Article Text

Abstract
Background Radiation therapy (RT) has the potential to enhance the efficacy of immunotherapy, such as checkpoint inhibitors, which has dramatically altered the landscape of treatments for many cancers, but not yet for pancreatic ductal adenocarcinoma (PDAC). Our prior studies demonstrated that PD ligand-1 and indoleamine 2,3-dioxygenase 1 (IDO1) were induced on tumor epithelia of PDACs following neoadjuvant therapy including RT, suggesting RT may prime PDAC for PD-1 blockade antibody (αPD-1) or IDO1 inhibitor (IDO1i) treatments. In this study, we investigated the antitumor efficacy of the combination therapies with radiation and PD-1 blockade or IDO1 inhibition or both.
Methods We developed and used a mouse syngeneic orthotopic model of PDAC suitable for hypofractionated RT experiments.
Results The combination therapy of αPD-1 and RT improved survival. The dual combination of RT/IDO1i and triple combination of RT/αPD-1/IDO1i did not improve survival compared with RT/αPD-1, although all of these combinations offer similar local tumor control. RT/αPD-1 appeared to result in the best systemic interferon-γ response compared with other treatment groups and the highest local expression of immune-activation genes, including Cd28 and Icos.
Conclusion Our RT model allows examining the immune-modulatory effects of RT alone and in combination with immune-checkpoint inhibitors in the pancreas/local microenvironment. This study highlights the importance of choosing the appropriate immune-modulatory agents to be combined with RT to tip the balance toward antitumor adaptive immune responses.
- radiotherapy
- gastroenterology
- oncology
- tumors
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Pancreatic ductal adenocarcinoma (PDAC) carries a very poor prognosis, with a 5-year overall survival of only 9% in the USA.1 More than 50% of the cases are diagnosed at an advanced stage, and the recurrence rate after surgical resection remains high despite adjuvant chemotherapy. Radiation therapy (RT) has been reported as a beneficial treatment for many types of cancers; but the benefit of adding RT to chemotherapy for PDAC treatment is still under debate.2–8 Given the controversy of RT for PDAC treatment in the clinical setting, one new application that RT holds great potential for is the combination of RT with other treatment modalities, such as immunotherapy.9 Immunotherapy, including checkpoint inhibitors, has dramatically altered the landscape for the treatment of many cancers, but not yet for pancreatic cancer.10–12 Animal and human studies suggest that the combination of immunotherapy and RT may have a synergistic and enhanced antitumor activity12–14 including studies in mouse models of PDAC.15 It has increasingly been recognized that RT may lead to an antitumor effect beyond its field of RT. This phenomenon, called the ‘abscopal effect’, is thought to be mediated by systemic immune responses induced locally by RT.16 17 RT causes the release of various cytokines and tumor-specific antigens from tumor cells that are undergoing necrotic cell death or apoptosis with caspase activation and leads to the activation of systemic immune responses.18 Human PDAC is known to have a non-immunogenic tumor microenvironment (TME).19 20 Thus, it will be intriguing to test whether RT can prime the non-immunogenic TME and sensitive PDAC for immunotherapy.
An attractive target for inhibition that can be combined with RT is the immune-checkpoint PD-1. Anti-PD-1 antibodies as a single agent are known to be ineffective for PDAC except for a rare subtype with microsatellite instability.12 We previously demonstrated that vaccine therapy can prime the PDAC tumors for future targeting by anti-PD-1 antibody therapy.19 Other priming mechanisms may also exist, including RT. However, whether and how RT can overcome the resistance of PDAC to anti-PD-1 antibody therapy has not been fully examined particularly in the syngeneic pancreatic orthotopic model of PDAC that recapitulates non-immunogenic TME of human PDAC better than the subcutaneous tumor model.21 The main hurdle for such preclinical studies was the lack of a mouse syngeneic pancreatic orthotopic tumor model convenient for treatment with RT.
Another attractive target for combination with RT is an intracellular enzyme associated with tumor immunosuppression and immune evasion, indoleamine 2,3-dioxygenase 1 (IDO1). It exerts its immunosuppressive effects by catalyzing the first and rate-limiting step of L-tryptophan catabolism, leading to the depletion of tryptophan and the influx of Kynurenine downstream catabolites that suppress the activation of T cells and enhance regulatory T cell differentiation.22–24 IDO1 expression is found in a variety of cancers, including PDAC, and its expression is associated with poorer prognosis.25 IDO1 expression is upregulated in metastatic PDAC cells as a mechanism for immunologic evasion.25 However, IDO1 inhibitor monotherapy for cancer has yielded disappointing results. Despite promising results in preclinical and early clinical studies, the phase 3 trial combining an IDO1 inhibitor with PD-1 inhibitor yielded negative outcomes—the IDO1 inhibitor did not provide additional benefit beyond that provided by PD-1 inhibition for the treatment of melanoma.26–28 However, employing IDO1 inhibition to remove the immunosuppressive brakes in cancer remains to be a viable option for combination therapy. Monjazeb et al reported that radiation increased IDO1 expression in a murine breast tumor model and that the combination of IDO1 inhibition and RT improved antitumor efficacy in various tumor models, including glioblastoma, breast adenocarcinoma, melanoma and sarcoma.29 Our recent study found that IDO1 expression is induced on tumor epithelia of PDAC from patients who received chemotherapy and RT.30 Therefore, it was of interest to test the combination of IDO1 inhibition and RT in the murine pancreatic tumor model.
In this study, we developed a mouse syngeneic orthotopic model of PDAC suitable for the hypofractionated RT experiment. Using this model, we investigated the antitumor efficacy of the combination therapies with radiation and PD-1 blockade or IDO1 inhibition or both. In this era of rapid drug development for cancer therapeutics and enthusiasm for immuno-oncological combination therapies, our results highlight the importance of performing diligent preclinical studies to help ensure that clinical trials are rationally designed and based on strong scientific evidence.31
Material and methods
Cell lines and medium
The KPC tumor cell line is a previously established murine PDAC tumor cell line that was derived from transgenic mice in a C57Bl6 background with tissue-specific Kras and p53 knock-in mutations.32 KPC cells were maintained at 37°C in 5% CO2 with RPMI 1640 media (Life Technologies) supplemented with 10% heat-inactivated fetal bovine serum (HI-FBS, Benchmark), 1% penicillin/streptomycin (pen/strep, Life Technologies), 1% MEM Non-Essential Amino Acids Solution (MEM-NEAA, Life Technologies), 1% sodium pyruvate (Sigma) and 1% L-glutamine (Life Technologies). Harvested tumor-infiltrating immune cells were processed in T-cell media, which consisted of RPMI 1640 media supplemented with 10% HI-FBS, 1% pen/strep, 1% HEPES (Life Technologies), 1% MEM-NEAA, 1% L-glutamine and 0.05% 2-mercaptoethanol (Sigma).
Mice and in vivo experiments
All animal experiments conformed to the guidelines of the Animal Care and Use Committee of the Johns Hopkins University. The animals were maintained in accordance with the Institutional Animal Care and Use Committee (IACUC) guidelines.
Procedures for the orthotopic model were modified from our previous report.33 2×106 PDAC cells of the KPC cell line were subcutaneously injected into the flanks of syngeneic female C57Bl/6 mice. After 1–2 weeks, the subcutaneous tumors were harvested and cut into 2–3 mm3 pieces. New syngeneic female C57Bl/6 mice, ages 8–10 weeks, were anesthetized. The abdomen was opened via a left subcostal incision and obtained open access to the body and tail of the pancreas. A small pocket was prepared in the middle of the pancreas using microscissors, and one piece of the subcutaneous tumor was implanted into the small pocket. The incision in the pancreas was closed with a 7–0 Prolene. On two sides of the implant point toward either the pancreas head or pancreas tail, Horizon Titanium Ligating Clips (small) were used as fiducial markers and implanted and symmetrically implanted 5–10 mm from the tumor with carefulness not to clip the pancreas body deeply. The abdominal wall of the skin was sutured using 4–0 sutures.
For RT, mice were anesthetized with isoflurane and pancreas tumors were irradiated with 8 Gy x 3 fractions delivered daily between day 6 and 8 after the surgery, at a dose rate of 3 Gy/minute using the Small Animal Radiation Research Platform (SARRP; Xstrahl). The isocenter was placed at the center of the fiducials (online supplementary figure S1). IDO1 inhibitor compound Bristol-Myers-Squibb (BMS-986205) was dissolved into Methocel vehicle via 30 min mixing and given to the tumor-bearing mice by oral gavage once a day at 60 mg/kg starting on day 6. Antimouse PD-1 antibodies (10 mg/kg; BMS) or IgG (10 mg/kg; BMS) were administered intraperitoneally starting on day 6 and continuing every 4 days for a total of six doses.
Supplemental material
Supplemental material
Tumor size was monitored weekly using small-animal ultrasound (Vevo770, VisualSonics). Blood was collected from facial vein (sub-mandibular) on day 9. After blood collection, blood was centrifuged at 10 000 g immediately and only serum was collected. For survival studies, the mice were monitored at least once a day. Mice with signs of distress, including hunched posture and lethargy, were euthanized and considered to have reached the “survival” endpoint in accordance with IACUC guidelines.
Immunohistochemistry
Tumor tissues for immunohistochemistry (IHC) staining were obtained from specimens collected from patients who underwent surgery following neoadjuvant chemotherapy and RT at our institution under IRB-approved protocol NA_00074221.19 34 Formalin-fixed paraffin-embedded tissue blocks were obtained from our pathology archive. Human PD ligand-1 (PD-L1) IHC staining of paraffin-embedded pancreatic tumor specimens was performed using the Dako Catalyzed Signal Amplification system as previously described.35 A PDAC was considered to be positive for PD-L1 expression if membranous staining was present in >5% of the neoplastic cells in the PDAC as previously described.36
Cell staining and flow cytometry
Following the isolation of tumor-infiltrating lymphocytes from the murine pancreas, cells were stained with the Live Dead Aqua Dead Cell Kit (Invitrogen). The lymphocytes were washed and subsequently blocked with mouse Fc antibody (BD Pharmingen) for 10 min on ice, followed by incubation or staining with cell-surface antibodies: CD3-PerCP-Cy5.5 (Biolegend), CD4-PacBlue (Biolegend), CD8a-PE/Cy7 (Biolegend), PD-1–FITC (eBioscience), CD11b-PE TexasRed (Invitrogen), MHC II (I-A, I-E)- Alexa Fluor 700 (Biolegend), CD11c-APC (Biolegend) and F4/80-PE (Invitrogen) for a 30 min incubation on ice. The cells were then washed twice and resuspended in FACs buffer, and flow cytometry was performed using CytoFLEX (Beckman Coulter). Flow data were analyzed using CytExpert software (Beckman Coulter).
Luminex assays
Mouse specific Luminex reagents were sourced from Millipore Sigma. 44 individual analytes (online supplementary table S1) were included in this study, being separated into three panels (catalog # MCYTMAG-70K-PX32, MHSTCMAG-70K, MTH17MAG-47K). Cytokine measurements were performed using manufacturer protocols that have been miniaturized to 384-well format (REF) at BMS.37 Lyophilized cytokine standard cocktails (Millipore Sigma) were prepared according to manufacturer instruction. Thirty μL of mouse serum were submitted for profiling in 96-well format at −70℃, being thawed on ice and diluted 1:1 in the Luminex kit assay buffer. Ten μL of diluted serum, standard and kit-provided control samples were transferred to 384-well assay plates (Greiner, catalog #781096) and mixed with 10 μL Luminex bead reagents. Plates were shaken overnight at 4℃ in the dark, being washed twice with 90 μL of 1X wash buffer using a magnetic plate washer (Biotek). Then, 10 μL of the detection antibody solution was added to each well and mixed. Plates were shaken for 1 hour at room temperature in the dark. After adding 10 μL of Streptavidin labeled Phycoerythrin to each well, the assay plates were incubated for 30 min at room temperature in the dark. After washing with 90 μL of 1X wash buffer, 80 μL of sheath fluid are added to each well. Plates were measured using the Bio-Plex 3D system (Bio-Rad) and data was analyzed using the Bio-Plex Software 3.0. 70 μL and a minimum of 50 bead events per analyte were acquired. The mean fluorescence intensity data were converted into pg/mL from the standard curve using a 4-parametric logistic fit model.
NanoString
Half of the mouse tumor tissues were submerged into RNA-later (invitrogen) after euthanization of the mouse and harvesting of the tumor. AllPrep DNA/RNA/Protein Mini Kit (Qiagen) was used to extract total RNA from the whole specimen as per manufacturer’s instructions. RNA was measured with the EPOCH2 spectrophotometer and equalized for NanoString hybridization using the Formulatrix Tempest. NanoString hybridization was performed using the murine PanCancer Immune panel codeset (XT_PGX_MmV1_CancerImm_CSO, cat. # 115000142) containing 750 target genes plus housekeeping and negative/positive control probes and data was analyzed using NanoString nSolver 3.0 and an internally developed NanoString Data Analyzer Rshiny app (BMS). Samples were run on the NanoString MAX system reader. The web application uses several CRAN and Bioconductor packages to clean, reformat and analyze the data. The dplyr, tidyr and reshape2 packages were used to match the sample annotations to the normalized data exported from NanoString nSolver V.3.0. Additionally, these packages were used to aggregate the data accordingly for calculating log2 normalization, mean and SE for the sample groups. Base package, t-test function was used to calculate the two-sided equal-variance p values. The multtest package was used to adjust the p values for false discovery rate analysis following the two-stage step-up method of Benjamini et al at an alpha value of 0.3.38
Statistical analyses
All statistical analyzes and graphing except for NanoString data were performed using GraphPad Prism software (GraphPad Software). The mean values of tumor sizes were analyzed using the unpaired t-test. Kaplan-Meier curves and log-rank tests were used to analyze survival outcomes among groups. For comparison of cell number and cytokine expression, the mean values were evaluated using unpaired t-test, and one-way analysis of variance was used for multiple comparisons of means. A p <0.05 was considered statistically significant.
Results
The addition of IDO1 inhibitor does not further improve the antitumor activity achieved by the combination of radiation and anti-PD-1 antibody therapies
We examined PD-L1 expression in resected PDACs from patients who received standard of care neoadjuvant chemotherapy including the FOLFIRINOX and gemcitabine/abraxane combinations and RT before they underwent surgical resection. Among 20 PDACs cases tested, IHC staining with anti-PD-L1 antibody showed that PD-L1 is expressed on the membrane of more than 5% of tumor epithelia in 8 (40%) of the cases, which is higher than 12.5%, as reported previously in patients who underwent upfront surgery (figure 1A,B).39 Moreover, among the same 20 PDACs cases, IDO1 expression on tumor epithelia was induced in 20% of the cases as previously reported, whereas IDO1 is essentially not expressed in untreated, resected PDACs.30 We reanalyzed this published result and noticed that two IDO1 positive PDACs in five cases (40%), who received minimal chemotherapy (less than three weekly doses of gemcitabine) among these 20 PDACs cases. This result suggests that the induction of IDO1 expression in PDAC is likely independent from chemotherapy. Therefore, we investigated if combination therapy with RT and IDO1 inhibitor and/or anti-PD-1 antibody could effectively slow down tumor growth in an orthotopic murine model of PDAC. In light of induction of PD-L1 expression by neoadjuvant chemotherapy and RT, we chose to focus on the study with anti-PD-1 antibody, not other immune-checkpoint inhibitors such as anti-CTLA4 antibody. Six days after surgical implantation of KPC tumors into the pancreas, the mice started to receive hypofractionated RT (three daily doses at 8 Gy), daily doses of IDO1 small-molecule inhibitor (IDO1i) and/or twice weekly doses of PD-1 blockade antibody (αPD-1) over a course of 21 days (figure 1C). Ultrasound imaging showed that αPD-1 slowed down the tumor growth significantly as compared with the IgG control during the treatment, before day 26 following tumor implantation (figure 1D). Mice in the control and RT groups started to reach the survival endpoint on day 26, possibly due to the development of disseminated metastatic diseases as observed at autopsy because their primary tumor sizes were not as big as those that reached the survival endpoint later during the course of experiment (online supplementary figure S2). Therefore, it would not be feasible to compare the tumor growth among all groups beyond day 26. We, thus, compared the tumor growth among only four groups (RT/αPD-1, RT/IDO1i, RT/αPD-1/IDO1i and αPD-1 alone) during the entire course of the experiment and found a significant difference in tumor growth between the groups treated with αPD-1 alone and αPD-1/RT (figure 1E). In an independent experiment, we confirmed that the IDO1i alone or the αPD-1/IDO1i combination did not show the tendency of suppressing tumor growth compared with the control group, and that the combination of RT with IDO1i slowed down tumor growth compared with the control, IDO1i alone and αPD-1/IDO1i combination groups (online supplementary figure S3). In this setting, we also found that the addition of αPD-1 to the RT/IDO1i combination further slowed down tumor growth. However, after a longer period of observation, the RT/αPD-1/IDO1i triple therapy showed similar tumor growth as the RT/αPD-1 dual therapy (figure 1E), suggesting that IDO1i did not add any significant antitumor activity to RT in the presence of αPD-1. This result suggests that the RT/αPD-1 combination has a stronger antitumor activity than the RT/IDO1i combination although there is no statistical significance in tumor growth between these two combination groups. Why IDO1i did not add any significant antitumor activity to RT in the presence of αPD-1 remains to be elucidated. This result is similar to our previous study of triple combination therapy of a cancer vaccine, αPD-1, and IDO1i in a PDAC murine model,30 showing that combining both immune-checkpoint inhibitors such as αPD-1 and IDO1i in the presence of vaccine therapy would suppress the tumor infiltration of CD4 +and CD8+T cells comparing to a single checkpoint inhibitor.
Supplemental material
Supplemental material
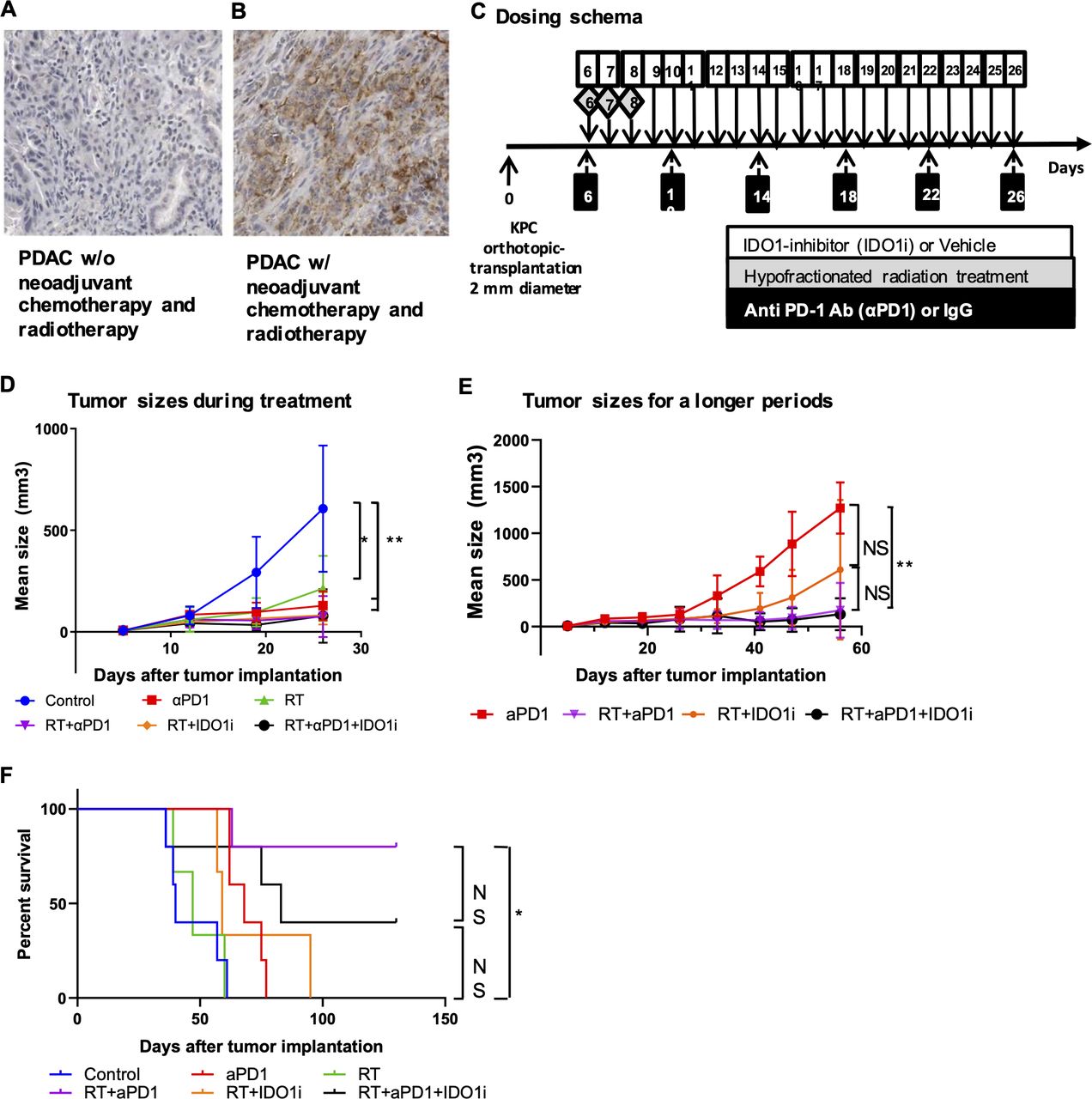
The addition of IDO1 inhibitor does not further improve the antitumor activity achieved by the combination of radiation and anti-PD-1 antibody therapies. (A, B) IHC staining of PD-L1 expression in PDACs from patients (n=20) treated without (A) and with (B) neoadjuvant treatment. (C) The treatment scheme. Mice underwent orthotopic implantation procedure receiving 2–3 mm KPC cancer tissues, followed by the combinational treatments with hypofractionated radiation treatment (3 fractions of 8 Gy during days 6–8), a small-molecule inhibitor of IDO1 (60 mg/kg, oral gavage during day 6–26) and/or anti PD-1 antibody (10 mg/kg, intraperitoneal injection for every 4 days, six times). (D, E) Growth curves of tumors evaluated by ultrasound imaging. (D) Growth curves of all treatment groups until day 26 following tumor implantation. (E) Growth curves of selected treatment groups during the entire course of the experiment (till day 56). Shown are the αPD-1 alone, RT/αPD-1, RT/IDO1i, and RT/αPD-1/IDO1i groups. The remaining groups are not shown because mice in these groups started to reach the survival endpoint on day 26. (F) Kaplan-Meier survival curves of mouse pancreatic cancer implantation model treated with combinations of radiation, anti-PD-1 antibody and IDO1 inhibitor. No mice were excluded for survival analysis. Data represent results obtained from experiments with five mice per group that was repeated at least twice. The error bars represent mean with SD. *P<0.05, **P<0.01; NS, not significant, by unpaired t-test or log-rank test. IDO1i, indoleamine 2,3-dioxygenase 1 inhibitor; IHC, immunohistochemistry; PDACs, pancreatic ductal adenocarcinomas; RT, radiation therapy.
The combination of RT and anti-PD-1 antibody, but not IDO1 inhibitor, extended survival significantly in the mouse model of PDAC
We next examined if the above treatment regimens improved survival. The addition of RT to IDO1i or αPD-1 led to significantly better tumor control, although the RT/αPD-1/IDO1i triple therapy did not show a significant difference in tumor growth compared with the RT/αPD-1 dual therapy (figure 1D,E). We hypothesized that the addition of RT to the combination of αPD-1 and IDO1i would contribute to a survival benefit. The results demonstrated that the combination of RT and αPD-1 extended survival significantly compared with control, αPD-1, RT or RT/IDO1i (figure 1F). However, there was no statistical difference in survival with the addition of IDO1i to the combination of RT and αPD-1 therapy; and in fact, there was a trend toward decreased survival with the triple therapy compared with the RT/αPD-1 dual therapy. By the end of the 130-day observation period, the survival rate of the triple combination therapy group was lower than the RT/αPD-1 therapy group (40% vs 80%). Interestingly, RT/IDO1i or RT/αPD-1/IDO1i did not offer significant survival benefit over RT alone even though these treatments offered better tumor growth control than RT. These data suggest adding IDO1i to RT/αPD-1 may not improve survival outcomes, but may, in fact, have detrimental effects on survival. Taken together, our study suggests that the combination of RT and αPD-1 is a better strategy of combining RT with an immune-oncology agent.
Various combinations of IDO1 inhibitor, anti-PD-1 antibody and radiation modulate the TME
To determine why the triple combination of RT/αPD-1/IDO1i did not improve survival and rate of tumor growth beyond RT/αPD-1, we investigated the effect the regimens had on the tumor immune milieu. Using the same murine orthotopic implantation model, we first extracted blood from the mice 1 day after the completion of RT treatment to analyze the serum for the early systemic effect of RT, and also harvested the tumors on day 14 to analyze the tumor-infiltrating leukocytes by flow cytometry (figure 2A). The flow cytometry data demonstrated that RT therapy alone significantly increases CD4 +T cells within the tumor compared with all groups except RT/αPD-1 (figure 2B). RT alone did lead to a statistically significant increase in CD4 +PD-1+T cells compared with the untreated control group (figure 2C). The numbers of CD4 +PD-1+T cells were significantly decreased in RT/αPD-1, RT/IDO1 and RT/αPD-1/IDO1i triple therapy group compared with RT alone. In contrast, tumor-infiltrating CD8 +T cells were scarce in all of the groups, suggesting that local response may not be mediated by CD8 +T cells (figure 2D). The number of tumor-associated macrophages (TAMs; F4/80+CD11b+CD11c- cells) was decreased in RT/αPD-1 and RT/IDO1i and significantly decreased in the RT/αPD-1/IDO1i triple therapy group compared with RT alone (figure 2E). In addition, the RT alone treatment increased the number of MHC II+CD11c+dendritic cells compared with the untreated control group (figure 2F). The addition of αPD-1 and/or IDO1i to RT led to decreased MHC II-expressing dendritic cells. These results suggest that RT-induced CD4 +T cells have regulatory functions, such as CD4 +PD-1+ and RT induce TAMs. However, IDOi and αPD-1 retain this negative impact of RT on the immune milieu. Interestingly, the addition of IDOi or αPD-1 also reversed the positive impact of RT to induce dendritic cells. The profiling of the immune subsets (figure 2) does not all explain the efficacy findings above, suggesting that the functionality analysis of the immune subsets is warranted in the future studies.
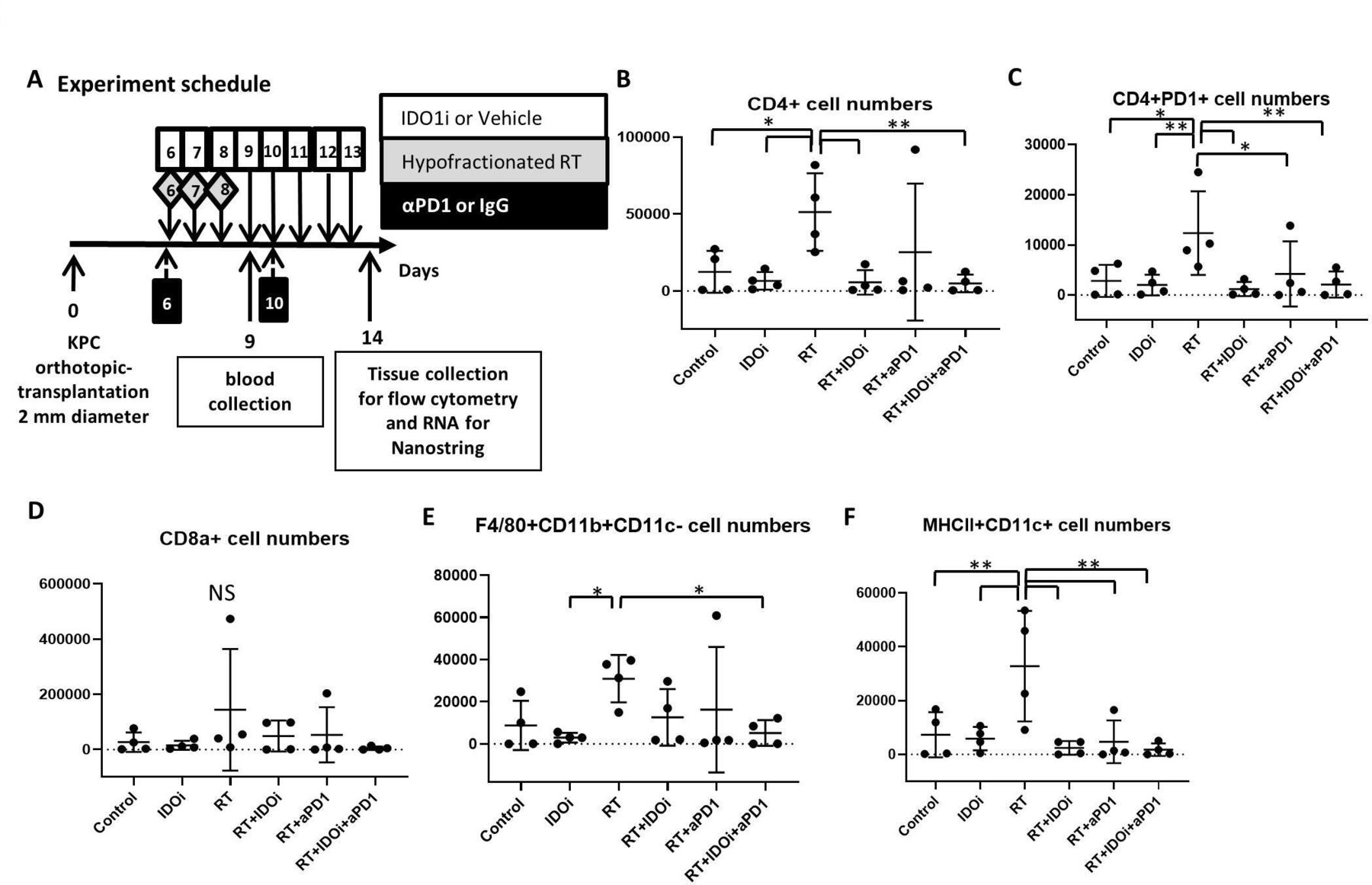
Various combinations between IDO1 inhibitor, anti-PD-1 antibody and radiation modulate the tumor microenvironment differently. (A) From KPC orthotopic implantation model mice, blood serum and tumor tissues were collected on day 9 and day 14. The normalized cell numbers of (B) CD4 +cells, (C) CD4 + PD-1 +cells, (D) CD8 +cells, (E) F4/80+CD11b+CD11 c negative cells, and (F) MHC-II +CD11c+cells as quantified by flow cytometry analysis in tumor-infiltrating cells. The cell numbers of immune cells per 1 million cells in the single cell suspensions of the tumors are shown in the histograms. Data represent mean±SEM of four mice per treatment group that was repeated twice. *P<0.05, **P<0.01; NS, not significant, by unpaired t-test and one-way ANOVA. ANOVA, analysis of variance; IDO1, indoleamine 2,3-dioxygenase 1; RT, radiation therapy.
Systemic interferon-γ was significantly induced by the RT/αPD-1 double therapy, but not the RT/αPD-1/IDO1i triple therapy
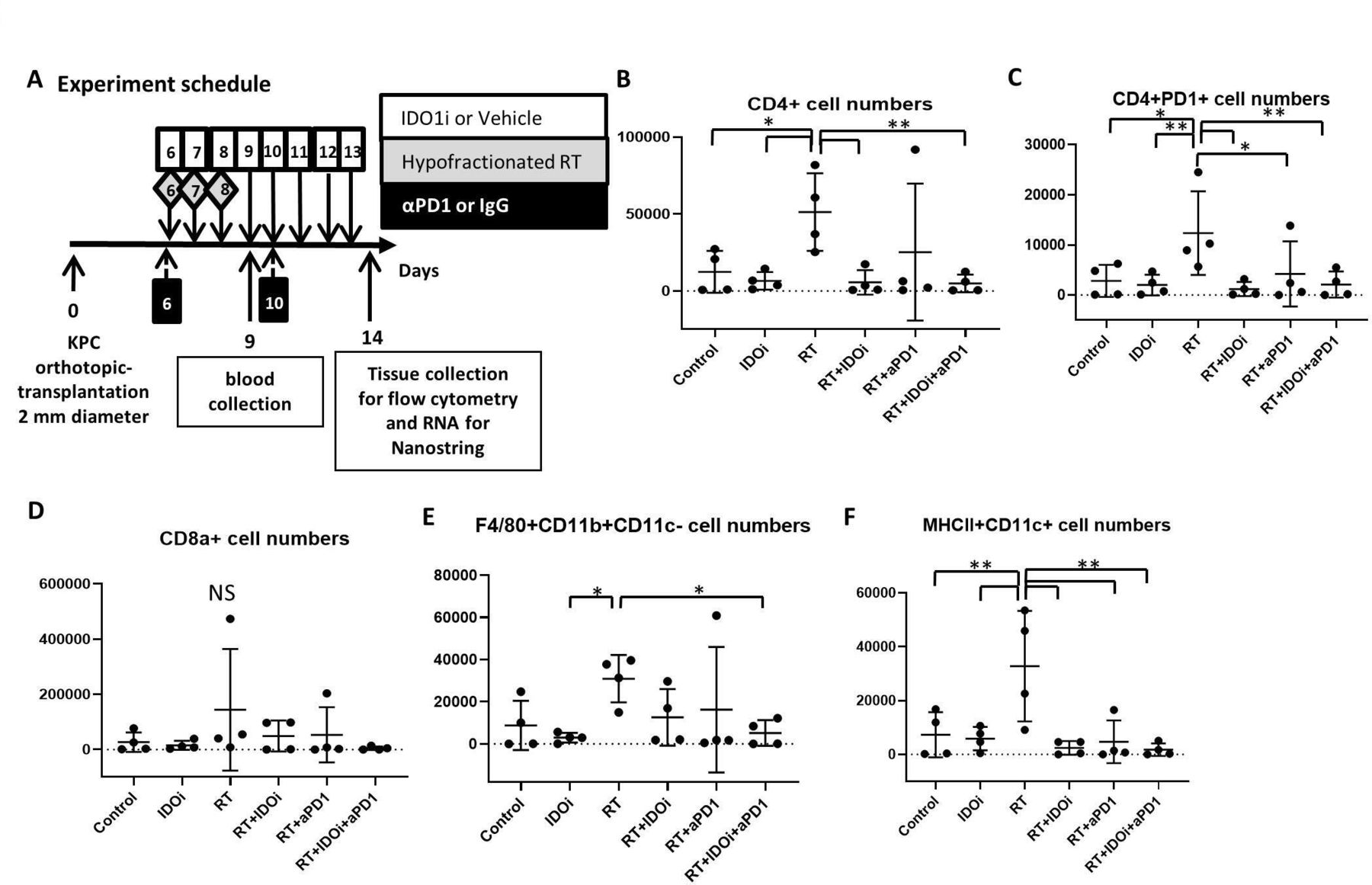
We thus analyzed the serum from the blood obtained 1 day after the completion of RT in order to evaluate the effect of RT-immunotherapy combo treatments (figure 2A). A comprehensive panel of 44 cytokines that are known to be pivotal for either antitumor or protumoral immune response (online supplementary table S1) were analyzed using a mouse-specific Luminex panel. Among these cytokines, interferon-gamma (IFN-γ), reported as being associated with the tumor suppression by RT, was only increased with RT/αPD-1 (figure 3A).40 Proinflammatory cytokine interleukin-22 (IL-22) increased significantly in three groups, RT, RT/αPD-1 and RT/IDO1i, and IL-22 was not significantly increased in the RT/αPD-1/IDO1i triple therapy compared with the vehicle control group (figure 3B). Analysis of other cytokines demonstrated that IL-5 was only increased with RT/αPD-1 and soluble CD40 ligand was only increased with IDOi alone (figure 3C,D). Interestingly, none of the 44 cytokines were increased in the RT/αPD-1/IDO1i triple therapy compared with vehicle control. As the cytokine changes represent the systemic response to the RT-immunotherapy combinations, this result may provide a clue on why there is worse survival in the RT/αPD-1/IDO1i group compared with RT/αPD-1.

Systemic IFN-γ was significantly induced by the RT/αPD-1 double therapy, but not the RT/αPD-1/IDO1i triple therapy. The blood serum collected from KPC orthotopic implantation model treated with combinational therapy was analyzed by Luminex cytokine profile. (A) IFN-γ, (B) IL-22, (C) IL-5, (D) sCD40L. Data represent mean±SEM of four mice per treatment group that was repeated twice. *P<0.05; NS, not significant, by unpaired t-est and one-way ANOVA. ANOVA, analysis of variance; IDO1, indoleamine 2,3-dioxygenase 1; IFN-γ, interferon-γ; IL-22, interleukin-22; RT, radiation therapy.
RNA profiling confirmed immune-modulatory effects of radiation and the induction of immune-activation pathways by the RT/αPD-1 double therapy
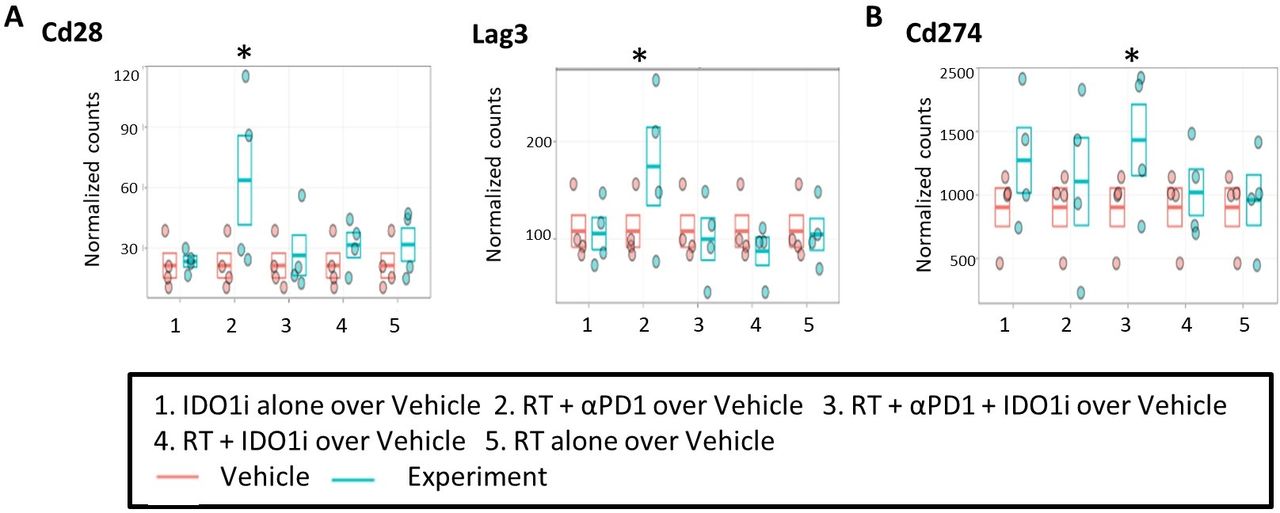
We used NanoString to analyze the RNA extracted from the mouse tumors collected on day 14 (figure 2A). The expressions of Cd28 and Lag3 were increased significantly in the RT/αPD-1 group compared with vehicle control (figure 4A). The expressions of Pdcd1, Icos and Tigit were also increased in the RT/αPD-1 group compared with vehicle control, but those were not statistically significant (online supplementary figure S4A). Cd274 expression only significantly increased in the triple therapy group compared with vehicle control (figure 4B). All these genes are known to be induced during the T cell activation process; and their induction may explain why RT/αPD-1 is associated with both the greatest tumor growth suppression and the best survival. We found that any treatment group receiving RT led to increased expression of Tlr7, Tlr9, Nlrp3, Ccr3, Ccr7, Tnfrsf4, Tnfrsf18, Tgf-b1, Pdcd1lg2, Ctla4, Havcr2, Irak3 and Sting and decreased expression of Tlr3 compared with vehicle control, although those were not statistically significant (online supplementary figure S4B). These results may suggest the immune-modulatory activities of RT, particularly its role in mediating innate immune responses by activating specific pattern recognition receptors, such as Tlr7, Tlr9 and Nlrp3, proinflammatory chemokine receptor Ccr3 and T cell trafficking chemokine receptor Ccr7. On the other hand, RT also induced certain immune activation pathway genes, Tnfrsf4 and Tnfrsf18, as well as immune-suppressive pathway genes, Tgf-b1, Pdcd1lg2, Havcr2 and Irak3. Ido1 expression decreased among all groups that contained RT except for the RT/αPD-1/IDO1i triple therapy group compared with vehicle control, but it was not statistically significant (online supplementary figure S4C). Overall, RT/αPD-1 is associated with the best activation of innate and adaptive immune responses locally in the TME. Nevertheless, the results of the Nanostring study warrant further validation to identify the biomarkers to guide the appropriate immune-modulatory agents to be combined with RT.
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
RNA profiling confirmed immune modulatory effects of radiation and the induction of immune-activation pathways by the combination therapy with radiation and anti-PD-1 antibody. The RNA derived from the tissues of KPC orthotopic implantation model treated with combinational therapy was analyzed by NanoString Analysis. The data showed the comparison of (A) Cd28, Lag3, and (B) Cd274 expression in the five treatment groups compared with the control group. n=4 for each treatment group. *False discovery rate (q=30%). IDO1, indoleamine 2,3-dioxygenase 1 inhibitor; RT, radiation therapy.
Discussion
To our knowledge, this is one of the first few studies investigating the effects of RT with IDO1i and/or αPD-1 combination therapy for the treatment of pancreatic cancer, particularly in a pancreatic orthotopic tumor model. Although the combination of RT and anti-PD-1 antibody is being tested in multiple clinical trials of PDAC, none of these clinical trials have reached any conclusion yet.12 Recently, several murine pancreatic orthotopic tumor models of PDAC were developed.41 42 The major differences among these models lie in how to guide the RT treatment more specifically to the tumor field. In one published study, tumors were visualized for RT via laparotomy and injection of cancer cells with lipiodol into the pancreas41; and in another published study, contrast reagent was injected into the abdomen of the mouse to enhance the shape of the pancreas to guide the RT treatment.42 In this study, we have developed a syngeneic pancreatic orthotopic implantation model by guiding the RT treatment more specifically to the tumor field within the pancreas by using the internally placed surgical clips. This RT model highly resembles stereotactic body radiation in the clinical practice.43 With this new RT preclinical model, our study showed that the combination therapy of αPD-1 and RT improved survival. The combination of RT/IDO1i did not improve survival compared with RT/αPD-1 although these two RT-based immunotherapy combinations offer similar local tumor growth control. In addition, the triple combination therapy of RT/αPD-1/IDO1i did not improve survival compared with RT/αPD-1, and in fact, trended toward worsening survival compared with RT/αPD-1. This study has demonstrated for the first time the immune-modulatory effects of RT in a preclinical model of PDAC. Our immune analysis found that triple therapy significantly suppressed the tumor infiltration of CD4 +T cells and TAMs induced by RT. However, this study also showed the complexity of the local and systemic effects of the RT-immunotherapy combinations. Understanding the immune-modulatory effects of RT in different combinations with immunotherapy provides the rationale for the optimal combination of RT-immunotherapy.
Previously, improved antitumor efficacy with combination therapies RT with αPD-1 and/or IDO1i has been shown for the treatment of non-PDAC malignancies.12 13 29 44 45 Our study confirms that RT/αPD-1 also improves survival in PDACs, but the addition of IDO1i to this already effective dual-therapy may be detrimental to survival. This result is similar to our previous study of triple combination therapy of a cancer vaccine, αPD-1, and IDO1i in a PDAC murine model.30 In this published study, IDO1i enhanced the antitumor efficacy of the cancer vaccine, but the addition of anti-PD-L1 antibody to the vaccine/IDO1i combination did not improve survival, and in fact, suppressed the tumor infiltration of CD4 +and CD8+T cells by inducing apoptosis of CD8 +T cells. A similar mechanism may exist in the current study to explain the less effectiveness of the RT/αPD-1/IDO1i triple therapy and remains to be explored. In addition, the current study showed that both RT/αPD-1 and RT/IDO1i dual therapies modulated certain immune pathways and the RT/αPD-1/IDO1i triple therapy failed to further modulate those pathways. Different from the prior study, the current study showed that RT/αPD-1 is the best RT-immunotherapy combination in the preclinical model of PDAC.
Among the different RT-immunotherapy combinations tested in this study, RT/αPD-1 appeared to result in the best systemic IFN-γ response and the highest local expression of immune-activation genes, including Cd28 and Icos. Liang et al reported that IFN-γ is associated with the tumor suppression by RT.40 These findings could partially explain the best survival with RT/αPD-1. RT/αPD-1 also resulted in systemic induction of IL-5, whose role in antitumor immune response remains to be explored. Therefore, our study supported that RT/αPD-1 leads to a systemic effect. This possibility deserves further investigation with an appropriate tumor model to study both primary tumor and distant metastases simultaneously.
As anticipated, RT, regardless of the immunotherapy combination, appeared to induce innate immune responses locally in the PDAC tumors by activating specific pattern recognition receptors such as Tlr7, Tlr9 and Nlrp3, proinflammatory chemokine receptor Ccr3, IL-1 receptor-associated kinase Irak3, and T cell trafficking chemokine receptor Ccr7, although those genes did not show significant difference. However, in the adaptive immune-response pathways, RT appears to be double edge by inducing certain immune-activation pathway genes, including Tnfrsf4 and Tnfrsf18, as well as immune-suppressive pathway genes, including Tgf-b1, Pdcd1lg2 and Havcr2. These results warrant further validations to identify the biomarkers to guide the appropriate immune-modulatory agents to be combined with RT to tip the balance toward antitumor adaptive immune responses.
This study found that intratumoral CD8 +T cells remained scarce among all of the treatment groups. This result suggests RT or RT-immunotherapy combinations do not induce CD8 +T cell infiltration in the PDACs. However, It has been well established that immune cells are indispensable for the anti-tumor effect of RT.10 46–48 It is possible that non-CD8 + immune cells such as CD4041 and/or the functionality of CD8 +T cells have played a role in sensitizing the tumors for RT. Whether αPD-1 or IDO1i can make the PDAC tumors more sensitive to RT and whether RT can enhance the immune response to αPD-1 and IDOi warrant further investigation.
We acknowledge that the preclinical RT model developed in this study has its limitations. First, the radiation field on the pancreatic tumor may not be homogenous. Second, the clips that are placed surrounding the implanted tumors may also induce inflammatory responses. Third, the tumors are implanted, but not spontaneously developed. Fourth, the surgery performed just days before the start of treatment may still influence the results. Fifth, the sample size in each treatment group within one individual experiment was kept small due to the large time consumption of ultrasound examination. However, each experiment has been repeated at least twice and was found to be reproducible. Some mice may die from metastases which may have become a confounding factor when the sizes of the primary pancreatic tumors were compared. Despite these limitations, this model represents the first syngeneic orthotopic preclinical model of PDAC for RT-immunotherapy. This model can be used for further testing RT with other immunotherapy agents and provides the strong rationale for bringing the RT-immunotherapy combination to clinical trial testing. The results from this preclinical study will need to be further validated in human patient studies.
References
Footnotes
Contributors KF, MTS, HJ, LC and LZ designed the research studies. KF, MTS, HJ, LC, ABB, DD, MW, JB, NC, JK and LZ developed methodology. KF, MTS, HJ, MZ, SM, AW, EB, LC, KL, NJ, ABB, MW, JB, NC and JK acquired data. KF, MTS, BH, ABB, DD, MW, JB, NC, JK and LZ analyzed and interpreted data. KF, MTS, AW, EB, LC, MW, JB, NC, JK and LZ wrote, reviewed and/or revised the manuscript. MW and LZ supervised the study.
Funding This study was funded by BMS II-ON grant (LZ), NIH grant R01 CA169702 (LZ), NIH grant R01 CA197296 (LZ), The Viragh Foundation and the Skip Viragh Pancreatic Cancer Center at Johns Hopkins (LZ), National Cancer Institute Specialized Programs of Research Excellence in Gastrointestinal Cancers grant P50 CA062924 (LZ), Sidney Kimmel Comprehensive Cancer Center grant P30 CA006973 (LZ).
Competing interests LZ receives grant support from Bristol-Meyer Squibb, Merck, iTeos, Amgen, NovaRock, Inxmed, and Halozyme, and received the royalty for licensing GVAX to Aduro Biotech. LZ is a paid consultant/Advisory Board Member at Biosion, Alphamab, NovaRock, Akrevia, Sound Biologics, FusunBiopharmaceutical, Foundation Medicine, Datarevive, and Mingruzhiyao. LZ holds shares at Alphamab and Mingruzhiyao.
Patient consent for publication Not required.
Ethics approval The Johns Hopkins Medical Institution (JHMI) Institutional Review Board (IRB) approved protocol (NA_00074221). All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request.