Article Text

Abstract
Background Cemiplimab, a high-affinity, potent human immunoglobulin G4 monoclonal antibody to programmed cell death-1 demonstrated antitumor activity in a Phase 1 advanced cutaneous squamous cell carcinoma (CSCC) expansion cohort (NCT02383212) and the pivotal Phase 2 study (NCT02760498). Here we report the primary analysis of fixed dose cemiplimab 350 mg intravenously every 3 weeks (Q3W) (Group 3) and provide a longer-term update after the primary analysis of weight-based cemiplimab 3 mg/kg intravenously every 2 weeks (Q2W) (Group 1) among metastatic CSCC (mCSCC) patients in the pivotal study (NCT02760498).
Methods The primary objective for each group was objective response rate (ORR) per independent central review (ICR). Secondary endpoints included ORR by investigator review (INV), duration of response (DOR) per ICR and INV, and safety and tolerability.
Results For Group 3 (n=56) and Group 1 (n=59), median follow-up was 8.1 (range, 0.6 to 14.1) and 16.5 (range, 1.1 to 26.6) months, respectively. ORR per ICR was 41.1% (95% CI, 28.1% to 55.0%) in Group 3, 49.2% (95% CI, 35.9% to 62.5%) in Group 1, and 45.2% (95% CI, 35.9% to 54.8%) in both groups combined. Per ICR, Kaplan–Meier estimate for DOR at 8 months was 95.0% (95% CI, 69.5% to 99. 3%) in responding patients in Group 3, and at 12 months was 88.9% (95% CI, 69.3% to 96.3%) in responding patients in Group 1. Per INV, ORR was 51.8% (95% CI, 38.0% to 65.3%) in Group 3, 49.2% (95% CI, 35.9% to 62.5%) in Group 1, and 50.4% (95% CI, 41.0% to 59.9%) in both groups combined. Overall, the most common adverse events regardless of attribution were fatigue (27.0%) and diarrhea (23.5%).
Conclusion In patients with mCSCC, cemiplimab 350 mg intravenously Q3W produced substantial antitumor activity with durable response and an acceptable safety profile. Follow-up data of cemiplimab 3 mg/kg intravenously Q2W demonstrate ongoing durability of responses.
Trial registration number Clinicaltrials.gov, NCT02760498. Registered May 3, 2016, https://clinicaltrials.gov/ct2/show/NCT02760498
- immunotherapy
- programmed cell death 1 receptor
- tumor biomarkers
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Cutaneous squamous cell carcinoma (CSCC) is the second most common skin cancer, and its incidence is increasing.1 2 Chronic sun exposure, advanced age, and immunosuppression are risk factors for CSCC.3 4 Most CSCC cases are diagnosed early,5 6 and patients with local disease are generally cured by surgery.4 7 Conversely, the prognosis is poor for patients with either locally advanced CSCC (laCSCC) not amenable to curative surgery or curative radiation or metastatic CSCC (mCSCC), collectively referred to as advanced CSCC, treated with cytotoxic chemotherapy or epidermal growth factor receptor inhibitors.8–10
Due to chronic skin damage from ultraviolet light, most CSCCs are hypermutated.11 12 Patients with high tumor mutational burden (TMB) solid tumors are more likely to derive clinical benefit from inhibition of immune checkpoints, such as programmed cell death (PD)-1.13 14 Intact immune surveillance is critical in CSCC prevention in immunocompetent individuals, as evidenced by the strong link between immunosuppression and increased CSCC risk.15 16 These considerations provided rationale for the study of PD-1 inhibition in advanced CSCC.
Cemiplimab is a high-affinity, highly potent human immunoglobulin G4 monoclonal antibody to the PD-1 receptor.17 Cemiplimab demonstrated substantial antitumor activity in a Phase 1 advanced CSCC expansion cohorts (NCT02383212) and produced an objective response rate (ORR) per independent central review (ICR) of 47.5% in the Phase 2 (NCT02760498) primary analysis of the weight-based dosing cohort for patients with mCSCC (Group 1) with emerging evidence of durable responses.18 Supported by these findings, cemiplimab-rwlc became the first therapy approved by the US Food and Drug Administration for the treatment of advanced CSCC.19 Subsequently, the European Commission granted conditional marketing authorization for cemiplimab for the treatment of advanced CSCC.20 The approved regimen is cemiplimab 350 mg every 3 weeks (Q3W) intravenously.
This article presents the primary analysis of the Phase 2 study of the approved fixed dose regimen (cemiplimab 350 mg intravenously Q3W; Group 3) in patients with mCSCC. At the time of the Group 3 primary analysis, an additional data cut with longer follow-up was performed in Group 1 (cemiplimab 3 mg/kg intravenously every 2 weeks (Q2W)) and reported here; results of the primary analysis of Group 1 have been previously reported.18 Exploratory TMB analyzes are also presented.
Methods
Patients
This is an open-label, non-randomized, multicenter, international, Phase 2 study of patients with distant or nodal mCSCC (Groups 1 and 3) (see online supplementary file 1, S1 for study sites and principal investigators). Enrollment for Group 3 opened after full enrollment of Group 1. The time point for the primary analysis of data from patients in Group 3 was reached.
Supplemental material
Eligible patients were aged ≥18 years with histologically confirmed diagnosis of invasive CSCC, an Eastern Cooperative Oncology Group performance status score of 0 or 1, adequate organ function, and at least one measurable lesion per Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST 1.1).21
Key exclusion criteria included ongoing or recent (within 5 years) autoimmune disease requiring systemic immunosuppression, prior treatment with an agent blocking the PD-1/PD ligand-1 (PD-L1) pathway, a history of solid organ transplantation, concurrent cancer (unless indolent or non-life-threatening), or hematologic cancer.
Study design
Patients were administered cemiplimab 350 mg intravenously over 30 min Q3W for up to 54 weeks, with the option to extend treatment to 96 weeks (Group 3) or cemiplimab 3 mg/kg intravenously over 30 min Q2W for up to 96 weeks (Group 1). The primary endpoint was ORR per ICR independently in each group. Tumor assessments were performed at the end of each treatment cycle (every 9 weeks for Group 3 and every 8 weeks for Group 1) (see online supplementary file 1, S2 for further details).
Secondary endpoints included ORR per investigator review (INV), duration of response (DOR) by ICR and INV, progression-free survival (PFS) by ICR and INV, overall survival (OS), complete response (CR) rate per ICR, adverse events (AEs), and quality of life. Durable disease control rate, defined as the proportion of patients with response or stable disease for at least 105 days was also examined. Safety assessments included treatment-emergent AEs (TEAEs), laboratory tests, vital signs, and physical examinations. The severity of TEAEs was graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.03).
Archived tumor samples from prior CSCC biopsies or surgeries were provided during the screening period. TMB was estimated in DNA samples extracted from formalin-fixed paraffin-embedded tumor biopsies. TMB was calculated as the total number of somatic single nucleotide variants and indels in the coding regions of targeted genes per megabase of analyzed genomic sequence (see online supplmentary file 1, S2 for further details).
The study also includes Group 4 and pilot Group 5, which explore alternative doses and/or schedules of cemiplimab, and Group 6 which is designed to confirm the results with 350 mg intravenously Q3W observed in Group 3. Groups 4 to 6 have not reached primary analysis and are not included in this report.
Statistical analysis
The primary endpoint analyses for each group were statistically independent. Fifty patients in each group were needed to provide at least 85% power to reject a null hypothesis of an ORR of 15% at a two-sided significance level of no more than 5%, if the true ORR was 34%. The primary efficacy analyses were undertaken using 95% binomial exact confidence intervals (CIs), which were generated using the Clopper-Pearson method.22 The secondary efficacy analyses of DOR, PFS, and OS are summarized by their medians and 95% CIs, which were generated by the Kaplan–Meier method. CR rates are summarized descriptively. The full analysis set, which included all patients who passed screening and were eligible for study participation, was used for the analysis of all efficacy endpoints. The safety analysis set included all enrolled patients who received any study drug. The primary efficacy analysis of both groups was performed 6 months after the first dose of cemiplimab had been administered to the last patient enrolled; the results from the primary efficacy analysis of Group 1 (cut-off date of October 27, 2017) have been previously published.18 In the present analysis, data are reported separately for Group 1 and Group 3, as well as for both groups combined. The cut-off date for the primary analysis of Group 3 and follow-up analysis of Group 1 was September 20, 2018.
Results
Patients
Results are presented for Group 3 primary analysis, and for Group 1 with approximately 11 months additional follow-up after the primary analysis. In total, 56 patients were enrolled and treated with cemiplimab 350 mg Q3W (Group 3) from July 2017 through March 2018, and 59 patients were enrolled and treated with cemiplimab 3 mg/kg Q2W (Group 1) from March 2016 through January 2017. Baseline characteristics were similar in regard to median age, gender, and extent of disease (nodal vs distant) (table 1). The median duration of follow-up was 8.1 (range, 0.6 to 14.1) months for Group 3 and 16.5 (range, 1.1 to 26.6) months for Group 1. Patients in Group 3 received a median of 11.5 (range, 1 to 20) doses of cemiplimab and were exposed to treatment for a median of 34.3 (range, 2.6 to 60.4) weeks. Patients in Group 1 received a median of 31.0 (range, 1 to 48) doses of cemiplimab and were exposed to treatment for a median of 65.0 (range, 2.0 to 96.1) weeks. Patient disposition is summarized in online supplementary file 2, figure S1.
Supplemental material
Baseline demographics and disease characteristics
Clinical activity
For the Group 3 primary analysis, ORR per ICR was 41.1% (95% CI, 28.1% to 55.0%). For the Group 1 update, ORR per ICR was 49.2% (95% CI, 35.9% to 62.5%). The combined ORR for both groups was 45.2% (95% CI, 35.9% to 54.8%), including 39 partial responses (PRs) and 13 CRs per ICR (table 2). The ORR per INV was 51.8% (95% CI, 38.0% to 65.3%) in Group 3 and 49.2% (95% CI, 35.9% to 62.5%) in Group 1 (see online supplementary file 3, table S1).
Supplemental material
Tumor response per independent central review
Per ICR, most evaluable patients in both Group 3 and Group 1 had a decrease from baseline in the target lesion diameters (figure 1), and most responses were evident at the first response assessment in both groups (see online supplementary file 2, figure S2). Durable responses are most evident in Group 1 due to longer follow-up and are emerging for Group 3 (see online supplementary file 2, figure S2). The median DOR had not been reached in either group at data cut-off. Per ICR, the Kaplan–Meier estimate for DOR at 8 months was 95.0% (95% CI, 69.5% to 99.3%) in responding patients in Group 3, and at 12 months was 88.9% (95% CI, 69.3% to 96.3%) in responding patients in Group 1 (table 2).

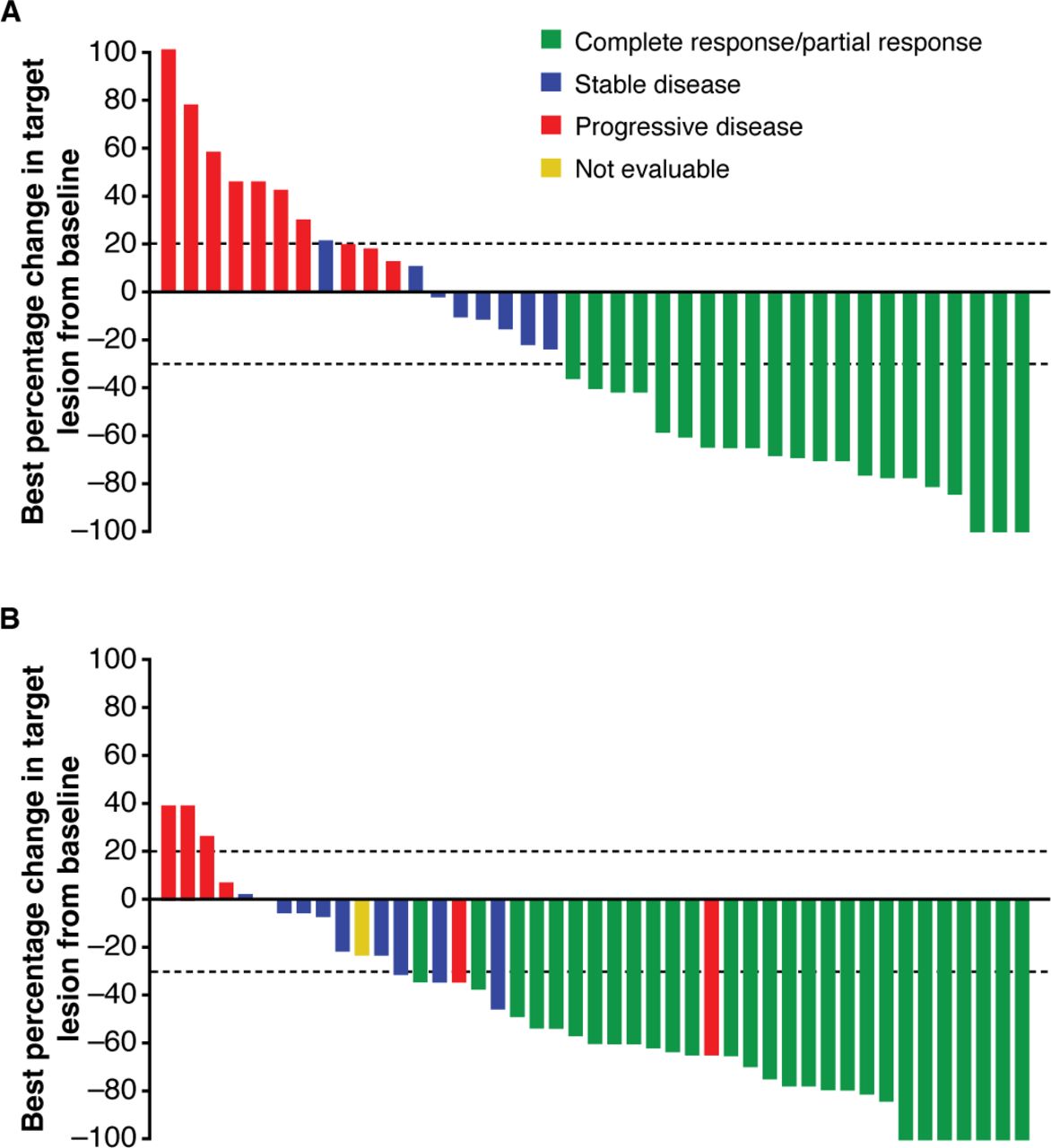{kind=link}
Best tumor response per RECIST 1.1 by independent central review for (a) Group 3 and (b) Group 1. This figure shows best percent change in the sum of tumor diameters for patients who had at least one post-baseline radiologic assessment (39 of 56 patients in Group 3 and 45 of 59 patients in Group 1). Lesion measurements after progression were excluded and patients who did not have at least one post-treatment radiologic assessment of target lesion(s) are not shown. The dashed lines indicate RECIST 1.1 criteria for partial response (≥30% decrease in sum of diameters) or progression (≥20% increase in sum of diameters) of target lesions. Patients with new lesions or unequivocal progression of non-target lesions are considered as progressive disease (red bars) regardless of target lesion response. Patients with a single assessment with ≥30% reduction of target lesion(s) are considered stable disease (blue bars) if there is not confirmatory assessment to establish partial response. One patient in Group 1 was not evaluable (NE) (yellow bar); this patient had radiologic and photographic data and was, therefore, reviewed by Independent Composite Review Committee and assessed as NE. Patients who did not have at least one evaluable post-baseline radiology assessment are not included in the figure but are included in the overall response analysis (table 2) per intention-to-treat. Increase in sum of target lesion diameters greater than 100% is reported as 100%. RECIST 1.1, Response Evaluation Criteria in Solid Tumors version 1.1.
The median time to response per ICR was 2.1 (range, 2.0 to 8.3) months for Group 3, 1.9 (range, 1.7 to 9.1) months for Group 1, and 2.1 (range, 1.7 to 9.1) months for both groups combined. The disease control rate per ICR was 64.3% (95% CI, 50.4% to 76.6%) in Group 3, 71.2% (95% CI, 57.9% to 82.2%) in Group 1, and 67.8% (95% CI, 58.5% to 76.2%) in both groups combined. The durable disease control rate per ICR was 57.1% (95% CI, 43.2% to 70.3%) in Group 3, 61.0% (95% CI, 47.4% to 73.5%) in Group 1, and 59.1% (95% CI, 49.6% to 68.2%) in both groups combined (table 2).
Median PFS had not been reached at the time of data cut-off. The median Kaplan–Meier estimated PFS per ICR was 10.4 (95% CI, 3.6 to not evaluable (NE)) months based on 44.6% event rate for Group 3, 18.4 (95% CI, 6.8 to NE) months based on 47.5% event rate for Group 1, and 18.4 (95% CI, 7.3 to NE) months based on 46.1% event rate for both groups combined. The Kaplan–Meier estimation of PFS at 12 months per ICR was 47.4% (95% CI, 29.6% to 63.3%) for Group 3, 52.9% (95% CI, 39.0% to 65.0%) for Group 1, and 51.2% (95% CI, 41.0% to 60.6%) for both groups combined (see online supplementary file 2, figure S3A).
Median OS had not been reached in either group at data cut-off. The Kaplan–Meier estimation of OS at 12 months was 76.1% (95% CI, 56.9% to 87.6%) for Group 3, 81.3% (95% CI, 68.7% to 89.2%) for Group 1, and 80.7% (95% CI, 71.9% to 87.1%) for both groups combined (see online supplementary file 2, figure S3B).
For Group 3, ORR per ICR was explored for patients with high body weight (>120 kg). Among four patients who weighed >120 kg at baseline, two experienced PR per ICR (see online supplementary file 3, table S2).
Safety
In both groups combined, 113 (98.3%) patients experienced at least one TEAE of any grade regardless of attribution, including 96.4% for Group 3% and 100.0% for Group 1 (table 3). The most common TEAEs in Group 3, Group 1, and both groups combined, were fatigue (28.6%, 25.4%, and 27.0%, respectively), diarrhea (17.9%, 28.8%, and 23.5%, respectively), and nausea (17.9%, 23.7%, and 20.9%, respectively). Grade ≥3 TEAEs regardless of attribution were reported in 45.2% of patients in both groups combined, with the most common in Group 3, Group 1, and both groups combined, being anemia (8.9%, 3.4%, and 6.1%, respectively), fatigue (5.4%, 1.7%, and 3.5%, respectively) and pneumonitis (0.0%, 5.1%, and 2.6%) (table 3). Treatment-related AEs (TRAEs) were reported in 71.3% (82/115) of patients, most commonly fatigue (13.0% (15/115)) (table 3; online supplementary file 3, table S2). TRAEs reported in ≥5% of patients in either treatment group are shown in online supplementary file 3, table S3. Immune-related AEs per INV are presented in online supplementary file 3, table S4.
Safety summary
Three (5.4%) patients in Group 3 discontinued treatment due to an AE (Grade 3 soft tissue necrosis, n=1; Grade 2 lethargy, n=1; and Grade 3 psoriasis, n=1). In Group 1, six (10.2%) patients discontinued treatment due to an AE; four of these were previously reported,18 and two occurred after the data cut for the Group 1 primary analysis. All AEs leading to treatment discontinuations were considered treatment-related, except for the patient in Group 3 with soft tissue necrosis on the head. One (1.8%) patient in Group 3 died due to arterial hemorrhage from their right lower extremity tumor which measured 12.5 cm in longest diameter at baseline. This death was not considered related to study treatment. No new AEs resulting in death were reported in the updated analysis of Group 1.
Biomarker analysis
Overall, 79 patients had pre-treatment tumor samples available for the analysis of associations between TMB and clinical activity of cemiplimab. Median TMBs were 61.4 and 53.2 mutations per megabase among responding patients in Group 3 and Group 1 and were 13.7 and 19.4 mutations per megabase among non-responding patients in Group 3 and Group 1, respectively (see online supplementary file 2, figure S4). Similar separations in median TMB were observed among patients who achieved durable disease control and those who did not (see online supplementary file 2, figure S5).
Discussion
The approved regimen of cemiplimab 350 mg intravenously Q3W (Group 3) is highly active therapy for mCSCC (ORR per ICR, 41.1%; 95% CI 28.1% to 55.0%). This result exceeds the prespecified statistical threshold for clinically meaningful ORR per ICR at time of primary analysis. The ORR per ICR for Group 1 at time of primary analysis was 47.5%,18 and has increased to 49.2% with longer follow-up in this report. In the combined analysis of all mCSCC patients (Group 3 and Group 1) in this report, ORR per ICR is 45.2%.
For the secondary endpoint of ORR per INV, numerical differences are smaller between the groups (51.8% in Group 3, 49.2% in Group 1). Discrepancies between ICR and INV are well described in oncology studies,23 24 and are more apparent in Group 3 than in Group 1 in this study. Despite these differences, the 95% CIs for ORR per ICR overlap broadly for the Group 3 primary analysis (28.1% to 55.0%) and the Group 1 update (35.9% to 62.5%). The characteristics of responses per ICR were similar for both groups in regard to the median time to response (2.1 and 1.9 months in Groups 3 and 1, respectively) and durability (estimated 8-month DOR of 95.0% and 12-month DOR of 88.9% in Groups 3 and 1, respectively). Numerical differences in point estimate of ORR per ICR between Group 3 and Group 1 are not attributed to differences between the two regimens, because the fixed-based and weight-based regimens display comparable pharmacokinetics,19 and exposure to cemiplimab in both groups was the same.25 The fixed-dose Group 3 regimen offers advantages such as a more convenient schedule for patients and less risk of dosing error or medication wastage. Cemiplimab 350 mg Q3W intravenously is the commercially-approved dose. Additional clinical data regarding the fixed-dose regimen among patients with advanced CSCC are being obtained in a confirmatory cohort (Group 6) in this study.
The 11-month update of Group 1 illustrates hallmarks of the potential clinical benefits of PD-1 blockade that become apparent with longer follow-up. Almost all the Group 1 responses illustrated in the primary analysis report18 are still ongoing at this update (see online supplementary file 2, figure S2B). Additionally, the quality of responses improved over time, with 10 CRs per ICR at the 11-month update versus only four at the time of the primary analysis.18 Because the responses are sustained, the median DOR has yet to be reached. Therefore, the protocol has been amended to allow another year of active follow-up with centrally reviewed imaging after completion of planned therapy. Patients in both groups continued in active follow-up after the data cut for this article, and long-term data continues to be collected so that the tail of the curve regarding survival and response duration can be more fully characterized in these groups.
Most TRAEs in both groups were Grades 1 to 2 and the discontinuation rate was low, regardless of attribution. The TEAE profile here is comparable to that of PD-1 checkpoint inhibitors pembrolizumab and nivolumab in patients with other cancer types such as head and neck squamous cell cancer.26 27 A larger proportion of patients in the present analyses was aged at least 75 years (36.5% vs 6.3% in pembrolizumab and 5.0% in nivolumab).26 27 The results presented here indicate that the safety profile with the anti–PD-1 class is not markedly different between younger and older patients. Overall, the safety profile for cemiplimab in this article continues to be consistent with that which has been reported for other anti–PD-1/PD-L1 inhibitors.28
As data accumulate regarding the treatment of advanced CSCC patients with cemiplimab, the distinctions from results obtained in earlier studies of conventional therapies become clearer. This includes studies of cytotoxic chemotherapy and epidermal growth factor receptor-targeted therapy.6 7 Durable responses to these agents are not common, and the TEAEs associated with these therapies can be difficult to manage among older patients with advanced CSCC. Although there are currently no clinical trials directly comparing cemiplimab with these therapies, and the study design of the conventional therapies are different from that of cemiplimab, the differences in efficacy, durability of response, and safety position highlight cemiplimab as the standard of care for patients with advanced CSCC. In recent congress proceedings, pembrolizumab demonstrated ORR of 34.3% (n=105; median follow-up of 9.5 months) in patients with recurrent/mCSCC.29 These data further support the clinical activity of PD-1 checkpoint inhibitors in advanced CSCC.
Higher median TMB was observed among responding patients than among non-responding patients in both Groups 3 and 1. Similar results were observed in exploratory analyses of laCSCC patients in Group 2.30 However, high TMB among some non-responders and low TMB among some responders preclude this assay from use as a patient selection tool. Future prospective data sets that integrate baseline TMB with other candidate biomarkers or clinical prognostic factors may better define features associated with clinical benefit among patients with advanced CSCC treated with cemiplimab.
In conclusion, cemiplimab 350 mg intravenously Q3W produced substantial antitumor activity. Durable responses have been observed in both weight-based and fixed-dosing groups. The safety profile was similar in both groups. Long-term follow-up of these patients is ongoing.
Acknowledgments
The authors would like to thank the patients, their families, all other study investigators, and all investigational site members involved in this study. Zhen Chen (Regeneron Pharmaceuticals, Inc) provided additional statistical analyses.
References
Footnotes
Contributors Responsibility for all opinions, conclusions, and data interpretation lies with the authors.
Funding This work was supported by Regeneron Pharmaceuticals, Inc. and Sanofi. Medical writing support, provided by Kate Carolan, PhD, of Prime (Knutsford, UK) according to Good Publication Practice guidelines (link), was funded by Regeneron Pharmaceuticals, Inc. and Sanofi.
Competing interests DR: institutional research grant and funding from Regeneron Pharmaceuticals, Inc., Sanofi, Roche, Merck Sharp & Dohme, Bristol-Myers Squibb, and GlaxoSmithKline; uncompensated scientific committee and advisory board from Merck Sharp & Dohme, Regeneron Pharmaceuticals, Inc., Sanofi, GlaxoSmithKline, and Bristol-Myers Squibb; travel and accommodation from Merck Sharp & Dohme and GlaxoSmithKline. MRM: honoraria and travel expenses from Regeneron Pharmaceuticals, Inc., Sanofi, Novartis, Genentech, Eli Lilly, and Sun Pharma; and institutional research funding from Regeneron Pharmaceuticals, Inc., Novartis, Genentech, and Eli Lilly. AML: uncompensated advisory board from Merck Sharp & Dohme and Bristol-Myers Squibb with travel and accommodation expenses. CDS: steering committee member for Castle Biosciences; a steering committee member and consultant for Regeneron Pharmaceuticals, Inc.; a consultant for Sanofi; has received research funding from Castle Biosciences, Regeneron Pharmaceuticals, Inc., Novartis, Genentech, and Merck, and is a chair for the National Comprehensive Cancer Network. NIK: grants from Regeneron Pharmaceuticals, Inc.; grants and advisory board fees from Bristol-Myers Squibb and HUYA Bioscience International; advisory board fees from EMD Serono, Regeneron Pharmaceuticals, Inc., Genentech, AstraZeneca (data safety monitoring committee), Merck, Array BioPharma, and Immunocore; grants from Merck, Novartis, GlaxoSmithKline, Celgene, and Amgen; honorarium from Sanofi; and common stock ownership of Bellicum Pharmaceuticals, Mazor Robotics, Amarin, and TransEnterix. BGMH: consulting or advisory roles at Merck Sharp & Dohme, Bristol-Myers Squibb, AstraZeneca, Pfizer, Roche, Eisai, Merck, and institutional research funding from Amgen. DS: institutional patients’ fees from Regeneron Pharmaceuticals, Inc.; advisory board honorarium fees from Amgen and Leo Pharma; speaker fee from Boehringer Ingelheim; advisory board, speaker honorarium and patients’ fees from Roche, Novartis, Bristol-Myers Squibb, and Merck-EMD; advisory board and speaker honorarium fees from Incyte and Pierre Fabre; advisory board honorarium and patients’ fees from Merck Sharp & Dohme, steering committee honorarium fees from 4SC, advisory board fees from AstraZeneca, Pfizer, and Array; and advisory board and patients’ fees from Philiogen. LAD: advisory role at Regeneron Pharmaceuticals, Inc., and research funding from Eisai, Pfizer, Regeneron Pharmaceuticals, Inc. LH-A: performed consulting and advisory roles at Massive Bio; speakers’ bureau roles at Sanofi and Regeneron Pharmaceuticals, Inc., and received travel, accommodations, and expenses from Regeneron Pharmaceuticals, Inc., Sanofi, and Bristol-Myers Squibb, and research funding from Bristol-Myers Squibb, Regeneron Pharmaceuticals, Inc., Immunocore, Merck Sharp & Dohme, Polynoma, Corvus Pharmaceuticals, Roche, Merck Serono, Amgen, MedImmune, and Takeda. ALSC: consulting and advisory roles at Regeneron Pharmaceuticals, Inc., Merck; research funding from Regeneron Pharmaceuticals, Inc., Novartis, Galderma, and Merck. BM: speaker’s honoraria, travel, accommodations and expenses from Regeneron Pharmaceuticals, Inc. and Sanofi. AH: institutional grants, speaker’s honoraria, and consultancy fees from Amgen, Bristol-Myers Squibb, Merck Sharp & Dohme/Merck, Pierre Fabre, Provectus, Roche, and Novartis; institutional grants and consultancy fees from Merck Serono, Philogen, and Regeneron Pharmaceuticals, Inc.; and consultancy fees from OncoSec. CU: honoraria, consulting, or advisory roles, speaker’s bureau role, research funding and travel, accommodation, and expenses from Novartis, Sanofi, Galderma, and Almirall. TE: consulting or advisory roles at Sanofi Genzyme, Bristol-Myers Squibb, Roche, Novartis and Merck Sharp & Dohme; speakers’ bureau role at Roche and Merck Sharp & Dohme and research funding from Novartis and Bristol-Myers Squibb. BS: consulting or advisory roles at Merck Sharp & Dohme and Merck KGaA Australia. ACP: honoraria and consulting or advisory roles at Bristol-Myers Squibb, Merck, Regeneron Pharmaceuticals, Inc., Array, Novartis, Seattle Genetics, Amgen; research funding from Bristol-Myers Squibb, Merck, Regeneron Pharmaceuticals, Inc., Celldex, and Forance and travel, accommodation, expenses from Regeneron Pharmaceuticals, Inc., Array, and Seattle Genetics. JLG: research institution support for the study from and advisory board for Regeneron Pharmaceuticals, Inc. RG: honoraria from Almirall Hermal GmbH, Amgen, AstraZeneca, Bristol-Myers Squibb, Merck Serono, Merck Sharp & Dohme, Novartis, Pierre Fabre, Regeneron Pharmaceuticals, Inc., Roche/Genentech, Sanofi, and Sun Pharma; consulting or advisory role for 4SC, Almirall Hermal GmbH, Amgen, Bristol-Myers Squibb, Incyte, LEO Pharma, Merck Serono, Merck Sharp & Dohme, Novartis, Pierre Fabre, Roche/Genentech, Sun Pharma, and Takeda; research funding from Amgen, Johnson & Johnson, Novartis, and Pfizer; travel, accommodations, expenses from Bristol-Myers Squibb, Merck Sorono, Pierre Fabre, and Roche. MA: consulting or advisory roles for Pulse Biosciences and Revance Therapeutics. EO, MM, VJ, ES, JB, SL, IL, MGF: employees and shareholders of Regeneron Pharmaceuticals, Inc. AG: personal fees and non-financial support (advisory board and travel support) from Bristol-Myers Squibb and Sun Pharma; personal fees (advisory board) from Merck KGaA, Eisai, and Pfizer; non-financial (travel) support from Astellas; and clinical trial unit support from PPD Australia.
Patient consent for publication Not required.
Ethics approval This study was conducted in accordance with the principles of the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice guidelines. The study protocol and all amendments were approved by the institutional review board/ethics committee at each participating study site. All patients provided written informed consent.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information. Qualified researchers may request access to study documents (including the clinical study report, study protocol with any amendments, blank case report form, statistical analysis plan) that support the methods and findings reported in this manuscript. Individual anonymized participant data will be considered for sharing once the indication has been approved by a regulatory body, if there is legal authority to share the data and there is not a reasonable likelihood of participant re-identification. Submit requests to https://vivli.org/.