Article Text

Abstract
Background Immune checkpoint inhibitors (ICIs) to date have demonstrated limited activity in advanced ovarian cancer (OC). Folate receptor alpha (FRα) is overexpressed in the majority of OCs and presents an attractive target for a combination immunotherapy to potentially overcome resistance to ICI in OCs. The current study sought to examine clinical and immunologic responses to TPIV200, a multiepitope FRα vaccine administered with programmed death ligand 1 (PD-L1) inhibitor durvalumab in patients with advanced platinum-resistant OC.
Methods Following Simon two-stage phase II trial design, 27 patients were enrolled. Treatment was administered in 28-day cycles (intradermal TPIV200 and granulocyte-macrophage colony-stimulating factor (GM-CSF) for 6 cycles and intravenous durvalumab for 12 cycles). Primary endpoints included overall response rate and progression-free survival at 24 weeks. Translational parameters focused on tumor microenvironment, PD-L1 and FRα expression, and peripheral vaccine-specific immune responses.
Results Treatment was well tolerated, with related grade 3 toxicity rate of 18.5%. Increased T cell responses to the majority of peptides were observed in all patients at 6 weeks (p<0.0001). There was one unconfirmed partial response (3.7%) and nine patients had stable disease (33.3%). Clinical benefit was not associated with baseline FRα or PD-L1 expression. One patient with prolonged clinical benefit demonstrated loss of FRα expression and upregulation of PD-L1 in a progressing lesion. Despite the low overall response rate, the median overall survival was 21 months (13.5–∞), with evidence of benefit from postimmunotherapy regimens.
Conclusions Combination of TPIV200 and durvalumab was safe and elicited robust FRα-specific T cell responses in all patients. Unexpectedly durable survival in this heavily pretreated population highlights the need to investigate the impact of FRα vaccination on the OC biology post-treatment.
- immunogenicity, vaccine
- antigens, neoplasm
- clinical trials, phase II as topic
- programmed cell death 1 receptor
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- immunogenicity, vaccine
- antigens, neoplasm
- clinical trials, phase II as topic
- programmed cell death 1 receptor
Introduction
Recurrent and platinum-resistant ovarian cancers (OC) are therapeutically challenging, with poor overall survival (OS) ranging from 12 to 14 months.1 Therapies are limited, and response rates to approved chemotherapy agents range from 10% to 15%.1 2 Platinum-refractory OC is particularly challenging with rare responses to chemotherapy; these patients are often excluded from clinical trials.3
Immune checkpoint inhibitors (ICIs) block inhibitory receptors on the surface of T cells or their corresponding ligands, preventing exhaustion and promoting activation of T cells to enhance tumor detection and destruction.4 Although immunotherapy is a promising treatment for recurrent OC,5 single-agent ICIs have only produced modest results in recurrent OC, with response rates ranging from 8% to 15%.6–8 The reasons for these disappointing results are not fully understood, but may include an immune suppressive tumor microenvironment, expression of multiple immune checkpoints, and a relatively low tumor mutational burden with a corresponding dearth of neoantigens in OC.9 10 Furthermore, marked intratumor and intertumor genetic and microenvironment heterogeneity presents an additional layer of complexity11 12 and may contribute to the lack of ICI response.13
Vaccination against tumor-associated antigens (TAAs) is a potential strategy to increase the therapeutic efficacy of ICIs through development or enhancement of antitumor T cell responses. Folate receptor alpha (FRα), also known as folate receptor 1 (FOLR1), is a glycosylphosphatidylinositol-linked protein which participates in embryonic neural tube formation.14 15 It is also overexpressed in a majority of OCs (70%–90%) but is rarely found in healthy adult tissues.16 17 Validity of FRα as a therapeutic target has been confirmed through trials using FRα-targeted antibody-drug conjugate mirvetuximab soravtansine, demonstrating single-agent efficacy in patients with tumor exhibiting positivity for FRα.18 Strategies for development of FRα-directed adoptive T cell therapy approaches are also in development.19 20 While it is a self-antigen, expression of FRα in adult tissues is highly restricted, potentially limiting self-tolerance; as a result several promiscuous epitopes of FRα have been identified to prompt robust and durable T cell response in over 70% of patients with ovarian and breast cancers.21 These features render FRα an attractive TAA candidate for vaccination as a means to increase the efficacy of ICI immunotherapy in OC.
Based on these observations, a multiepitope anti-FRα vaccine (huFR-1 or TPIV200, Marker Therapeutics) was developed by synthesizing the five most highly antigenic human leukocyte antigen (HLA) class II-binding peptides from FRα identified in a group of unvaccinated patients with OC and breast cancer, as compared with normal volunteers. In women in remission from OC or breast cancer, intradermally administered TPIV200 admixed with granulocyte-macrophage colony-stimulating factor (GM-CSF) increased FRα-specific T cell immune responses in all evaluable patients.22 All five of the constituent peptides were found to be immunogenic, and all patients appeared to have developed immune responses to at least two, and in the majority more than three, of the five vaccine peptides.
Based on these findings, we hypothesized that, in advanced platinum-resistant OC, combination of TPIV200 and durvalumab would result in clinical benefit through generation of FRα-specific T cell responses coupled with inhibition/prevention of T cell exhaustion through programmed death ligand 1 (PD-L1) blockade.
Materials and methods
Study design
This was an investigator-initiated, single-center (Memorial Sloan Kettering Cancer Center, MSKCC), non-randomized, open-label, phase II study to evaluate the safety and clinical activity of TPIV200 in combination with durvalumab in patients with recurrent or persistent platinum-resistant OC. The study was conducted at MSKCC between May 2016 and February 2018. All patients signed written informed consent.
Patients
Women >18 years of age with recurrent or persistent platinum-resistant, epithelial, non-mucinous, high-grade ovarian carcinomas with measurable disease per response evaluation criteria in solid tumors (RECIST 1.1.) and adequate organ function were eligible. Further inclusion criteria were Eastern Cooperative Oncology Group (ECOG) performance status of 0–1, adequate marrow (absolute neutrophil count (ANC) ≥1.5), hepatic (bilirubin ≤1.5 × upper limit of norma (ULN); aspartate aminotransferase (AST) and alanine aminotransferase (ALT) ≤2.5 × ULN), and renal function (creatinine clearance >40 mL/min by Cockcroft-Gault). Patients with previous treatment with a PD-1 or PD-L1 inhibitor were excluded, as were patients requiring immunosuppressive medication within 28 days before the first dose, or with active or prior clinically significant autoimmune disease within the preceding 2 years. Patients with a history of pneumonitis or other lung injury from prior FRα-targeting therapy or prior grade ≥3 immune-related adverse event (irAE) while receiving previous immunotherapy agent or any unresolved irAE grade >1 were excluded.
Protocol treatment
During 28-day cycles, patients received a flat dose of durvalumab 750 mg intravenously on days 1 and 15 in cycles 1–12, and TPIV200 (500 µg per peptide; Marker, ref IB) admixed with GM-CSF (125 µg; Sargramostim) via three intradermal injections in the upper extremities on day 1 in cycles 1–6. Patients received study treatment until disease progression, intolerable toxicity, elective withdrawal from the study, or study completion. Efficacy assessments were performed every three cycles (or 12 weeks) from the initiation of study treatment until disease progression.
Response criteria
Patients who discontinued treatment for reasons other than progression continued efficacy assessments every three cycles (or 12 weeks) until progression was demonstrated. Patients who demonstrated radiologic progression by RECIST 1.1 criteria were considered for continued therapy (but not primary efficacy analysis) if they were deemed to be clinically benefitting, according to predetermined permitted criteria (section 12.4 of the protocol; online supplementary material).
Supplemental material
Safety
Safety was evaluated by monitoring all serious and non-serious adverse events (AEs) and irAEs, graded according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events (V.4.03). A safety stopping rule was established to terminate the study early in the case of excess toxicity. We assumed a 20% rate of clinically significant treatment-related AEs, such as persistent or unexpected renal injury or pulmonary toxicity, or clinically significant laboratory abnormality to be unacceptable. The trial would be terminated at any point if at least 7 out of 40 patients (per a graded stopping rule; online supplementary material) experience a grade 3/4 protocol-related AE unique to the combination. Frequencies of toxicities were tabulated to specify grade 3 or 4 treatment-related non-hematologic/non-laboratory toxicities.
Statistical methods
The primary objective was overall response rate (ORR) by RECIST and progression-free survival (PFS) rate at 6 months. ORR, defined as complete response (CR) and partial response (PR), was the primary endpoint. A prespecified 10% ORR was deemed to be low and 25% or higher to be considered promising for further studies. Forty patients would provide 90% power and type I error of 10% using a Simon two-stage design. There was an interim analysis after 27 patients; if 3 or more responders (CR+PR) were observed, then the study would continue to the second stage. At the end of the study, 7 or more responders out of 40 patients would be required to declare this study positive and the combination worthy for further investigation. PFS at 6 months as 15% would be considered low, vs 35% which would be considered promising, if the ORR cut-off were also met. Disease control rate (DCR) is defined as the percentage of patients with CR+PR+stable disease (SD) ≥12 weeks from the start of the treatment. PFS is defined from the date of start of treatment to the investigator-determined date of progression or death. OS is defined from the date of start of treatment to the date of death or last follow-up date. Event-free survival (EFS) for postimmunotherapy treatments is calculated from the start of treatment until next treatment, death, or loss of follow-up, whichever comes first. Descriptive statistics were provided. All CIs were provided as one-sided 90% CI. The median PFS/OS/EFS and the PFS/OS/EFS rates at prespecified time points were estimated using the Kaplan-Meier method.
For biomarker analyses, patients were dichotomized into benefit versus no benefit groups on the basis of PFS at 24 weeks or OS at 21 months (based on the median OS) as the outcome measure. Patients lost to follow-up before 21 months (n=1) were excluded from the latter analysis. Statistical significance between pretreatment and on-treatment enzyme-linked immunosorbent spot (ELISPOT) values was determined by Wilcoxon matched-pairs signed-rank test. Statistical comparisons of ELISPOT values and tumor microenvironment parameters between the benefit and no benefit groups were performed using Wilcoxon two-sample t-test.
Multiplex tissue staining and imaging
Primary antibody staining conditions were initially optimized using standard immunohistochemical staining on the Leica Bond RX automated research stainer with diaminobenzidine (DAB) detection (Leica Bond Polymer Refine Detection DS9800). Using 4 µm formalin-fixed, paraffin-embedded tissue sections and serial antibody titrations, the optimal antibody concentration was determined followed by transition to a seven-color multiplex assay with equivalency. Multiplex assay antibodies and conditions are described in table 1.
Antibody clones and dilutions used for tissue staining.
Seven-color multiplex imaging assay
At 62°C, 4 µm formalin-fixed paraffin-embedded (FFPE) tissue sections were baked for 3 hours with subsequent deparaffinization performed on the Leica Bond RX followed by six sequential cycles of staining, with each round including a 30 min combined block and primary antibody incubation (Perkin Elmer antibody diluent/block ARD1001). For all antibodies other than CD68, detection was performed using a secondary horseradish peroxidase (HRP)-conjugated polymer (Perkin Elmer Opal Polymer HRP Ms+Rb ARH1001; 10 min incubation). Detection of the CD68 primary antibody was performed using a goat antimouse Poly-HRP Secondary Antibody (Invitrogen B40961; 10 min incubation). PanCK, CK7 and Cam5.2 primary antibodies were used as a cocktail. The HRP-conjugated secondary antibody polymer was detected using fluorescent tyramide signal amplification using Opal dyes 520, 540, 570, 620, 650 and 690 (Perkin Elmer FP1487a, FP1494a, FP1488a, FP1496a, FP1495a, FP1497a). The covalent tyramide reaction was followed by heat-induced stripping of the primary/secondary antibody complex using Perkin Elmer AR9 buffer (AR900250ML) at 100°C for 20 min preceding the next cycle. After six sequential rounds of staining, sections were stained with Hoechst (Invitrogen 33342) to visualize the nuclei and mounted with ProLong Gold antifade reagent mounting medium (Invitrogen P36930).
Multispectral imaging, spectral unmixing and cell segmentation
Seven-color multiplex stained slides were imaged using the Vectra Multispectral Imaging System V.3 (Perkin Elmer).23 Scanning was performed at 20× (200× final magnification). Filter cubes used for multispectral imaging were 4′,6-diamidino-2-phenylindole (DAPI), fluorescein isothiocyanate (FITC), Cy3, Texas Red and Cy5. A spectral library containing the emitted spectral peaks of the fluorophores in this study was created using the Vectra image analysis software (Perkin Elmer). Using multispectral images from single-stained slides for each marker, the spectral library was used to separate each multispectral cube into individual components (spectral unmixing) allowing for identification of the seven marker channels of interest using Inform V.2.4 image analysis software. Images were exported to Indica Labs Halo image analysis platform, and cell segmentation and signal thresholding were performed separately on each case using a supervised algorithm.
IFNγ ELISPOT assay
Peripheral blood mononuclear cells (PBMCs) were collected using mononuclear cell preparation tubes (CPT) prior to treatment initiation and at 3 and 6 weeks and were cryopreserved in liquid nitrogen at 5×106 cells per vial in cryopreservation media containing 10% dimethyl sulfoxide (DMSO). The procedure for the FRα ELISPOT assay has been described previously.22 Briefly, 2.5×105 PBMCs/well were plated in triplicate into 96-well round bottom plates in 200 µL of RPMI1640 containing L-glutamine, penicillin, streptomycin, and 10% serum (T cell medium), along with cyclin D1 (control) peptide (MELLLVNKLKWNLAA), FR30, FR56, FR76, FR113, FR238, FRα protein (Sino Biological), tetanus toxoid (List Biological Laboratories), or Phytohemagglutinin (PHA; Sigma-Aldrich). Peptides were plated at 10 µg/mL, PHA was used at 1 µg/mL, FRα protein and tetanus were used at 100 ng/mL, and FRα protein was used at 100 ng/mL. The cells were incubated at 37°C at 5% CO2. After 24 hours, the cells were transferred to nitrocellulose plates coated the day before with 10 µg/mL anti-IFNγ antibody (MabTech) and incubated at 37°C for another 24 hours. After a wash, the plate was incubated with 5 µg/mL biotinylated anti-IFNγ Ab (in 100 µL), washed three times with phosphate buffered saline (PBS), and further incubated with 100 µL/well streptavidin–alkaline phosphatase at a dilution of 1:1000 in PBS for 2 hours at room temperature. After washing three times in PBS, the plate was incubated with 100 µL/well alkaline phosphatase (AP) colorimetric substrate for 20–30 min, rinsed with cool tap water, and allowed to dry completely. After drying overnight, the plates were read on an AID ELIspot reader, which provides quantitative spot information based on the number of stimulated cells that secrete IFNγ. Antigen-specific T cell frequencies were defined as the average number of spots elicited by a given antigen minus the average number of spots with no added antigen (presented as normalized spots in the results).
Results
Patient demographics
Between May 2016 and February 2018, 27 patients were enrolled. Baseline demographic and tumor data are summarized in table 2. The median age at trial enrollment was 64 years (42–76). The majority (23, 85%) had high-grade serous histology. All patients had disease that progressed during or within 6 months of last platinum therapy, with median platinum-free interval (PFI) of 1 month (mean 1.9; range 0–6). Twelve (44%) patients had platinum-refractory disease, defined by disease progression while receiving the last platinum-based regimen. The median number of prior lines of therapy was 4 (range 1–8). Five patients had deleterious germline (2) or somatic (3) BRCA1/2 mutations identified; germline or somatic BRCA1/2 mutation status was unknown in 7 and 20 patients, respectively.
Baseline characteristics (N=27)
Safety
TPIV200-related AEs were generally mild and primarily consisted of injection site reactions (all grade 1) both immediate and delayed, with some persisting for many months with a waxing and waning course, often coinciding with durvalumab infusions (table 3). Most durvalumab-related AEs encountered were grade 1–2, with few grade 3–4 toxicities deemed to be related to treatment. There were two irAEs of interest, including one patient with new onset of type 1 diabetes mellitus and one patient with immune-mediated thrombocytopenia; however, there was no evidence to suggest these toxicities were unique to the combination.
Treatment-related adverse events (N=27)
TPIV200-specific immune responses
PBMCs were collected prior to treatment initiation and at 6 weeks on therapy. Matched pretreatment and on-treatment PBMCs were available for analysis from 24 out of 27 patients. In all 24 patients, an increased response to at least one of the five FR peptides or full-length FR protein was observed (figure 1A); the majority of the patients developed increased responses to all peptides (figure 1B). Minimal changes in response to unrelated tetanus or cyclin D1-derived peptides were observed (online supplementary figure 1).
Supplemental material

T cell responses to vaccination. (A) Overall ELISPOT heatmap. (B) ELISPOT responses to individual peptides. Comparisons of pretreatment and on-treatment responses to individual peptides were performed using Wilcoxon matched-pairs signed-rank test. ELISPOT, enzyme-linked immunosorbent spot. ***p<0.001, ****p<0.0001.
Clinical efficacy
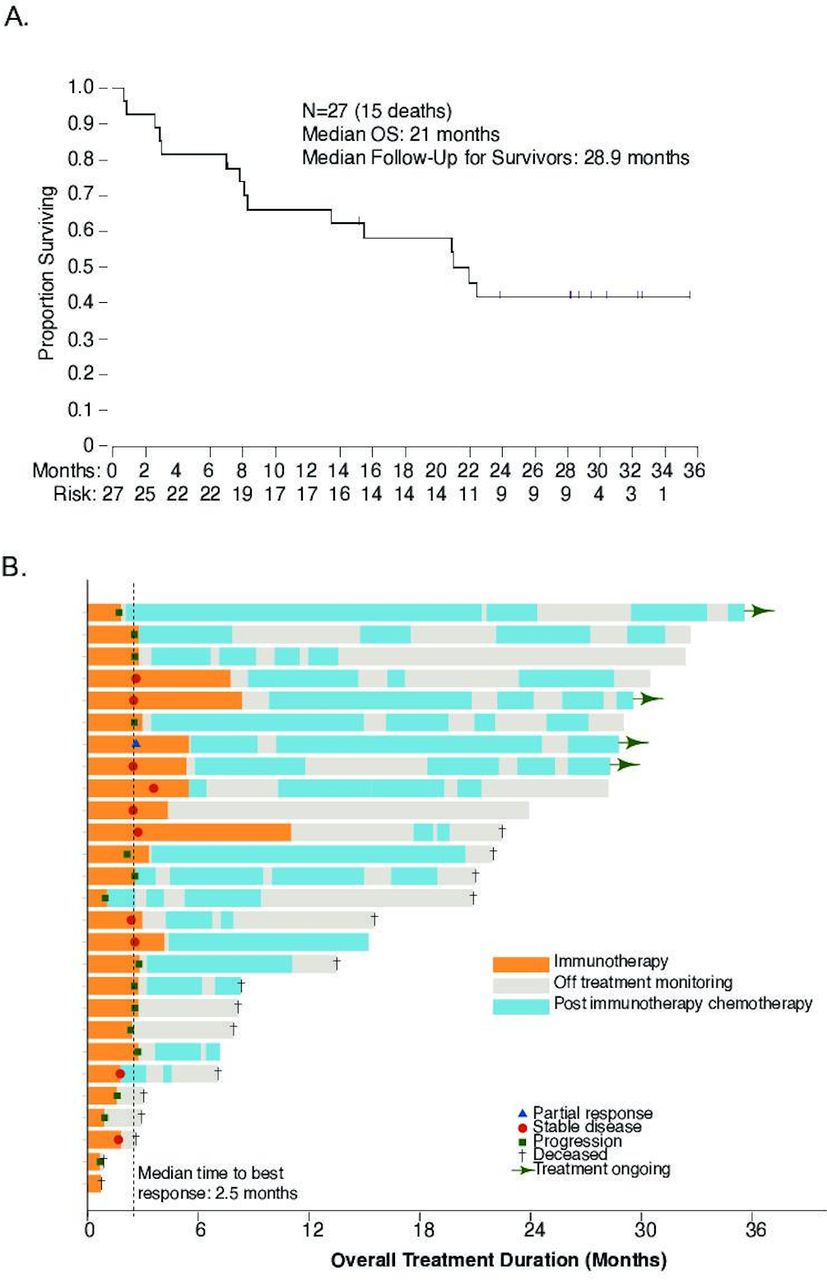
The efficacy cohort included 27 patients, all of them were evaluable for safety and efficacy after having received at least a single dose of the study medications. There was one unconfirmed PR after stage 1, which did not meet the prespecified criteria to proceed to stage 2 of the analysis (figure 2A). Nine (33%) patients had SD as the best response, with a DCR of 37% (24.4%–100%) (figure 2A). The median PFS was 2.8 months (2.5–∞), with a 6-month PFS rate of 11.1% (4.9%–100%) (figure 2B). Since all of the patients eventually experienced disease progression, no patient was censored for PFS analysis.

Overall response, duration of response and progression-free survival. (A) Spider plot demonstrating responses and response duration per Response Evaluation Criteria in Solid Tumors (RECIST) 1.1. (B) Progression-free survival. PR, partial response; SD, stable disease; PD, progressive disease.
Despite the limited efficacy of the combination, the observed median OS was 21 months (13.5–∞), with a median follow-up for survivors at 29 months (figure 3A). The estimated OS rate at 12 months was 66% (52.9%–100%), which was markedly higher than expected for this heavily pretreated patient population mostly with platinum-refractory disease. To better understand the reasons for the apparently improved survival, analysis of postimmunotherapy treatments was undertaken in all patients, focusing on up to four lines of treatment (figure 3B). Twenty (74%) patients underwent at least one subsequent line of therapy. The median postimmunotherapy EFS for each line of therapy is summarized in online supplementary figure 2 and presented in figure 3B. Types of chemotherapy varied across patients (online supplementary figure 3A); in general, a combination of chemotherapy with bevacizumab appeared to lead to longer EFS (online supplementary figure 3B), although this was only apparent during the first line of treatment postimmunotherapy (online supplementary figure 3C,D).

Overall survival and duration of postimmunotherapy treatments. (A) Overall survival (OS) at a median of 28.9-month follow-up. (B) Swimmer plot demonstrating duration of immunotherapy and duration of subsequent treatments.
Association of clinical benefit with vaccine-specific immune response
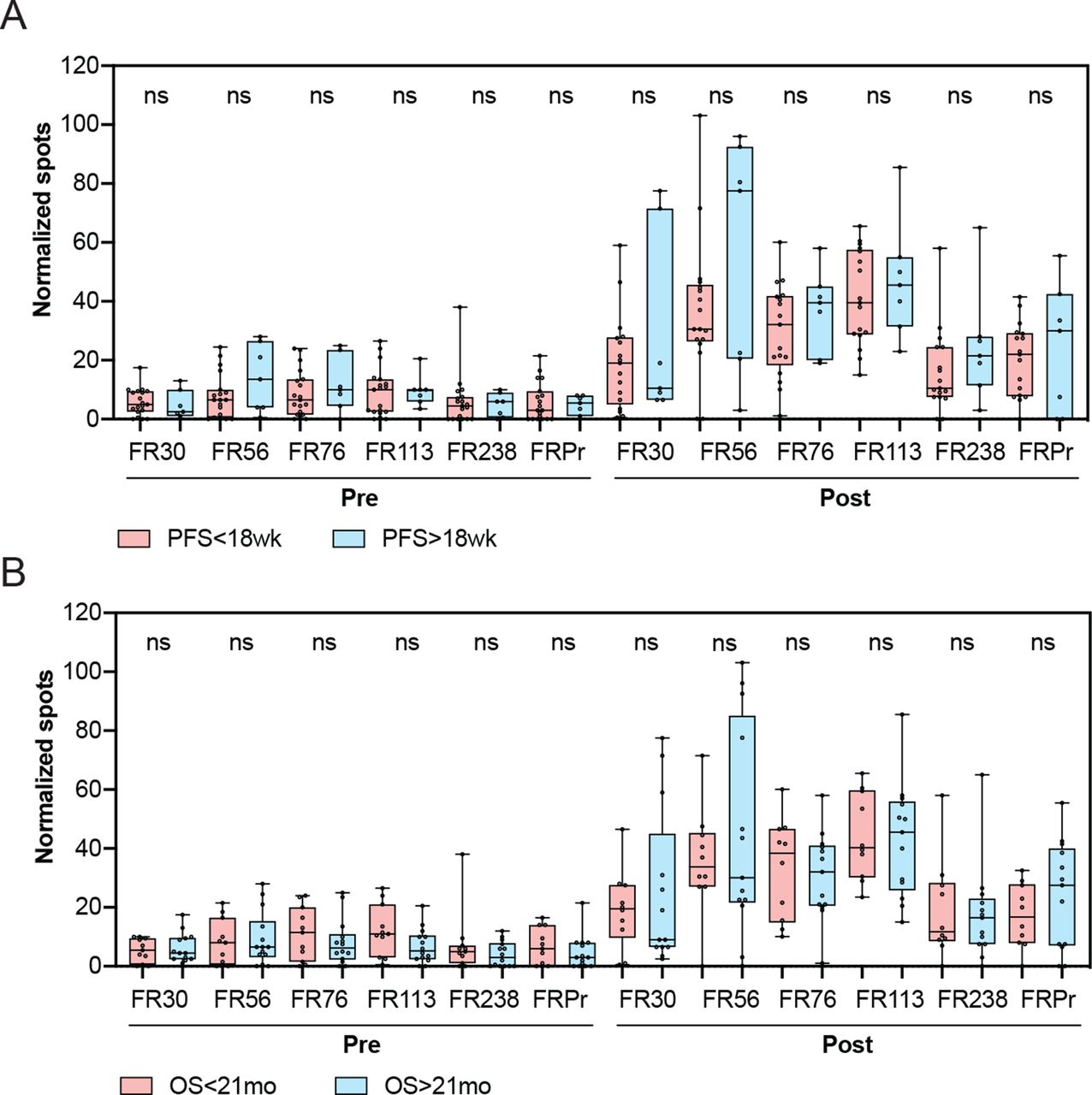
There was no statistically significant correlation between the degree of response to either of the individual peptides and clinical benefit, which was defined as PFS of 18 weeks or longer (figure 4A). Similarly, there was no association of the degree of response to the peptides and OS, based on a median OS of ≥21 months (figure 4B). Similarly, no association was observed between responses to the control cyclin D1 and tetanus peptides and PFS or OS (online supplementary figure 4). Overall, these data demonstrate that while vaccination with TPIV200 in combination with durvalumab led to vaccine-specific responses in all patients, the relative magnitude of response was not associated with PFS or OS.
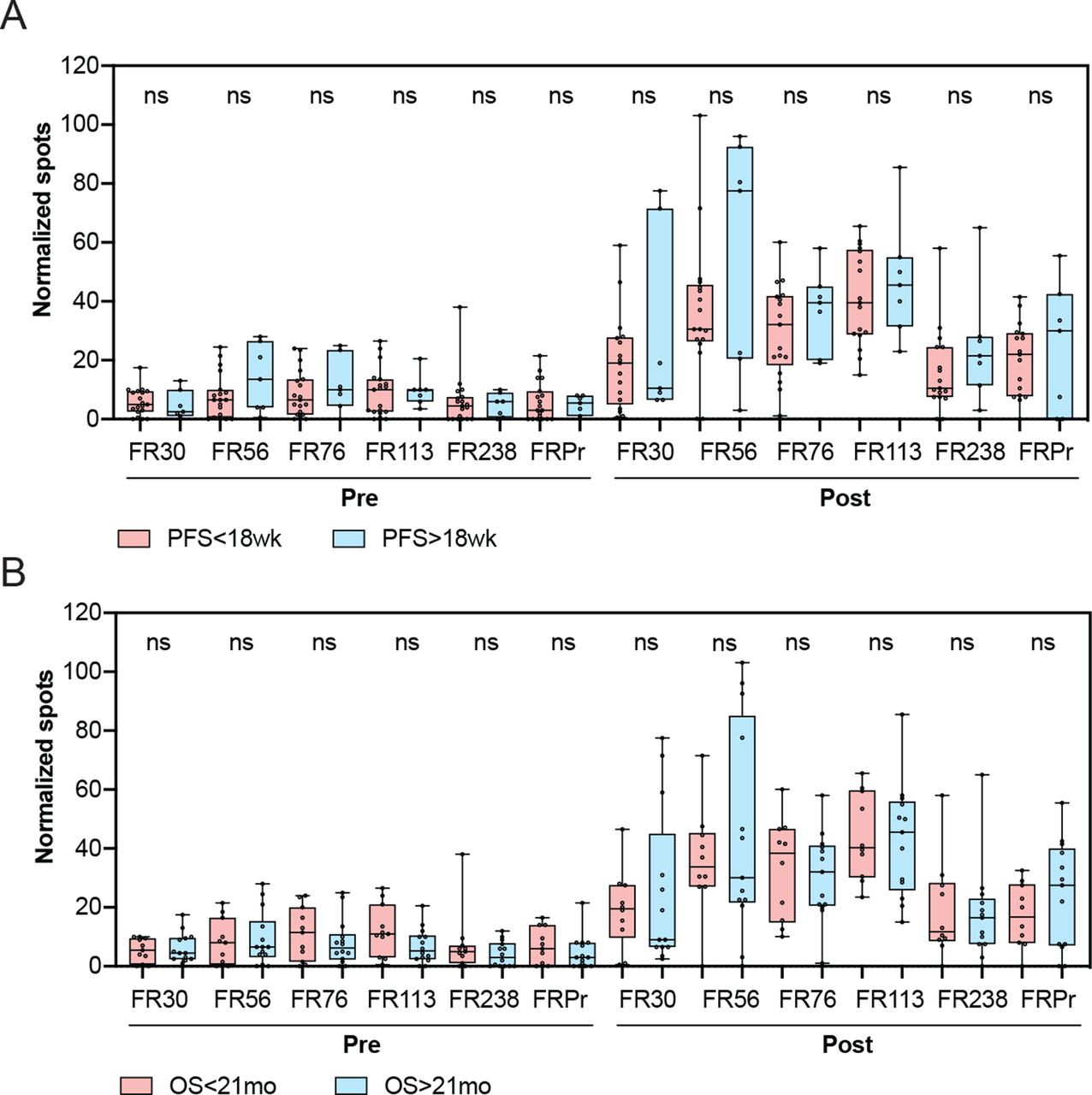
Association of clinical benefit and overall survival with T cell response to vaccination. (A) Association of pretreatment and on-treatment responses to individual peptides with progression-free survival (PFS). (B) Association of pretreatment and on-treatment responses to individual peptides with overall survival (OS). Comparisons of responses to individual peptides between groups were performed using Wilcoxon two-sample t-test. ns, non-significant.
Association of clinical benefit with tumor microenvironment parameters and FOLR1 expression
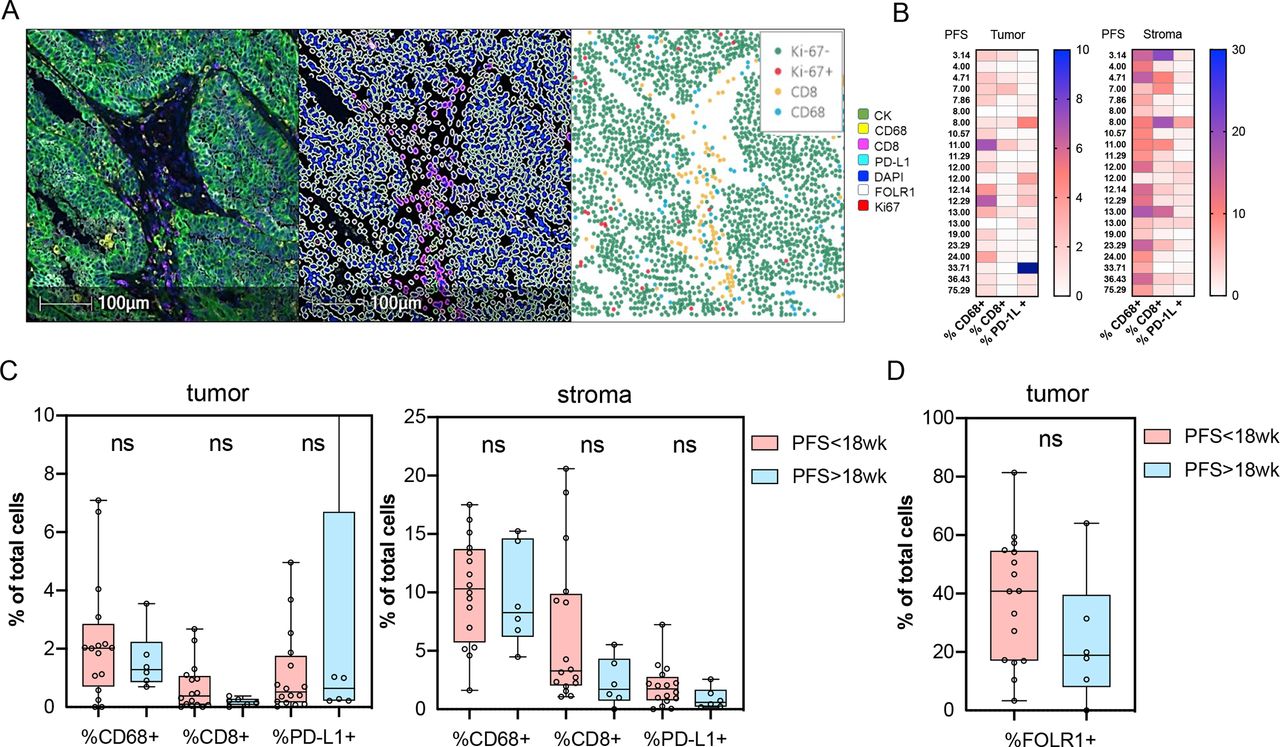
Archival tissue was available for testing from 22 out of 27 patients. Using multiparameter immunofluorescence microscopy, the tumors were stained for markers delineating tumor cells (CK), macrophages (CD68), cytotoxic T cells (CD8), as well as Ki-67, FRα (FOLR1), and PD-L1, and the percentages of the individual cell populations were quantified in tumor and stroma (figure 5A,B). There was no association between clinical benefit (defined by PFS at ≥18 weeks vs not) and the relative percentages of CD68+, CD8+, or PD-L1+ cells in tumor or stroma (figure 5C). Similarly, there were no significant differences in the percentages of FRα expression between the patients who derived clinical benefit and those who did not (figure 5D). One patient with prolonged clinical benefit (PFS of 75 weeks) developed disease progression and underwent a biopsy of one of the progressing lesions. Interestingly, the lesion demonstrated an almost complete loss of FRα expression and a significant increase in tumor infiltrating CD8+ T cells, macrophages, and upregulation of PD-L1 (online supplementary figure 5A,B). These findings suggest that immunoediting could have been a potential mechanism of tumor escape in this patient, and imply that FRα-specific T cell response may have contributed to prolonged disease control.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Association of clinical benefit with baseline tumor microenvironment parameters. (A) Sample image (left), segmentation mask (center), and individual cell counts (right) used for quantification. (B) Heatmap of overall percentages of the indicated cell populations in tumor and stroma ordered by PFS. (C) Association of the indicated cell populations and programmed death ligand 1 (PD-L1) expression in tumor and stroma with PFS. (D) Association of baseline FRα (FOLR1) expression in tumor with PFS. Comparisons of the individual cell populations were performed using Wilcoxon two-sample t-test. ns, non-significant; PFS, progression-free survival.
Discussion
Cancer immunotherapy with ICIs to date has demonstrated limited efficacy in patients with advanced platinum-resistant OC. There is thus clearly an unmet need for development of rational therapeutic combinations relevant to OC biology.
Vaccination against TAAs is a potential strategy to enhance antitumor immunity through the development of novel tumor-reactive T cells or through amplification of pre-existing T cells reactive to the same antigens. FRα is overexpressed in the majority of OCs and presents a compelling antigenic target for immunotherapy. Prior studies targeting FRα with antibody-drug conjugate mirvetuximab soravtansine provided clinical rationale for therapeutic targeting of FRα using additional therapeutic modalities, such as adoptive T cell therapies and vaccines.24 25 Indeed, prior study using FRα-directed vaccination in patients with ovarian and breast cancer in remission demonstrated new or augmented FRα-specific T cell responses in 90% of the vaccinated patients.22
Several vaccines have been previously explored in OC, primarily focusing on targeting of cancer-germline antigens (eg, NY-ESO-1) and proteins known to be overexpressed in OC (eg, p53, survivin, MUC1).26–40 The majority of the studies unfortunately failed to yield substantial evidence of clinical response. Recent studies have highlighted that despite the development of vaccine-specific immune responses, the generated tumor-specific T cells are still likely subject to exhaustion and other immunosuppressive mechanisms active within the tumor microenvironment.4 Studies in preclinical models indeed highlight that combination of tumor vaccines with immune checkpoint blockade can be superior to either approach alone,41–48 thus generating a rationale for exploration of similar strategies in human trials.
Based on these findings, in the current study, we sought to evaluate whether addition of FRα vaccine TPIV200 to PD-L1 blockade could lead to improved outcomes. Combination immunotherapy was deemed to be safe, and no new safety signals were observed. Biomarker analyses revealed development of robust FRα-specific T cell responses, with the majority of patients responding to all of the peptides. Despite these findings, the study failed to meet its prespecified clinical efficacy endpoint and was stopped after stage I accrual. These findings highlight that development of vaccine-specific T cell responses even in the setting of a therapy targeting T cell exhaustion was still not sufficient to drive tumor regression and caution against labeling cancer vaccines as ‘effective’ based on immunologic endpoints alone.
Several potential mechanisms could be responsible for the observed apparent lack of radiographic response. First of all, in this heavily pretreated population with weakened immune system, the magnitude of T cell response might not have been sufficient to elicit tumor regression. Second, even with development of sufficient number of tumor-specific T cells, PD-L1 blockade might not be sufficient to overcome the immunosuppressive mechanisms active in the OC tumor microenvironment. Finally, a possibility exists that the tumors lacked sufficient expression of FRα for recognition by the newly generated T cells. In this study, FRα expression was detectable in most patients; however, we did not observe any association between the relative levels of tumor FRα expression and clinical benefit. While this could potentially be explained by the use of archival tissue to assess FRα expression, prior studies demonstrated high concordance in expression between archival tissue and fresh biopsy samples,16 suggesting that in the absence of therapeutic selection pressure, temporal and spatial variation in FRα expression is likely not very high. Notably, in the studies of antibody-drug conjugate mirvetuximab soravtansine, detection of FRα expression in archival tissue was sufficient to predict responses to the drug in advanced recurrent disease setting.49 Interestingly, in a patient who had initial prolonged disease stabilization followed by progression, we saw significant reduction in tumor cell FRα expression in the progressing tumor (online supplementary figure 5), which supports the hypothesis that FRα was indeed a possible target of T cell recognition.
Intriguingly, despite the apparent limited response rate and PFS, at the median follow-up of 29 months, the median OS was 21 months. Platinum-resistant OC overall portends poor prognosis, with an average reported survival of approximately 12 months.1 2 Standard therapies in this setting include single-agent chemotherapies with or without bevacizumab. In the phase III AURELIA trial, which evaluated single-agent chemotherapy with or without bevacizumab in patients with platinum-resistant disease and one to two prior lines of therapy, the median PFS with addition of bevacizumab was reported at 6.7 months, with resultant OS of 16.6 months,50 thus establishing this regimen as a standard of care for platinum-resistant OC. Interestingly, the patient population in the current study represented a more heavily pretreated patient population than that evaluated in the AURELIA study, with a median of 4 prior lines of therapy, median PFI of 1 month, and with 12 out of 27 patients having platinum-refractory disease. Postimmunotherapy follow-up was suggestive of improved clinical benefit from standard chemotherapies, particularly in the patients who received chemotherapy in combination with bevacizumab during the immediate postimmunotherapy treatment course. Exploration of cancer vaccines in OC in combination with chemotherapy and bevacizumab has previously demonstrated evidence of clinical benefit,51 thus providing rationale for potential evaluation of TPIV200 in combination with chemotherapy. Overall, these data demonstrate that vaccination against FRα may have a potential to alter patient outcomes even in the absence of apparent clinical benefit based on the standard response assessment criteria, and highlight the rationale to investigate the impact of FRα vaccination on the OC biology post-treatment.
Acknowledgments
We thank Patrick Yeramian and Glynn Wilson of Marker Therapeutics for their input and advice.
References
Footnotes
Twitter @SWalderich
Contributors DZ and JAK conceived and conducted the study, analyzed the results, and composed the manuscript. SW, AH, LC, YL, TJH, RG, CE, MB, KK, REO, and CA collected the data and participated in manuscript review. QZ and AI performed the statistical analyses. TM mentored SW and AH and was involved in manuscript review.
Funding The clinical trial was supported by AstraZeneca and Marker Therapeutics. The study was supported in part by the MSK Cancer Center Support (grant P30 CA008748) and in part by the Mayo Clinic SPORE in Ovarian Cancer (P50-CA136393), to KK and MB. DZ is a member of the Parker Institute for Cancer Immunotherapy at MSKCC. DZ is supported by the Ovarian Cancer Research Foundation Liz Tilberis Award and the Department of Defense Ovarian Cancer Research Academy (OC150111).
Competing interests KK is listed as a coinventor on a patent entitled 'Immunity to folate receptors', which is owned by the Mayo Clinic and licensed to Marker Therapeutics. MB and KK report receiving commercial research support from Marker Therapeutics. DZ reports personal/consultancy fees from Merck, Synlogic Therapeutics, Biomed Valley Discoveries, Trieza Therapeutics, Tesaro, and Agenus, outside of the scope of the submitted work.
Patient consent for publication Not required.
Ethics approval The study was approved and was annually reviewed by the internal institutional review board.
Provenance and peer review Not commissioned; externally peer reviewed.