Article Text

Abstract
Immune checkpoint blockade is arguably the most effective current cancer therapy approach; however, its efficacy is limited to patients with “hot” tumors, warranting an effective approach to transform “cold” tumors. Oncolytic viruses (especially properly armed ones) have positive effects on almost every aspect of the cancer–immunity cycle and can change the cancer–immune set point of a tumor. Here, we tested whether oncolytic vaccinia virus delivering tethered interleukin 12 (IL-12) could turn a “cold” tumor into a “hot” tumor while avoiding IL-12’s systemic toxicity. Our data demonstrated that tethered IL-12 could be maintained in the tumor without treatment-induced toxic side effects. Moreover, the treatment facilitated tumor infiltration of more activated CD4+ and CD8+ T cells and less Tregs, granulocytic myeloid-derivedsuppressor cells, and exhausted CD8+ T cells, with increased interferon γ and decreased transforming growth factor β, cyclooxygenase-2, and vascular endothelial growth factor expression, leading to transformed, immunogenic tumors and improved survival. Combined with programmed cell death 1 blockade, vaccinia virus expressing tethered IL-12 cured all mice with late-stage colon cancer, suggesting immediate translatability to the clinic.
- cytokines
- immunotherapy
- oncolytic virotherapy
- oncolytic viruses
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Cancer immunotherapy is joining surgery, chemotherapy, and radiotherapy as a standard treatment modality in the war against cancer. It relies on tipping the cancer–immune set point1; that is, changing the equilibrium between factors that stimulate or inhibit anticancer immunity to restore anticancer effects. These tumor cell-intrinsic or cell-extrinsic factors within the tumor microenvironment (TME) are coordinated at each step of the cancer–immunity cycle.2 3 Immune checkpoint blockade is arguably the most important approach during the past decade to modulate the cancer–immunity cycle for cancer therapy. Such blockades include application of antibodies to target the cytotoxic T-lymphocyte antigen 4 (CTLA-4)/CD284 and programmed cell death 1 (PD-1)/PD-ligand 1 (PD-L1) axes.5 While dramatic, durable, and therapeutic responses are observed after immune checkpoint inhibitor therapy, these successes are still limited to a small percentage of patients.6 This is largely attributed to the lack or paucity of T cell infiltration in most tumors, defining the so-called non-T-cell-inflamed or “cold” tumors.7 Therefore, approaches that can convert non-T-cell-inflamed or “cold” tumors into T-cell-inflamed or “hot” tumors, mainly characterized by an abundance of tumor-infiltrating T cells, are urgently needed.
Oncolytic viruses can selectively replicate in and destroy tumor cells and stromal cells while exposing antigens for cross-presentation and elucidating danger signals, triggering innate and adaptive immunity.8 Oncolytic viruses have positive effects on almost every aspect of the cancer–immunity cycle and can be further armed with chemokines to improve tumor T cell infiltration; with cytokines to reprogram immune cells; or with ligands or antibodies to block immune checkpoints and relieve immunosuppression.9 Thus, oncolytic viruses might be perfect candidates to modulate the TME and convert “cold” tumors into “hot” tumors. Cytokines have gained researchers’ continuous interest for improving the therapeutic effects of oncolytic viruses.10 However, uncontrolled cytokine expression and secretion by a live, replicating virus may lead to toxicity and cytokine release syndrome. Recently, we generated an oncolytic vaccinia virus expressing membrane-bound interleukin 2 (IL-2) to diminish the severe toxicity associated with systemic IL-2 exposure and to modulate the TME. This virus not only reduced toxic side effects but also tipped the cancer–immune set point in the TME and treated several murine tumor models. In combination with PD-1/PD-L1 blockade, this virus cured most of the mice with a high tumor burden.11 IL-12 is another cytokine with great expectations for cancer therapy based on the therapeutic success of IL-12 treatment in a variety of murine tumor models.12 However, these expectations have been greatly diminished because systemic IL-12 in clinical trials evoked severe side effects such as leukopenia and thrombocytopenia.13–15 Several IL-12 modifications have been explored to improve its safety while preserving its activity.16–21 The goal of this study was to investigate whether and how an oncolytic vaccinia virus expressing membrane-bound IL-12 modulates the TME and further improves antitumoral effects alone or in combination with immune checkpoint blockade.
Results and discussion
To reduce the severe toxic side effects caused by the systemic application of IL-12, we used vvDD, a double viral gene-deleted (tk− and vgf−) vaccinia virus, to deliver membrane-bound IL-12 into the tumor bed. This replicating virus is tumor-selective22 and its safety has been demonstrated in clinical trials.23 24 We constructed vvDD-IL-12, vvDD-IL-12-FG, and vvDD-IL-12-RG to express secreted IL-12 or membrane-bound IL-12 after infection in tumor cells, respectively. Membrane association is realized by the glycosylphosphatidylinositol (GPI) anchor form of human CD16b. The difference between vvDD-IL-12-FG and vvDD-IL-12-RG is that between the IL-12p40 subunit and GPI anchor, the former has a flexible linker (G4S)3 and the latter has a rigid linker (A(EA3K)4AAA)25 (online supplementary figure S1). When MC38-luc, AB12-luc, and B16 cells were infected with these three IL-12-armed viruses or control virus vvDD at a multiplicity of infection (MOI) of 1, viral housekeeping gene (A34R) mRNA levels were similar among the viruses, consistent with similar in vitro infection and replication of the different viruses. In addition, the IL-12 mRNA levels were similar in IL-12-armed viruses, as expected (figure 1A). We also measured IL-12 expression at the protein level using ELISA and flow cytometry. The amount of IL-12 in the supernatant from vvDD-IL-12-infected tumor cells was significantly higher than the other constructs (figure 1B), while cell-surface IL-12 expression was significantly more prevalent in vvDD-IL-12-FG-infected or vvDD-IL-12-RG-infected cells (figure 1C; online supplementary figure S2). This demonstrates the successful realization of membrane association by GPI anchored to one subunit of IL-12. This was further confirmed by measuring the amount of IL-12 cleaved by phosphatidylinositol-specific phospholipase C (PI-PLC) from membrane-associated GPI-anchored IL-12. As expected, membrane-bound IL-12 correlated with virus MOI (figure 1D). We further demonstrated that the GPI-anchored IL-12 is functional in vitro. Con A-activated mouse splenic T cells were co-cultured with mitomycin C (MMC)-inactivated MC38 cells, which were mock-infected or infected with the viruses overnight and washed thoroughly with phosphate-buffered saline (PBS) before co-culture. Only vvDD-IL-12-FG-infected or vvDD-IL-12-RG-infected MC38 cells which had membrane-associated IL-12 could stimulate the proliferation of activated T cells. This stimulation was significantly attenuated by IL-12 antibody neutralization (figure 1E). In a previous report, a single-chain IL-12 fused with GPI anchor from human folate receptor and a flexible linker from pyruvate ferredoxin oxidoreductase was used for membrane association; however, the percentage of IL-12+ cells was moderate and a large amount of IL-12 was released into the culture supernatant after adenovirus delivery.17 This indicates that the delivery vector, GPI anchor sequence, and linker sequence might affect the membrane association of GPI-anchored proteins. In fact, we previously showed that a rigid linker between the IL-2 and GPI anchor is better for IL-2 membrane association than a flexible linker,11 whereas here a flexible linker was better for IL-12 membrane association suggesting an effect of the nature of targeted proteins on the design of a GPI-anchored protein. The cytotoxicity of IL-12-armed viruses was tested in four murine tumor cells. The results demonstrated that they had similar in vitro cytotoxicity compared with the parental virus vvDD (figure 1F).
Supplemental material
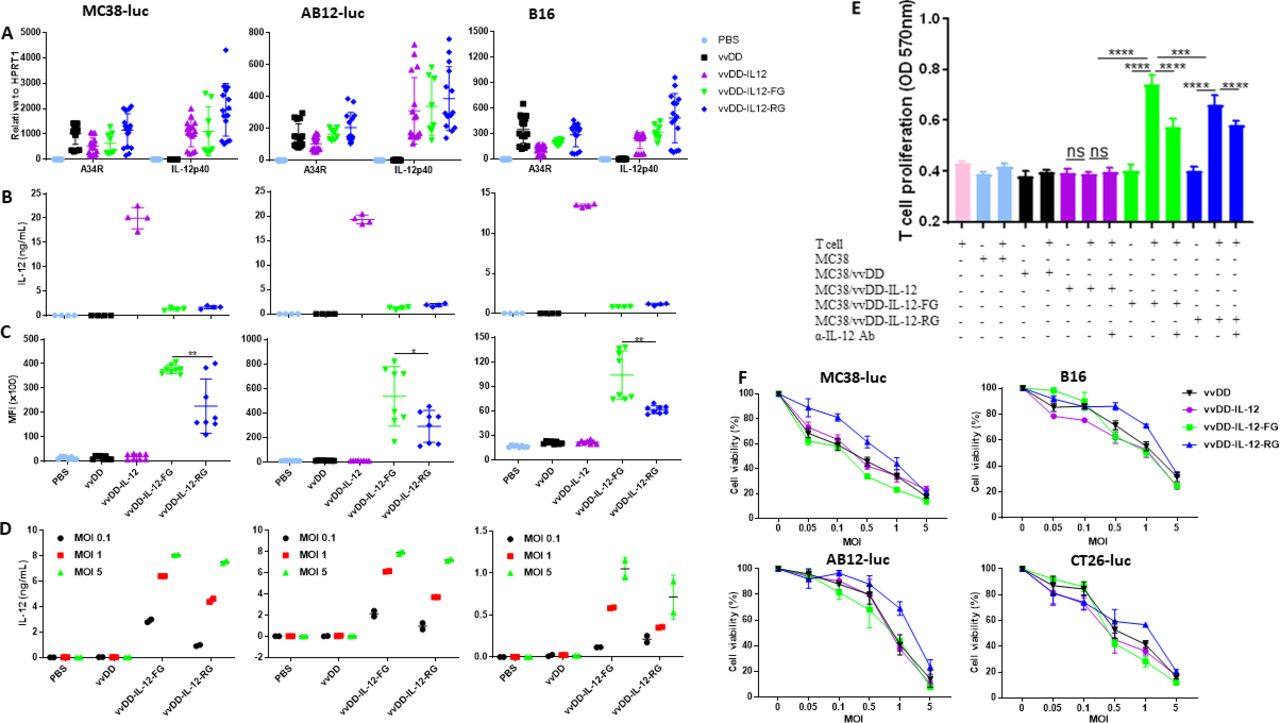
Tethered IL-12 variants show functional IL-12 membrane association and similar cytotoxicity. (A) Tumor cell MC38-luc (3×105 cells), B16 (2×105 cells), or AB12-luc (3×105 cells) were mock-infected or infected with vvDD, vvDD-IL-12, vvDD-IL-12-FG, or vvDD-IL-12-RG at an MOI of 1. The cell pellets were harvested to measure A34R or IL-12 expression at 24 hours using RT-qPCR. The culture supernatants were harvested to measure secreted IL-12 using ELISA (B) and the cell pellets were also harvested to measure membrane-bound IL-12 using flow cytometry (cell surface staining) (C) 24 hours post-infection. (D) MC38-luc (3×105 cells), B16 (2×105 cells), or AB12-luc (3×105 cells) were mock-infected or infected with vvDD, vvDD-IL-12, vvDD-IL-12-FG, or vvDD-IL-12-RG at MOIs of 0.1, 1, and 5. The cell pellets were harvested to measure membrane-bound IL-12 using ELISA after PI-PLC cleavage 24 hours post-infection. (E) Naïve B6 splenocytes were activated and stimulated with IL-12 variant-infected MC38 cells (Responder: stimulator=1:5) in the absence/presence of α-IL-12 Ab, and T cell proliferation was measured using MTT assay 48 hours after coculture. (F) MC38-luc (1×104 cells), B16 (5×103 cells), AB12-luc (5×103 cells), or CT26-luc (1×104 cells) were infected with IL-12 variants at indicated MOIs and cell viability was measured using Cell Counting Kit-8 or MTS assay 48 hours post-infection. Data represent two independent experiments. *P<0.05; **P<0.01; ***P<0.001; and ****P<0.0001. MOI, multiplicityof infection; NS, not significant; PI-PLC, phosphatidylinositol-specificphospholipase C.
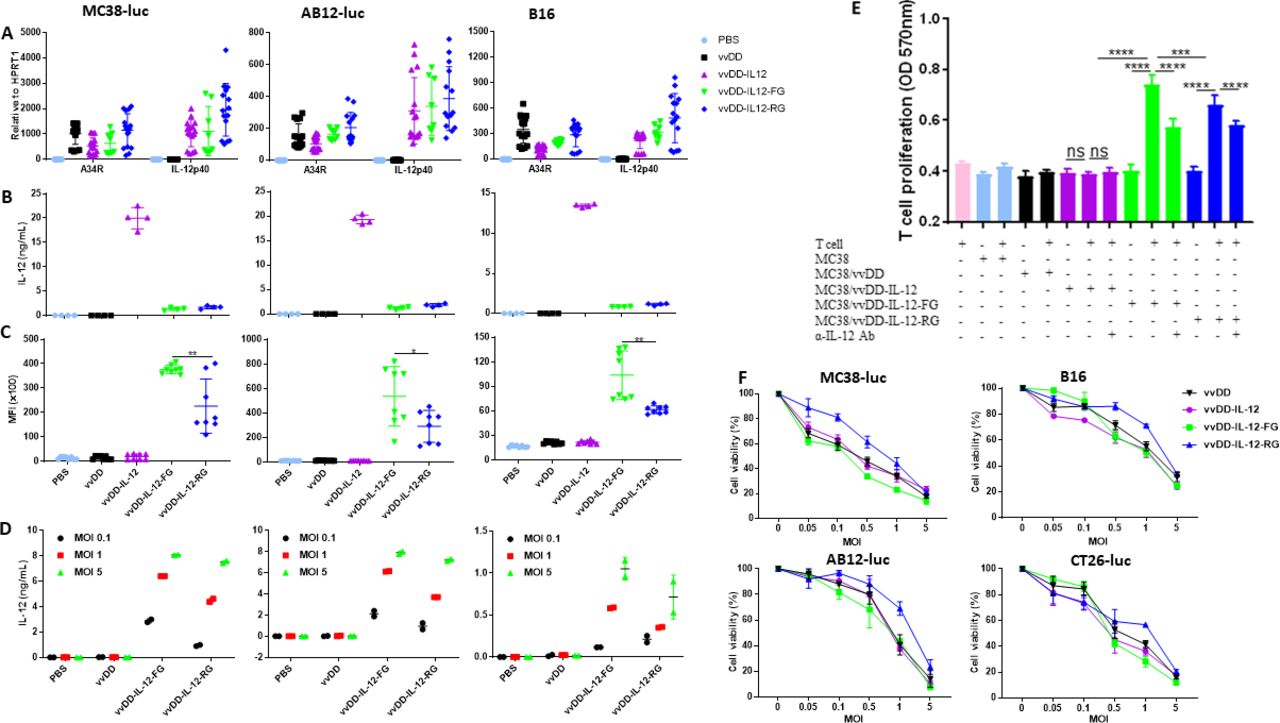
To investigate the toxicity induced by these viruses, we first measured the levels of IL-12 and interferon γ (IFN-γ) in mouse sera and tumor nodules. The IL-12 levels were significantly higher in sera from mice treated with vvDD-IL-12 compared with those treated with membrane-bound forms (online supplementary figure S3A); however, as a main mediator of IL-12-induced antitumor effects,26 IFN-γ levels in sera were similar after the different IL-12-armed virus treatments (online supplementary figure S3B). Therefore, membrane-bound IL-12 might have a similar function with a lower risk of toxicity. The levels of either IL-12 or IFN-γ in tumor nodules receiving vvDD-IL-12-FG treatment were significantly higher compared with other treatments (figure 2A,B). We next investigated the membrane association of IL-12 in vivo using flow cytometry. There were significantly more IL-12+ cells in the tumor in mice receiving vvDD-IL-12-FG treatment than in those receiving other treatments (online supplementary figure S3C). This further confirmed that vvDD-IL-12-FG could retain IL-12 at the tumor site, compared with other constructs. We also found that only vvDD-IL-12 treatment (1×109 plaque-forming units (PFU)/mouse) induced lung and kidney edema as evidenced by an increase in water content in lungs and kidneys after treatment (figure 2C,D), and liver toxicity as evidenced by an increase in aspartate transaminase (AST) and alanine transaminase (ALT) in the sera (figure 2E,F). While the mice tolerate doses up to 1×109 PFU and primates have been shown to be more permissive to WR strain vaccinia virus infection,27 we anticipate that a similar dose expressing membrane-tethered IL-12 may be potent in humans. Collectively, our data demonstrate that vvDD-IL-12-FG can effectively maintain IL-12 in the TME and is therefore safer. Hence, we selected vvDD-IL-12-FG for further investigation in the remainder of this study.

vvDD-IL-12-FG treatment produces tethered IL-12 in tumors and is safe and effective in therapeutic tumor models. B6 mice were intraperitoneally inoculated with 5×105 MC38-luc cells and treated with PBS, vvDD, vvDD-IL-12, vvDD-IL-12-FG, or vvDD-IL-12-RG at 1×109 PFU/mouse 9 days post-tumor inoculation. The mice treated above were sacrificed at day 5 after treatment. Tumor nodules were collected to measure the amount of IL-12 (A) and IFN-γ in tumor (B); the lungs and kidneys were collected to monitor pulmonary tissue edema (C and D); sera were collected to measure AST (E) and ALT (F) in sera. (G) B6 mice were intraperitoneally inoculated with 5×105 MC38-luc cells and treated with PBS, vvDD, vvDD-IL-12, or vvDD-IL-12-FG at 2×108 PFU/mouse 5 days post-tumor inoculation (n≥8). The vvDD-IL-12-FG cured mice were subcutaneously re-challenged with MC38 (H) or LLC (I). B6 mice were intraperitoneally inoculated with 5×105 MC38-luc cells and treated with PBS or indicated viruses at 2×108 PFU/mouse 9 days post-tumor inoculation (n≥23). The survival curve was shown (J). Some of these treated mice were sacrificed and splenocytes were restimulated with mitomycin C-inactivated MC38 cells to monitor IFN-γ production (K). (L) BALB/c mice were intraperitoneally inoculated with 4×105 AB12-luc cells and treated with PBS or indicated viruses at 2×108 PFU/mouse 9 days post-tumor inoculation (n≥10). A log-rank (Mantel–Cox) test was used to compare survival rates. *P<0.05; **P<0.01; ***P<0.001; and ****P<0.0001. ALT, alanine transaminase; AST, aspartate transaminase; IFN-γ, interferon γ; IL-12, interleukin 12; NS, not significant; PBS, phosphate-buffered saline; PFU, plaque-forming units.
To evaluate the antitumoral efficacy of vvDD-IL-12-FG, we intraperitoneally injected virus at the dose of 2×108 PFU/mouse to treat B6 mice bearing 5-day-old peritoneal murine colon cancer (MC38-luc). Survival results demonstrated that vvDD-IL-12-FG and vvDD-IL-12 elicited potent antitumoral effects compared with PBS or vvDD treatment (figure 2G). Impressively, vvDD-IL-12-FG treatment cured all mice that received the treatment, though there was no significant difference in survival between the different IL-12-armed virus treatments. All the mice bearing peritoneal MC38-luc cured by vvDD-IL-12-FG treatment received a subcutaneous re-challenge of either MC38 or an irrelevant tumor control, Lewis lung cancer (LLC). MC38 tumor growth was retarded in the cured mice (figure 2H), but LLC growth was not (figure 2I), compared with naïve mouse control, suggesting that a systemic tumor-specific antitumor immunity was elicited. We explored the therapeutic efficacy of vvDD-IL-12-FG using 2×108 or 1×108 PFU/mouse to treat BalB/c mice bearing 5-day-old peritoneal murine colon cancer (CT26-luc; online supplementary figure S3D) or murine mesothelioma (AB12-luc; online supplementary figure S3E) with similar results, respectively. We also evaluated the antitumoral efficacy of vvDD-IL-12-FG in 9-day tumor-bearing mouse models, which are more akin to metastatic human tumors, characterized by heavier tumor burden and increased immunosuppressive factor expression in the TME (PD-1, PD-L1, CTLA-4, transforminggrowth factor β (TGF-β), CD105, and vascular endothelial growth factor (VEGF)).11 Both IL-12-armed virus treatments significantly improved survival compared with PBS or vvDD treatment in this 9-day MC38 model (figure 2J). The splenocytes from 9-day MC38-bearing mice receiving IL-12-armed virus treatment secreted significantly more IFN-γ after restimulation with MMC-inactivated MC38, suggesting systemic antitumor immunity in the late-stage tumor model (figure 2K). Similar survival results were obtained using a 9-day AB12-bearing mouse model (figure 2L). Occasionally, a few mice that received vvDD-IL-12 treatment, but not vvDD-IL-12-FG treatment, died earlier than those that received PBS treatment (figure 2L), consistent with the IL-12-induced toxicity.
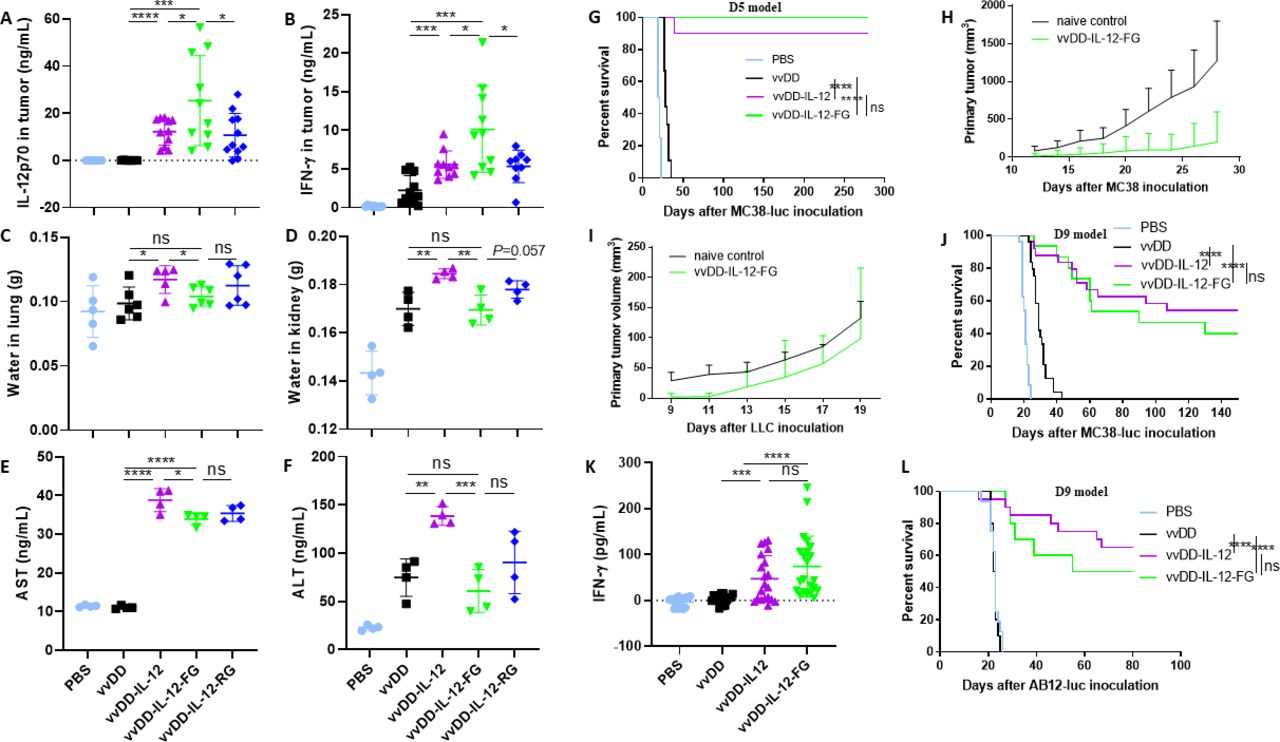
To explore the mechanism by which vvDD-IL-12-FG treatment elicits antitumoral immune activity in the profoundly immunosuppressive advanced tumor model, we investigated the immune cell profile in the TME using the late-stage tumor model. The percentages of CD4+Foxp3− and CD8+ T cells from tumors receiving IL-12-armed virus treatment were increased compared with those treated with PBS or vvDD (figure 3A,B) and the IFN-γ secretion from both CD8+ and CD4+ T cells was increased (figure 3C,D). The results also showed that the exhausted PD1+CD8+ T cells, more severely exhausted PD1+Tim-3+CD8+, PD1+TIGIT+CD8+, and PD1+LAG-3+CD8+ T cells in the tumor-infiltrating CD8+ T-cell population decreased after IL-12-armed virus treatment (figure 3E–H). We examined myeloid-derived suppressor cells (MDSCs) in tumors after virus treatment. As reported by a previous study,28 we found that granulocytic MDSCs (G-MDSCs, CD11b+Ly6G+Ly6Clow) were increased after vvDD treatment; however, they were decreased after IL-12-armed virus treatment (figure 3I). We also examined regulatory T cells (Tregs, CD4+Foxp3+) and found that the percentage of CD4+Foxp3+ T cells in tumor-infiltrating CD4+ T cells was decreased after IL-12-armed virus treatment (figure 3J), implying suppression of tumor-induced Treg proliferation by the IL-12/IFN-γ axis,29 owing to the significantly higher IL-12 and IFN-γ levels in tumors that received IL-12-armed virus treatments (figure 2A,B; online supplementary figure S4A). However, the increase of IFN-γ in the TME did not upgrade the expression of PD-1 on T cells (figure 3E; online supplementary figure S4B-C) or PD-L1 on CD45− or CD11b+ cells (online supplementary figure S4D-F), suggesting that IL-12 did not reinforce adaptive immune resistance in vvDD-related therapy.30 We next found a significant decrease in the expression of pro-cancer factors, including TGF-β, cyclooxygenase-2 (COX-2), and angiogenesis markers (VEGF and CD105) in tumors after IL-12-armed virus treatment compared with other treatments (figure 3K–M; online supplementary figure S4G). It is interesting that although the IL-12-armed virus treatment did not decrease the TGF-β expression in CD11b+ cells (online supplementary figure S4H), it did decrease the TGF-β expression in Tregs cells (figure 3N) and CD45− cells (figure 3O), which suggests a potential synergy with PD-1/PD-L1 blockade since the reduction of TGF-β signaling in stromal cells can enhance the antitumor immunity of anti-PD-1 antibody treatment.31 We further depleted IFN-γ, NK1.1+ cells, and CD4+ and CD8+ T cells by antibodies post vvDD-IL-12-FG treatment (figure 3P; online supplementary figure S5) and found that the antitumor effect elicited by vvDD-IL-12-FG treatment was IFN-γ-dependent and CD8+ T-cell-dependent, but not CD4+ T-cell-dependent or NK1.1+ cell-dependent (figure 3Q). Collectively, these data demonstrated that vvDD-IL-12-FG treatment, as well as vvDD-IL-12 treatment, tipped the cancer–immune set point in tumor-bearing mice and turned “cold” tumors to “hot” tumors, which significantly extended the survival of mice receiving IL-12-armed virus treatment. Therefore, we hypothesized that the combination with immune checkpoint blockade would enhance the antitumor effects. We previously reported that the anti-PD-1 antibody either alone or combined with vvDD could not elicit an effective antitumor response in the late-stage tumor model.11 We tested whether the combination of vvDD-IL-12-FG and anti-PD-1 antibody could improve the therapeutic effects using the late-stage tumor model. The survival results showed that in combination with anti-PD-1 antibody, vvDD-IL-12-FG cured all the advanced tumor-bearing mice (figure 3R). Thus, the safe delivery of tethered IL-12 into the TME via oncolytic virus, combined with immune checkpoint blockade, makes it possible to revisit IL-12 as a cancer immunotherapy agent.32

{kind=link}
{kind=link}
{kind=link}
IL-12-variant treatments modify the tumor microenvironment and improve the therapeutic effect with PD-1 blockade. B6 mice were intraperitoneally inoculated with 5×105 MC38-luc cells and treated with PBS, vvDD, vvDD-IL-12, or vvDD-IL-12-FG at 2×108 PFU/mouse 9 days post-tumor inoculation. Tumor-bearing mice were sacrificed 5 days post-treatment and primary tumors were collected and analyzed using flow cytometry to determine CD4+Foxp3− (A) and CD8+ T cells (B), IFN-γ+CD8+ (C), IFN-γ+CD4+ (D), exhausted CD8+ T cell (E–H), G-MDSCs (I), or regulatory T cells (CD4+Foxp3+) (J), TGF-β+Treg (N), TGF-β+CD45− (O), using RT-qPCR to determine TGF-β, COX-2, and VEGF (K–M). In a separate experiment, B6 mice were intraperitoneally inoculated with 5×105 MC38-luc cells and treated with vvDD-IL-12-FG or PBS 9 days post-tumor inoculation. Anti-CD8 Ab (250 µg/injection), α-CD4 Ab (150 µg/injection), PK136 (300 µg/injection), α-IFN-γ Ab (200 µg/injection), or α-PD-1 Ab (200 µg/injection), (n≥7) were intraperitoneally injected into mice to deplete CD8+ T cells, CD4+ T cells, or NK1.1+ cells, neutralize circulating IFN-γ, or enhance virotherapy with α-PD-1 Ab (P), and a log-rank (Mantel–Cox) test was used to compare survival rates (Q and R), respectively. *P<0.05; **P<0.01; ***P<0.001; and ****P<0.0001. COX-2,cyclooxygenase-2; IFN-γ,interferon γ; IL-12, interleukin 12; G-MDSC, granulocytic myeloid-derived suppressor cells; NS, not significant; PBS, phosphate-buffered saline; PFU, plaque-forming units; TGF-β, transforming growth factor β; VEGF, vascular endothelial growth factor.
Conclusions
Our data demonstrate that vvDD-IL-12-FG treatment can deliver IL-12 to the tumor bed and tether IL-12 on cell membranes, which is safe and effective in modifying the cancer–immune set point and producing an immune-favorable microenvironment that leads to improved efficacy as a monotherapy. Impressively, in profoundly immunosuppressive, advanced stage disease, vvDD-IL-12-FG synergizes with anti-PD-1 antibody therapy, leading to the cure of all late-stage MC38 tumors. Our data suggest that vvDD-IL-12-FG as a new form of IL-12 immunotherapy represents a treatment for cancers that have historically been unresponsive to immune checkpoint blockade-based immunotherapy.
Materials and methods
Mice and cell lines
Female C57BL/6 (B6 in short) and BalB/c mice were purchased from The Jackson Laboratory (Bar Harbor, Maine) and housed in specific pathogen-free conditions in the University of Pittsburgh Animal Facility. Mouse colon cancer MC38-luc, colon cancer CT26-luc, and mesothelioma AB12-luc cells were generated by the infection of parental tumor cells with firefly luciferase-carrying lentivirus and antibiotic blasticidin selection. Normal African green monkey kidney fibroblast CV1, mouse melanoma B16, and Lewis lung cancer cells were obtained from American Type Culture Collection. Primary T cells were grown in RPMI-1640 supplemented with 10% fetal bovine serum (FBS), 1 mM sodium pyruvate, 2 mM L-glutamine, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 50 µM β-mercaptoethanol, 1×non-essential amino acid, and 1× penicillin/streptomycin (Invitrogen, Carlsbad, California). Other cell lines were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS, 2 mM L-glutamine, and 1× penicillin/streptomycin in a 37°C, 5% CO2 incubator.
Virus generation
VSC20, a vgf gene-deleted Western Reserve strain vaccinia virus, was used as the parental virus for homologous recombination. Shuttle vectors pCMS1-IRES, pCMS1-IRES-FG, and pCMS1-IRES-RG have been described previously.11 Murine IL-12p35 and IL-12p40 cDNAs were inserted into pCMS1-IRES, pCMS1-IRES-FG, or pCMS1-IRES-RG to get shuttle plasmids pCMS1-IL-12p35-IRES-IL-12p40, pCMS1-IL-IL-12p35-IRES-IL-12p40-FG, or pCMS1-IL-12p35-IRES-IL-12p40-RG, respectively. The primers for plasmid cloning based on PCR are listed (online supplementary table S1). All these shuttle vectors were used for homologous recombination of murine IL-12 variants into the tk locus of the vaccinia viral genome. To make the new viruses vvDD-IL-12, vvDD-IL-12-FG, and vvDD-IL-12-RG, CV-1 cells were infected with VSC20 at a MOI of 0.1 and then transfected with the shuttle plasmids, resulting in virus mixture. Selection of the new recombinant viruses was based on expression of yellow fluorescent protein in CV1 cells 24 hours post-infection of relative virus mixture. vvDD-YFP, or vvDD for short, a double viral gene-deleted (tk− and vgf−) vaccinia virus carrying yfp cDNA at the tk locus, was the control virus for this work.
Supplemental material
Viral replication and IL-12 expression in vitro
MC38-luc (3×105), B16 (2×105), or AB12-luc (3×105) cells were seeded in 24-well plates overnight and infected with vvDD, vvDD-IL-12, vvDD-IL-12-FG, or vvDD-IL-12-RG at a MOI of 1 in 0.15 mL 2% FBS-containing DMEM for 2 hours. Of note, 0.35 mL 10% FBS-containing DMEM was added to cells and the mixture was cultured until harvest at 24 hours post-viral infection. The culture supernatants were harvested to measure IL-12 using ELISA (BD Bioscience, San Jose, California) and the cell pellets were applied either to measure membrane-bound IL-12 using flow cytometry or to extract RNA to measure the viral house-keeping gene A34R to monitor viral replication and transgene IL-12 expression by RT-qPCR, respectively. To further confirm the membrane association of IL-12, the tumor cells were infected with indicated viruses at MOIs of 0.1, 1, and 5 and harvested 24 hours post-infection to measure membrane-bound IL-12 using ELISA after cleavage of PI-PLC (Sigma, P5542; 8 units per mL PBS).
Cytotoxicity assay in vitro
Tumor cells were plated at 1.0×104 (except B16 cells, which were plated at 5×103) cells per well in 96-well plates and infected with indicated viruses the next day at different MOIs. Cell viability was determined at 48 hours after infection using CellTiter 96 Aqueous Nonradioactive Cell Proliferation Assay (Promega, Madison, Mississippi) or Cell Counting Kit-8 (Boster Biological Technology, Pleasanton, California).
Primary T cell proliferation assay
Splenic T cells were isolated from naïve B6 mice with a pan T cell isolation kit (Miltenyi Biotec, Auburn, CA) and cultured in T-cell medium mentioned above containing 4 µg/mL Con A and 200 U/mL IL-2 at a density of 2×106/mL for 2 days (0.1 mL per well in 96-well plates). On the same day, MC38 (3×105) cells were seeded in 24-well plates overnight and infected with indicated viruses at an MOI of 5 in 0.15 mL 2% FBS-containing DMEM for 2 hours. And then 0.35 mL 10% FBS-containing DMEM was added to cells and the mixture was cultured until harvest at 24 hours post-viral infection. The mock or virus-infected MC38 cells were harvested and treated with MMC (StressMarq Biosciences: SIH-246) (200 µg/mL) in a 37°C, 5% CO2 incubator for 2 hours and washed extensively for use. The MMC-inactivated tumor cells were resuspended in T-cell medium at a density of 1×107/mL per well and 0.1 mL was added to T-cell-containing 96-well plates if needed. For some wells, MMC-inactivated cells were pre-incubated with anti-mIL-12 antibody (5 µg/mL; BioLegend: #505304) for half an hour before coculture. The proliferation of activated T cells was measured using MTT assay 2 days after coculture.
Rodent tumor models
B6 mice were intraperitoneally inoculated with 5×105 MC38-luc cancer cells or BalB/c mice were intraperitoneally inoculated with 4×105 AB12-luc or CT26-luc cancer cells, respectively, and divided into required groups at the indicated day post-tumor cell inoculation according to tumor size based on live animal IVIS imaging, performed using a Xenogen IVIS 200 Optical In Vivo Imaging System (Caliper Life Sciences, Hopkinton, Massachusetts). Grouped mice were intraperitoneally injected with indicated viruses, antibodies, the combinations, or PBS. In some experiments, anti-CD8 Ab (clone 53–6.7; Bio X Cell; 250 µg/injection), anti-CD4 Ab (clone GK1.5, Bio X Cell; 150 µg/injection), anti-NK1.1 Ab (clone PK136, Bio X Cell; 300 µg/injection), or anti-IFN-γ Ab (clone XMG1.2, Bio X Cell; 200 µg/injection) were intraperitoneally injected into mice to deplete CD8+ T cells, CD4+ T cells, or NK1.1+ cells or neutralize circulating IFN-γ, respectively. Anti-PD-1 Ab (clone RMP1-14; Bio X Cell; 200 µg/injection) was intraperitoneally injected into mice for combination therapy. In some experiments, mice were sacrificed 5 days after treatments to harvest peritoneal tumor nodules for further analysis. In some experiments, mice were sacrificed to harvest spleens 8 days after treatments and splenocytes (4×105) were isolated and restimulated with MMC-inactivated MC38 cells (4×104 cells) in 200 µL T-cell medium mentioned above at 37 °C, 5% CO2 for 2 days. The concentration of IFN-γ in the culture supernatants was measured by ELISA. MC38-luc-tumor-bearing B6 mice treated with vvDD-IL-12-FG, which had survived for more than 250 days, were subcutaneously challenged with 5×105 MC38 or 1×106 Lewis lung cancer cells per mouse. Naïve B6 mice also received the same dose tumor challenge as a control. Subcutaneous tumor size was measured using an electric caliper in two perpendicular diameters.
Assessment of treatment-related toxicity
MC38-luc-bearing B6 mice receiving virus treatment at day 9 after tumor inoculation with the viral dose of 5×108 or 1×109 PFU/mouse were used to monitor the toxicity. Mouse blood samples were collected daily from mock-treated or virus-treated mice and kept for 2 hours at room temperature and sera were separated by centrifugation. These mice were sacrificed at day 5 after treatments to collect blood, tumor nodules, lungs, and kidneys. Tumor nodules were weighed, homogenized in lysis buffer (RayBiotech, CODE: EL-lysis) in the presence of protease inhibitor cocktail (Sigma, #l11836170001) at 10 µL lysis buffer per milligram tumor using a FastPrep Cell Disrupter (Model FP120) (Qbiogene, Carlsbad, California), and centrifuged to collect supernatant. Tumoral supernatant and sera were used to measure IL-12 and IFN-γ; sera were also used to measure ALT and AST, using commercialized kits (BioLegend and G-Biosciences, respectively), according to the vendors’ instructions. Water content was used to monitor tissue edema. Briefly, wet tissue was weighed and dehydrated overnight over 100 °C in a chemical fume hood. The weight difference between wet tissues and dry tissues was calculated.
Flow cytometry
Collected tumor tissues were weighed and incubated in RPMI 1640 medium containing 2% FBS, 1 mg/mL collagenase IV (Sigma: #C5138), 0.1 mg hyaluronidase (Sigma: #H6254), and 200U DNase I (Sigma: #D5025) at 37°C for 1–2 hours to make single cells. In vitro virus-infected cells or single cells from tumor tissues were blocked with α-CD16/32 Ab (clone 93, eBioscience: #14-0161-85; 1:1000) and then stained with antibodies against mouse CD45 (PerCP-Cy5.5 or FITC, clone: 30-F11, BioLegend: #103 132 or 103108; 1:300), CD4 (APC, clone: RM4-5, eBioscience: #17-0042-81; FITC, clone: GK1.5, BD Biosciences: #553729, 1:300), Foxp3 (PE, clone: FJK-16s, eBioscience: #12-5773-82; 1:100), CD8 (PE or APC, clone: 53–6.7, eBioscience: #12-0081-85 or 17-0081-83; 1:300), PD-1 (PerCP-Cy5.5 or Brilliant Violet 785, clone: 29F.1A12, BioLegend: #135 208 or #135225; 1:300), TIM-3 (Biotin-TIM-3, clone: RMT3-23, BioLegend: #119720; 1:300+PE SA, eBioscience: #12-4317-87; 1:1000), TIGIT (Biotin-TIGIT, clone: 1G9, BioLegend: #142113; 1:300+PE SA, eBioscience: #12-4317-87; 1:1000), LAG-3 (PE, clone: C9B7W, BioLegend: #125208; 1:300), CD11b (FITC or PE, clone: M1/7, BioLegend: #101 206 or #101208; 1:300), Ly-6G (APC, clone: 1A8, eBioscience: #17-9668-82; 1:300), Ly-6C (FITC, clone: HK1.4, BioLegend: #128006; 1:300), PD-L1 (APC, clone: 10F.9G2, BioLegend: #124312, 1:300), TGF-β (Brilliant Violet 421, clone: TW7-16B4, BioLegend: #141408, 1:100), and IL-12p40 (PE, clone: C17.8, eBioscience: #12-7123-82; 1:300). The intracellular staining kit for Foxp3, TGF-β, and IFN-γ staining was purchased from BioLegend. Samples were collected on a BD Accuri C6 cytometer or BD LSR Fortessa I, and data were analyzed using BD Accuri C6 cytometer software or FlowJo software (Tree Star Inc., Ashland, Oregon).
RT-qPCR
Total RNA was extracted from viral-infected cells or tumor tissues using the RNeasy Kit (Qiagen, Valencia, California). One microgram of RNA was used for cDNA synthesis, and 25 to 50 ng of subsequent cDNA was used to conduct mRNA expression TaqMan analysis on the StepOnePlus system (Life Technologies, Grand Island, New York). All primers for the analysis were purchased from Thermo Fisher Scientific (Waltham, Massachusetts). Gene expression was normalized to the housekeeping gene HPRT1 and expressed as fold increase (2−ΔCT), where ΔCT=CT(target gene) − CT (HPRT1).
Statistics
Statistical analyses were performed using unpaired Student’s t-test (GraphPad Prism V.7). Data are means±SD. Animal survival is presented using Kaplan–Meier survival curves and was statistically analyzed using a log-rank test (GraphPad Prism V.7). Values of p<0.05 were considered statistically significant, and all p values were two-sided. In the figures, standard symbols are used: *p<0.05; **p<0.01; ***p<0.001; and ****p<0.0001.
Acknowledgments
The authors thank Christine Burr, BA (Scientific Writer, Department of Anesthesiology and Perioperative Medicine/Department of Surgery, University of Pittsburgh School of Medicine) for her editorial assistance with the manuscript.
References
Footnotes
ZL and DLB are joint senior authors.
YG and HW contributed equally.
Contributors YG, HW, JR, WL, LC, HC, JY, ED, CM, and ZL conducted the experiments and collected the data; ZL, ZSG, and DLB conceived the project; ZL and DLB designed the experiments, interpreted the data, and wrote the manuscript. SJ provided advice and made suggestions for the manuscript.
Funding This project was supported by UPMC funds (Retention funds and ITTC funds) and used UPMC Hillman Cancer Center shared facilities supported in part by NIH grant award P30CA047904.
Competing interests The University of Pittsburgh has filed a patent application directed to the subject matter of this manuscript.
Patient consent for publication Not required.
Ethics approval The animal studies have been approved by the Institutional Biosafety Office, and Institutional Animal Care and Use Committee at the University.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement The data used or analyzed during this study are included in this article and available from the corresponding authors upon reasonable request.