection of CaSR-activating antibodies matching the patient’s clinical course further supports a diagnosis of autoimmune hypoparathyroidism caused by the development of CaSR antibodies that could inhibit PTH release from the parathyroid even at below optimum levels of blood calcium.14
A similar case of hypoparathyroidism in the setting of ICIs has been reported. Piranavan et al described a case with similar clinical presentation and development of CaSR antibodies.10 In that case also, antibody-activation of the CaSR was demonstrated by increased intracellular IP1 levels in a CaSR-expressing cell line following treatment with the patient’s IgG. In our case, we were able to confirm that the patient had negative CaSR antibodies before their hypoparathyroidism diagnosis and to demonstrate that the patient became CaSR antibody-positive at the time of hypoparathyroidism diagnosis and remained so with long-term follow-up. Hypoparathyroidism appears to be a long-term sequela, however, so continuous follow-up is needed to assess possible recovery, although this is challenging for patients with advanced malignancies who have received ICIs late in their disease course.
While T cell-mediated immune destruction of healthy tissue is typically considered the overarching mechanism for irAEs associated with ICIs,21 22 it is increasingly appreciated that humoral immunity may play a role in their development. Some irAEs such as hypothyroidism, bullous pemphigoid, myasthenia gravis, and nephritis have been observed with similar autoantibody profiles traditionally associated with their autoimmune disease counterparts.7 23–25
Seronegative antibody-mediated irAEs have also been described with ICIs. Wilson et al reported novel IgG and IgM reacting to relevant tissue preparations in a patient with seronegative transverse myelitis secondary to pembrolizumab and another with seronegative myasthenia gravis due to ipilimumab-nivolumab administration,26 suggesting that ICIs uncover previously unrecognized autoreactive antibodies. This is corroborated by Gowen et al, demonstrating that patients who later developed severe irAEs to ICIs possessed a distinct pretreatment antibody pattern extending beyond the known autoantibodies, such as antinuclear, antismooth muscle, and antithyroid antibodies.27
Recently, Das et al showed that combined ICIs led to a decrease in circulating B cells, an increase in CD21 B cells, and an increase in plasmablasts after the first cycle of therapy.28 More importantly, the magnitude of early B cell changes correlated with both the time to onset and the grade of subsequent irAEs, and the patient cohort with early B cell changes had much higher rates of grade 3 or higher irAEs at 6 months. The use of anti-CD20 antibodies in the management of irAEs from ICIs has also been reported.29 Altogether, this evidence supports the notion of a B cell-mediated mechanism for irAEs that exists alongside the T cell-dependent model. It remains unclear whether B cell and antibody changes represent the direct impact of ICIs or the indirect effect of self-tolerance dysregulation secondary to ICIs or both.
In conclusion, hypoparathyroidism is a rare but life-threatening endocrinopathy associated with ICIs. Thus, calcium levels should be monitored during treatment. The underlying mechanism of ICI-related hypoparathyroidism likely involves the formation of CaSR-activating antibodies. Investigation into the B cell compartment may allow for a better understanding of irAEs pathophysiology and open new avenues for identifying patient subsets at high risk for irAEs and for developing novel treatment options for these.
Acknowledgments
The authors thank the patient for allowing their case to be researched.
References
Footnotes
Twitter @DrSapnaPatel
Contributors EHK: Collected and analyzed data, created figures, edited and approved the final manuscript; EMB: Collected data, edited and approved the final manuscript; HAT: Collected data, critically appraised, edited and approved the manuscript; JMS: Collected data, edited and approved the final manuscript; RD: Developed the concept, collected data, critically appraised, edited and approved the final manuscript; SPP: Collected data, edited and approved the final manuscript; TDC: Collected data, edited and approved the final manuscript; TER: Created figures, was involved with case presentation, edited and approved the final manuscript; VAT: Collected data, edited and approved the final manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval The patient’s serum samples were collected longitudinally under an Institutional Review Board approved protocol. The patient gave informed consent for their samples to be collected and their case to be researched.
Provenance and peer review Not commissioned; externally peer reviewed.