Article Text

Abstract
Accumulation of extracellular adenosine within the microenvironment is a strategy exploited by tumors to escape detection by the immune system. Adenosine signaling through the adenosine 2A receptor (A2AR) on immune cells elicits a range of immunosuppressive effects which promote tumor growth and limit the efficacy of immune checkpoint inhibitors. Preclinical data with A2AR inhibitors have demonstrated tumor regressions in mouse models by rescuing T cell function; however, the mechanism and role on other immune cells has not been fully elucidated.
Methods We report here the development of a small molecule A2AR inhibitor including characterization of binding and inhibition of A2AR function with varying amounts of a stable version of adenosine. Functional activity was tested in both mouse and human T cells and dendritic cells (DCs) in in vitro assays to understand the intrinsic role on each cell type. The role of adenosine and A2AR inhibition was tested in DC differentiation assays as well as co-culture assays to access the cross-priming function of DCs. Syngeneic models were used to assess tumor growth alone and in combination with alphaprogrammed death-ligand 1 (αPD-L1). Immunophenotyping by flow cytometry was performed to examine global immune cell changes upon A2AR inhibition.
Results We provide the first report of AZD4635, a novel small molecule A2AR antagonist which inhibits downstream signaling and increases T cell function as well as a novel mechanism of enhancing antigen presentation by CD103+ DCs. The role of antigen presentation by DCs, particularly CD103+ DCs, is critical to drive antitumor immunity providing rational to combine a priming agent AZD4635 with check point blockade. We find adenosine impairs the maturation and antigen presentation function of CD103+ DCs. We show in multiple syngeneic mouse tumor models that treatment of AZD4635 alone and in combination with αPD-L1 led to decreased tumor volume correlating with enhanced CD103+ function and T cell response. We extend these studies into human DCs to show that adenosine promotes a tolerogenic phenotype that can be reversed with AZD4635 restoring antigen-specific T cell activation. Our results support the novel role of adenosine signaling as an intrinsic negative regulator of CD103+ DCs maturation and priming. We show that potent inhibition of A2AR with AZD4635 reduces tumor burden and enhances antitumor immunity. This unique mechanism of action in CD103+ DCs may contribute to clinical responses as AZD4635 is being evaluated in clinical trials with IMFINZI (durvalumab, αPD-L1) in patients with solid malignancies.
Conclusion We provide evidence implicating suppression of adaptive and innate immunity by adenosine as a mechanism for immune evasion by tumors. Inhibition of adenosine signaling through selective small molecule inhibition of A2AR using AZD4635 restores T cell function via an internal mechanism as well as tumor antigen cross-presentation by CD103+ DCs resulting in antitumor immunity.
- tumors
- oncology
- pharmacology
- adaptive immunity
- adenosine
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Advances in the development of immune checkpoint inhibitors such as antiprogrammed death-ligand 1 (anti-PD-L1) and anticytotoxic T-lymphocyte-associated protein 4 have led to unprecedented responses in patients with advanced-stage malignancies. However, not all patients benefit from currently approved inhibitors, leaving many patients in need of alternative therapies. Efforts have therefore focused on the development of next-generation immune-modulating agents to enhance clinical response rates and extend long-term survival. Accumulation of extracellular adenosine within the tumor microenvironment has been identified as a key mechanism exploited by tumors to escape immune surveillance.1–3
Adenosine is a purine nucleoside and binds to four cell surface receptors (A1, A2A, A2B and A3) that are expressed on multiple immune subsets including T cells, natural killer (NK) cells, macrophages and dendritic cells (DCs).4 5 Extracellular adenosine accumulates within the tumor microenvironment through the coordinated activity of nucleosidases CD39 and CD73 on the cell surface, which catabolize extracellular ATP generated during cell stress, cell turnover and hypoxia. Although normally expressed on stromal cells and certain leukocytes, CD73 expression is also expressed at high levels on tumor cells in some cancers, and has been shown to be a negative prognostic factor in several types of cancer including gastric, colorectal and triple negative breast cancer.6–8 Although efforts to quantify intratumoral adenosine levels have been challenging due to the extremely short half-life9 of adenosine, studies using microdialysis probes in solid tumors suggest that adenosine levels are higher than in adjacent normal tissues and a gradient of adenosine exists,10 although radio-labeled adenosine as a standard is required to accurately quantitate adenosine levels. Adenosine signaling through the high-affinity adenosine 2A receptor (A2AR) on antigen-presenting cells and lymphocytes elicits a range of immunosuppressive effects including suppression of antitumor activity of cytotoxic CD8+ T cells,11–14 induction of immune suppressive cytokines by DCs11 12 and inhibition of antigen presentation by DCs.11 13
The non-redundant role of adenosine-A2AR on immune regulation and antitumor responses became evident with the in vivo use of A2a and A2a/A2b antagonists or genetic elimination of A2AR in T cells which liberated CD8+ tumor-reactive T cells from tumor-induced immunosuppression.14 These studies demonstrate that A2AR-deficient mice spontaneously regress allografted tumors through a CD8+ T cell-mediated response, leading to durable tumor regressions.2 Additionally, reduction of adenosine levels through the inhibition of CD39/CD73 activity with targeted antibodies has been shown to induce regression in preclinical models of breast cancer, colorectal cancer and melanoma.15–18 A recent report of small molecule inhibition of A2AR has shown restoration of T cell function contributing to antitumor immunity in syngeneic tumor models, however the effect on other immune cell types was not addressed.19
DCs are critical for the initiation of adaptive immune responses and are therefore sentinels of the immune system. In murine models, a rare subset of basic leucine zipper transcription factor (Batf3)-dependent DCs termed CD103+ DCs are critical for cross-presentation of tumor antigens enhancing cytotoxic T lymphocyte (CTL) activity tumor growth control.20 21 Recent studies have implicated the requirement of intratumoral CD103+ DCs for tumor-reactive effector T cell recruitment through CXCL9 and CXCL10.20 Tumor intrinsic mechanisms such as oncogenic signaling through β-catenin in melanoma have been found to suppress the recruitment of intratumoral CD103+ DCs preventing tumor antigen T cell recruitment and priming.20 In humans, the transcriptional signatures of the homologous population CD141+ DCs were found to be elevated in cancers, correlating with better clinical outcome18 as well as with responsiveness to anti-PD-1 immunotherapy.19
Here, we describe the characterization of AZD4635, a novel, orally bioavailable A2AR antagonist in its effect on antitumor immunity alone as single agent and in combination with an anti-PD-L1 antibody. In several syngeneic mouse tumor models, AZD4635 reduced tumor burden and enhanced antitumor immunity alone and in combination with anti-PD-L1. Functional assays demonstrate that AZD4635 treatment leads to a broad spectrum of immune modulation, including increase in T cells, importantly tumor antigen-specific CD8+and CD4+ T cells, as well as T cell activation and proliferation. Furthermore, AZD4635 reverses adenosine-mediated immune suppression in T cells, it also reactivates CD103+ DCs a unique finding. In vivo studies also support that increases in antigen-specific immunity contribute to tumor efficacy with AZD4635 in combination with PD-L1 checkpoint blockade. To our knowledge, this is the first report implicating A2AR blockade in rescuing the cross-priming function of CD103+ DCs, providing a novel mechanism of action for adenosine signaling blockade. Collectively, our data demonstrate that A2AR inhibition could reverse the immune suppressive mechanism by adenosine, leading to enhanced antitumor immunity. Our preclinical evidence supports the development of AZD4635 as a single agent and in combination with durvalumab (anti-PD-L1 Ab) in patients with solid malignancies which is currently in clinical trials (https://www.clinicaltrials.gov/ct2/show/NCT02740985?term=AZD4635&rank=3).
Materials and methods
Radioligand binding competition assay
The binding affinity of AZD4635 was determined for human A1, A2A, A2B and A3 receptors using transfected Chinese hamster ovary (CHO; hA1R), and HEK-293T (hA2AR, hA2BR and hA3R) cells (Eurofins Cerep). A radioligand binding assay was performed using eight agonist concentrations ranging from 30 nM to 100 µM. The inhibition constant (Ki) of each compound was calculated by the Cheng-Prusoff equation: Ki=IC50/(1+[L]/Kd), where IC50 is the concentration of compound that displaces the binding of radioligand by 50%, L is concentration of radioligand in the assay and Kd is affinity of the radioligand for the receptor. IC50 values were determined by non-linear regression analysis of the competition curves generated with mean replicate values using Hill equation curve fitting.
StaR generation
The thermostabilization of the human A2A receptor (resulting in A2A-StaR2) using a mutagenesis approach,22 has been previously described.23
Expression, membrane preparation and protein purification
The truncated A2A-StaR2-b RIL562 construct has been described previously24 and harbors eight thermostabilizing mutations (A54L2.52, T88A3.36, R107A3.55, K122A4.43, L202A5.63, L235A6.37, V239A6.41 and S277A7.42) as well as a mutation to remove a glycosylation site (N154A). The construct further comprises an apocytochrome b RIL562 fusion between transmembrane helices 5 and 6 and a C-terminal decahistidine tag. The receptor was expressed using the Bac to Bac Expression System (Invitrogen) in Trichoplusia ni Tni PRO cells using ESF 921 medium (Expression Systems) supplemented with 5% (v/v) fetal bovine serum (Sigma-Aldrich) and 1% (v/v) penicillin/streptomycin (PAA Laboratories). Cells were infected at a density of 2.6×106 cells/mL with virus at an approximate multiplicity of infection of 1. Cultures were grown at 27°C with constant shaking and harvested by centrifugation 48 hours postinfection. All subsequent protein purification steps were carried out at 4°C unless otherwise stated. For each protein preparation, cells from 2 L cultures were resuspended in 40 mM TRIS buffer at pH 7.6 supplemented by 1 mM EDTA and Complete EDTA-free protease inhibitor cocktail tablets (Roche). Cells were disrupted at ~15 000 psi using a microfluidizer (Processor M-110L Pneumatic, Microfluidics). Membranes pelleted by ultracentrifugation at 200,000 g for 50 min, were subjected to a high salt wash in a buffer containing 40 mM Tris pH 7.6, 1 M NaCl and Complete EDTA-free protease inhibitor cocktail tablets, before they were centrifuged at 200,000 g for 50 min. Washed membranes were resuspended in 50 mL 40 mM Tris pH 7.6 supplemented with 10 µM AZD4635 and Complete EDTA-free protease inhibitor cocktail tablets and stored at −80°C until further use. Membranes were thawed, resuspended in a total volume of 150 mL with 40 mM Tris-HCl pH 7.6, Complete EDTA-free protease inhibitor cocktail tablets (Roche), 20 µM AZD4635 and incubated for 2 hours at room temperature. Membranes were then solubilized by addition of 1.5% n-Decyl-β-D-maltopyranoside (DM, Anatrace), and incubation for 2 hours at 4°C, followed by centrifugation at 145,000 g for 60 min to harvest solubilized material. The solubilized material was applied to a 5 mL nickel-nitrilotriacetic acid Superflow cartridge (Qiagen) pre-equilibrated in 40 mM Tris pH 7.4, 200 mM NaCl, 0.15% DM, 10 µM AZD4635. The column was washed with 25 column volumes of buffer 40 mM Tris pH 7.4, 200 mM NaCl, 0.15% DM, 70 mM imidazole, 10 µM AZD4635 and then the protein was eluted with 40 mM Tris pH 7.4, 200 mM NaCl, 0.15% DM, 280 mM imidazole, 10 µM AZD4635. Collected fractions were analyzed by SDS-PAGE and fractions containing A2 a -StaR2-b RIL562 were pooled and concentrated using an Amicon Ultra Ultracell 50K ultrafiltration membrane to a final volume of ~800 µL. The protein sample was ultracentrifuged at 436,000 g for 10 min before being applied to a Superdex200 size exclusion column (GE Healthcare) pre-equilibrated with 40 mM Tris pH 7.4, 200 mM NaCl, 0.15% DM, 10 µM AZD4635. Eluted fractions containing the protein were analyzed by SDS-PAGE, pooled and concentrated to ~35 mg/mL using an Amicon Ultra Ultracell 50K ultrafiltration membrane and subjected to an ultracentrifugation at 436,000 g prior to crystallization. Protein concentrations were measured using the DC assay (Bio-Rad), and confirmed using quantitative amino acid analysis.
Crystallization
The A2A-StaR2-b RIL562 in complex with AZD4635 was crystallized in lipidic cubic phase at 20°C. Concentrated protein was mixed with monoolein (Nu-Chek) supplemented with 10% (w/w) cholesterol (Sigma-Aldrich) and 10 µM AZD4635 using the twin-syringe method.25 The final protein:lipid ratio was 40:60 (w/w); 40 nL boli were dispensed onto 96-well Laminex Glass Bases (Molecular Dimensions) using a Mosquito LCP crystallization robot (TTP Labtech) and overlaid with 800 nL precipitant solution. Glass bases were sealed using Laminex Film covers (Molecular Dimensions); 60–80 µm long plate-shaped crystals grew within 2 weeks in 0.1 M tri-sodium citrate pH 5.3–5.4, 0.05 M sodium thiocyanate, 29%–32% PEG400, 2% (v/v) 2,5-hexanediol and 10 µM AZD4635.
Diffraction data collection and processing
X-ray diffraction data were measured on a Pilatus 6M detector at beamline I24 (Diamond Light Source) using a 6×9 µm beam size of for crystals of A2A-StaR2-b RIL562 in complex with AZD4635. A complete dataset was acquired using data from three crystals and using an unattenuated beam and 0.2° oscillation per frame, with an exposure of 0.1 s per degree of oscillation. Data were integrated using XDS 26 merged and scaled using AIMLESS 27 from the CCP4 suite.28 Data collection statistics are reported in table 1.
Data collection and refinement statistics
Structure solution and refinement
The structure of A2A-StaR2-b RIL562-AZD4635 was solved by molecular replacement (MR) with Phaser 29 using the A2A-StaR2-b RIL562-theophylline complex structure as the search model (PDB code: 5MZJ). Iterative rounds of model refinement performed using phenix.refine,30 were interspersed with manual model building in COOT.31 Both X-ray and B-factor restraint weights were optimized in phenix.refine, and 2 TLS groups corresponding to the receptor and to the b RIL562 respectively were defined during refinement. Refinement was with positional and individual isotropic B-factor refinement. The final models were validated using MolProbity.32 The final refinement statistics are presented in table 1. Structure figures were generated using PyMOL.33
cAMP accumulation inhibition assay
pIRES_neo3 expression vectors were generated for human A2AR (UniProt P29274) or mouse A2AR (UniProt Q60613) modified with an N-terminal preprolactin signal peptide to aid functional expression (plasmid sequences available on request); 6 µg of plasmid was transfected into CHO-K1 cells using PEI transfection agent, and single cell clones were selected over 22 days in the presence of 0.5 mg/mL G418 (Sigma, G8168). Selected single cell clones for human A2AR (CHO-K1 PPL-SP-hA2AR) and mouse A2AR (CHO-K1 PPL-SP-mA2AR) were cultured in Ham’s F12 medium supplemented with 0.5 mg/mL G418. Cells were harvested to a seeding density of 4×105 cells/mL in Ham’s F12 media in the absence of G418, supplemented with 0.2 U/mL adenosine deaminase (Sigma, 52544) and 100 µL/well seeded into 96-well plates. Following overnight incubation (18 hours), the monolayer was washed twice with phosphate-buffered saline (PBS), then washed once in Krebbs-Ringer Bicarbonate Buffer (KRBG; Sigma) to remove adenosine deaminase. Cells were incubated in 40 µM Rolipram (Sigma) prepared in KRBG for 10 min at room temperature before addition of a concentration response of test antagonist. Following 10 min incubation under tissue culture conditions, the indicated concentrations of adenosine (Sigma, A9251) were added for 10 min under tissue culture conditions. Media was removed and cells lysed in Cell Lysis Buffer (Molecular Devices). cAMP was quantified using the CatchPoint Cyclic-AMP Fluorescent Assay Kit (Molecular Devices) following manufacturer’s instructions.
Animal studies
All animal studies were performed according to AstraZeneca Institutional Animal Care and Use Committee guidelines. B16F10-OVA, MC38, MC38-OVA and MCA205 cells were confirmed free of mycoplasma and mouse pathogens by PCR as part of a rodent pathogen testing panel (IMPACT, IDEXX Bioresearch). Thawed cells were cultured in DMEM (B16F10-OVA, MC38, MC38-OVA) or RPMI 1640 (MCA205) supplemented with 10% heat-inactivated fetal bovine serum and 1% L-glutamine (Sigma-Aldrich) at 37°C in a humidified incubator maintained at 5% CO2. Cell viability counts were determined by trypan blue exclusion counting prior to implantation using an automated counter (Nexcelom) prior to implantation. For subcutaneous implants, B16F10-OVA (5×105 cells/mouse), MC38/MC38-OVA (5×105 cells/mouse) and MCA205 cells (1×106 cells/mouse) were re-suspended in sterile PBS and injected subcutaneously into the right flanks of female C57BL/6 mice aged 6–8 weeks (B16F10-OVA, MC38, MC38-OVA, MCA205) or NOD-scid IL2Rgnull (NSG) mice (Jackson Labs) in a total volume of 0.1 mL/mouse. For in vivo treatment studies, mice were randomized into treatment groups at a starting tumor volume of 50–90 mm3, 80–100 mm3 for MC38-OVA. AZD4635 nanosuspension formulation (Aptuit Verona) was reconstituted in sterile water and dosed orally twice daily at 50 mg/kg. Mouse-antimouse PD-L1 (clone 80, mIgG1 D265A, AZ) and isotype control antibody (NIP228 mIgG1 D265A, AZ) were resuspended in sterile PBS and dosed intraperitoneally at 10 mg/kg twice weekly. Tumor volume and body weight were measured twice weekly after randomization. Tumor growth inhibition (%TGI) was determined during the dosing period using the formula: %TGI=(1–[Tt/T0/Ct/C0]/1–[C0/Ct])×100, where Tt=median tumor volume of treated at time t, T0=median tumor volume of treated at time 0, Ct=median tumor volume of control at time t and C0=median tumor volume of control at time 0. For disseminated tumor implants, mice were injected intravenously with MCA205 cells (5×105 cells/mouse) in a total volume of 0.1 mL/mouse. Mice were treated daily with AZD4635 (50 mg/kg twice daily) and anti-PD-L1 (clone 80, mIgG1 D265A, AstraZeneca; 10 mg/kg intraperitoneally twice weekly) as indicated from day 0. At 15 days after injection of tumor cells, lungs were harvested and tumor nodules counted under a dissecting microscope. In addition to the tumor efficacy studies, OT-I transgenic mice (C57BL/6-Tg(TcraTcrb)1100Mjb/J) were sourced from Jackson Labs (cat# 003831) for OVA/Kb-SIINFEKL antigen-specific assays requiring splenic CD8+ T cells. Additionally, bone marrow was aseptically harvested from female C57BL/6 mice aged 8 weeks for in vitro assays where indicated. To investigate role of CD8 T cells in AZD4635+PD-L1 treatment, MC38-OVA tumor-bearing C57BL/6 mice were injected with either rat-IgG2b isotype control (cat# BP0090, Bioxcell, Lebanon, New Hampshire, USA) or antimouse CD8 antibody (cat# BE0061, Bioxcell) every 3 days and treated with AZD4635 and anti-PD-L1 as described above.
Tumor immunophenotyping flow cytometric analysis
Following 14 days of dosing, tumors were harvested for PD immunophenotyping, immune cell sorting and ex vivo stimulation following dissociation using the Miltenyi Mouse Tumor Disassociation Kit (Miltenyi Biotec). Following RBC lysis with ACK lysis buffer (ThermoFisher Scientific, A1049201), cells were blocked with purified antimouse CD16/CD32 for FcR blocking and stained with Live/Dead Blue (Life Technologies; L23105), and antibodies in flow cytometry staining buffer (ThermoFisher Scientific cat# 00-4222-26), as indicated in the online supplementary methods. Non-specific antibody binding blocking for intracellular staining procedures included unconjugated matched species normal serum (2% in 1×permeabilization buffer). Samples were acquired using a LSRFortessa SORP flow cytometry system with FACSDiva software (BD Biosciences for both), and data were analyzed using FlowJo software V.10.5.3. Panel-specific FMO controls and single color controls were prepared for each experiment For MC38-OVA ex vivo tumor-infiltrating lymphocyte (TIL) analysis, as an adjunct to conventional gating in two out of four studies, T-distributed stochastic neighbor embedding (tSNE) analysis was performed as follows by two independent analysts (CMB, LBP): an opt-SNE dimensionality reduction was performed using FlowJo V.10.5.3 with the described plugin enabled.34 35 The following parameters were used: input of 1,168,929 single, live, CD45+ events; clustering channels (all biexponentially transformed) for Side Scatter and antibodies from the flow cytometry panel described in the online supplementary methods, set for a maximum total number of 1000 iterations, learning rate 81825, perplexity 30.
Supplemental material
Ex vivo stimulation cytokine flow cytometric analysis from tumors
For ex vivo cytokine production analysis single cells suspensions of tumors were prepared using the Miltenyi Mouse Tumor Disassociation Kit (Miltenyi Biotec) and were plated in flat-bottomed, TC-treated 96-well plates at 2-4e5 cells per well based on live counts in a final volume of 200 µL/well complete T cell medium. eBioscience Cell Stimulation Cocktail (1×) (ThermoFisher Scientific cat# 00-4975-03), containing phorbol myristate acetate (PMA) and ionomycin plus the protein transport inhibitors, monensin and brefeldin A, or dimethyl sulfoxide (DMSO) as a control, and anti-mouse CD107a antibody, was added for 12 hours incubation at 37°C/ 5% CO2 atmosphere. Cells were then collected, washed and intracellularly stained for accumulated cytokines using fluorescently conjugated antibodies per the panel detailed in online supplementary methods in conjunction with components from the BD Bioscience CytoFix/Cyto Perm Solution Kit (cat# 554714) per the manufacturer’s instructions.
ELISpot assay
Lymph node (LN) cells were isolated from mice from different treatment groups (7 days post-treatment) and analyzed for tumor antigen-specific CD8 T cell responses by ELISpot assay. Briefly, 50,000 LN cells were co-cultured with 50,000 CD3 cell-depleted spleen cells and stimulated with 1 μg/mL of OVA peptide (SIINFEKL) or p15E peptide (KSPWFTTL) for 72 hours (MBL International, Woburn, Massachusetts, USA). After the 72 hours, the frequency of interferon (IFN)-γ-producing cells was determined by ELISpot according to standard protocols (R&D Systems, Minneapolis, Minnesota, USA) using an ELISpot reader (CTL Immunospot S4 analyzer, Cellular Technology, Cleveland, Ohio, USA).
Gene expression analysis
RNA was extracted from snap frozen tumor samples using the RNeasy Mini kit (Qiagen cat# 74104) according to manufacturer’s instructions. A multiplex gene expression array with 770 genes from 24 different immune cell types, common checkpoint inhibitors, CT antigens and genes covering both the adaptive and innate immune response (PanCancer Mouse Immune Profiling Panel) was performed using the nCounter MAX Analysis System. Raw counts were normalized using nSolver V.4.0 software (Nanostring Technologies). T test was performed on normalized and log2 transformed gene expression between ‘Vehicle group’ and ‘AZD4635 treatment group’. Significantly expressed genes were identified using p value cut-off of 0.05 and fold change of 2. Hierarchical clustering was then performed across the samples in ‘Vehicle group’ and ‘AZD4635 treatment group’.
CD103+ DC mouse bone marrow differentiation cultures
The generation of CD11c+ CD103+ CLEC9A+ DC was described previously.36 Briefly, bone marrow was aseptically isolated from femurs and tibiae of untreated C57BL/6 mice and cultured in complete T cell medium for 16 days with the addition of 200 ng/mL rmFLT3L and 5 ng/mL rmGM-CSF (R&D Systems cat# 427-FL-005 and cat# 415 ML, respectively). Where indicated, after 10 days of initial culture with growth cytokines, AZD4635 at 3 µM, or DMSO as control was added and allowed to incubate for 30 min prior to addition of 5 µM 5′-(N-ethylcarboxamido) adenosine (NECA) (Tocris). The cultures were then continued with growth cytokines for the remaining 6 days prior to cell harvest. Immature dendritic cells (iDC) were induced to a mature phenotype with 1 µg/mL lipopolysaccharide (InvivoGen cat# tlrl-eblps) for 24 hours. Both iDC and mature DC were used for experiments where indicated. Before use in subsequent assays such as T cell co-cultures, DC were washed with Dulbecco’s PBS (DPBS), harvested with Accutase (Sigma cat# A6964) and then washed and resuspended in complete T cell media or flow cytometry staining buffer (ThermoFisher Scientific cat# 00-4222-26).
Pinocytosis assay
The pinocytic activity of iDCs was assessed by uptake of Alexa 488-labeled ovalbumin (ThermoFisher Scientific cat# O34781). 1e6 iDC were resuspended in 500 µL of complete T cell medium with fluorescent ovalbumin added at a final concentration of 50 µg/mL and incubated for 30 min at 37°C, or at 4°C as negative control. Cells were then washed twice with ice-cold DPBS and kept chilled until flow cytometric analysis.
Analysis of antigen presentation
CD103+ iDC cultures, washed free of NECA and compounds, were incubated with endotoxin-free ovalbumin protein (InvivoGen cat# vac-pova) at a final concentration of 1 mg/mL overnight prior to maturation. Where SIINFEKL/OVA 257–264 peptide (InvivoGen cat# vac-sin) pulsing conditions were used, peptide was added at 1 µg/mL 2–4 hours prior to maturation stimuli addition. Antigen presentation was then assessed flow cytometrically using clone 25-D1.16, an antibody specific for major histocompatibility complex (MHC) class I molecules Kb bound to SIINFEKL (BioLegend cat# 141603) and H-2Kb MuLV p15E Tetramer-KSPWFTTL-PE (MBL International) at a concentration of 0.125 μg/1e6 cells in a volume of 100 µL staining buffer after first blocking the cells for 15 min with mouse FcR block (Miltenyi Biotec cat# 130-092-575). For assessing T cell function as a result of stimulation by SIINFEKL peptide or OVA protein pulsed CD103+ DC populations, DC were co-cultured at a 1:5 ratio with MACS-isolated CD8+ T cells from OT-I mice for 12 hours in the presence of brefeldin-A and antimouse CD107a and then fixed, permeabilized and intracellularly stained for accumulated IFN-γ using reagents from the BD Bioscience CytoFix/CytoPerm Solution Kit. In some assays, killed MC38-OVA cells were added to DC cultures and surface antigen presentation of Kb-bound SIINFEKL peptide was determined after cross-presentation of tumor cell-associated antigen. For tumor cell, killed MC38-OVA cells were transfected for 5 min with a final concentration of 1 µg/mL 5’ triphosphate hairpin RNA (3p-hpRNA, InvivoGen cattlrl-hprna) using lipofectamine RNAi-MAX transfection reagent (Invitrogen cat# 13778030) according to the manufacturer’s protocol. Cells were then incubated for 42 hours at 37°C/5% CO2, a condition that results in ≥60% loss of viability and is associated with an immunogenic mode of death37 prior to addition to OT-1 CD8+ T cells.
Ex vivo CD103+ DC isolation and OT-I T cell co-culture studies
TdLN from MC38-OVA tumor-bearing mice were dissected post-mortem, pressed through 100 µM cell strainers (Corning), washed in T cell complete medium and enriched for CD11c+ cells using Mouse CD11c+ Microbeads (Miltenyi Biotec cat# 130-108-338) followed by sorting for live (propridium iodide negative) CD11c+ CD3− CD8a− CCR7+ CD103+ CLEC9A+ DC per the antibody panel in the online supplementary methods on a SONY Biotechnology SH800 cell sorter using a 70 µM nozzle chip. Isolated CD103+ DC were co-cultured at a 1:20 ratio with CFSE-labeled (CellTrace CFSE ThermoFisher Scientific cat# C34534), negatively selected CD8+ T cells (Miltenyi Mouse CD8+ T cell Isolation Kit, cat# 130-104-075) in 200 µL T cell complete medium for 4 days with subsequent proliferation evaluated by dye dilution analysis using FlowJo V.10 software following acquisition on a MACSQuant-10 flow cytometer (Miltenyi Biotec).
Antigen-specific priming of HLA-A*0201 Melan-A CD8+ T cells
AZD4635 was tested for effects in reversing adenosine-mediated defects in antigen-specific priming of Melan-A (ELAGIGILTV)-specific T cells from the naïve repertoire of HLA-A*0201 individuals. As previously described,38 naïve CD8+ T cells were co-cultured with autologous monocyte-derived DCs, both cell populations isolated from PBMC from HLA-A*0201 positive healthy donors (AllCells, Research Blood Components). Monocytes were exposed to 5 µM NECA alone or 3 µM AZD4635 for 30 min prior to NECA in the differentiation cultures starting 24 hours after initial plating. DC were washed free of NECA prior to addition of CD8+ T cells, such that any effects on priming would purely be based on immune modulation of the antigen-presenting cells not a combination of both innate and adaptive immune cell effects. The following modifications of the published protocol were adapted for our studies: purified human monocytes, obtained by untouched antibody-labeled magnetic bead separation (Miltenyi Biotec cat# 130-096-537) were used for Mo-DC generation instead of panning for adherent cell enrichment, a Melan-A MHC-CL.I (HLA-A*0201)-restricted recognizing the ELAGIGILTV epitope peptide tetramer (MBL cat# TB-0009) was used instead of a dextramer product for identifying Melan-A-specific CD8+ T cells at the end of culture, and in lieu of irradiation, DCs were treated for 30 min with 25 µg/mL mitomycin C (Sigma cat# M4287), followed by extensive washing with complete T cell medium prior to T cell addition in the presence of peptide and cytokines, according to the protocol.
Statistical analysis
Statistical calculations were performed using two-sided unpaired Student’s t-test and Mann-Whitney U test with significance accepted at a 95% confidence level (p<0.05).
Results
AZD4635 is a potent and specific A2AR antagonist
AZD4635 (figure 1A) is a novel, small-molecule inhibitor targeting A2AR which was derived from structure-based design.37 38 The affinity of AZD4635 for human adenosine receptors was evaluated using radioligand binding competition assays and cells stably transfected with human A1R, A2AR, A2BR and A3R. A concentration-response binding competition curve was generated using eight agonist concentrations ranging from 30 nM to 100 µM and binding affinity (Ki) was calculated using the Cheng-Prusoff equation. As shown in figure 1B, AZD4635 is a potent and selective competitive antagonist of A2AR with a Ki of 1.7 nM and binding affinity for other adenosine receptors of 160 nM for A1 (selectivity of 94-fold), 64 nM for A2BR (selectivity of 37-fold) and >10 000 nM for A3 (selectivity >5882-fold).

AZD4635 is a potent A2AR antagonist. (A) Chemical structure of AZD4635. (B) AZD4635 binds to human A2AR with a Ki of 1.7 nM and with >30-fold selectivity over other adenosine receptors as measured by radioligand receptor occupancy assay. (C) Structure of A2AR bound to AZD4635 (potential hydrogen bonds represented as dashed red lines). (D) Chinese hamster ovary (CHO) cells stably expressing human A2AR were incubated with adenosine in the presence of AZD4635. AZD4635 is capable of inhibiting adenosine-mediated cAMP accumulation in human A2AR-expressing cells (n=3, error bars represent SD). (E) CHO cells stably expressing human and mouse A2AR were incubated with EC80 concentrations of 5′-(N-ethylcarboxamido) adenosine (NECA) in the presence of AZD4635. AZD4635 is capable of inhibiting adenosine-mediated cAMP accumulation in both human and mouse A2AR-expressing cells (n=3, error bars represent SD).
Binding mode and co-complex crystal structure of A2AR-AZD4635
The overall structure of the A2A-StaR2-AZD4635 complex is consistent with previously solved A2A-StaR2 antagonist structures, with the receptor displaying the canonical fold and molecular hallmarks of an inactive conformational state (figure 1C). Statistics for data collection and refinement are given in table 1. The co-crystal structure demonstrates clear positive omit density at 3.0σ (data not shown) for the presence and position of the ligand in the orthosteric binding pocket as expected. The structure of A2A-StaR2-AZD4635 (figure 1C) indicates that the ligand makes critical H-bonding interactions with the side chain of Asn2536.55. In addition, Phe168 on extracellular loop 2 (ECL2) forms a π-π stacking interaction with the ligand, and the side chain of Met2707.35 makes a hydrophobic interaction from the opposite face. A complex network of water-mediated interactions revealed by the high-resolution data complete the receptor interactions around this region of the ligand. The fluorophenyl substituent of the ligand sits on one side of the base of the pocket (as defined by Trp2466.48) and occupies a hydrophobic pocket deeper inside the receptor (flanked by Leu843.32, Leu853.33, Met1775.38, Asn1815.42, Trp2466.48, Leu2496.51 and His2506.52). The second aromatic substituent occupies the ribose binding pocket (of the natural agonist adenosine) defined by His2787.43 and Ser2777.42. From this substituent, the methyl group occupies a hydrophobic subpocket (Ala632.61 and Ile662.64) thought to be important for subtype selectivity. Finally, a water molecule at the back of the ribose pocket forms important polar interactions with N4 of the pyridine and the side chain of His2787.43.
AZD4635 is a potent inhibitor of A2AR signaling which is impacted by adenosine concentrations
The A2A receptor is a member of the G-coupled receptor family with activation leading to increased intracellular levels of cAMP. The potency of AZD4635 in inhibiting A2AR signaling was determined in a cAMP accumulation assay using CHO cells stably transfected with human A2AR (figure 1D) and stimulated with adenosine (100, 10, 1 and 0.1 µM). Owing to the competitive binding mechanism of inhibition, the antagonist activity (IC50) of AZD4635 was impacted by the adenosine concentration. As the concentration of adenosine was increased, the potency of AZD4635 antagonist activity was reduced, with a linear relationship. AZD4635 demonstrated an IC50 of 0.000794, 0.0123, 0.155 and 2.69 µM at concentrations of 0.1, 1, 10 and 100 µM adenosine, respectively, with comparable antagonist activity against mouse A2AR (figure 1E).
AZD4635 reverses adenosine-mediated immune suppression in vitro
Previous studies have demonstrated that activation of A2AR signaling on T cells inhibits activity by suppressing IFN-γ production.39 To determine whether AZD4635 can reverse adenosine-mediated T cell suppression, CD8+ T cells were incubated with NECA, a stable analog of adenosine and stimulated with CD3/CD28 beads. NECA caused potent suppression of IFN-γ secretion in T cells with 1 µM NECA leading to 80% suppression of IFN-γ secretion online supplementary figure 1A. Incubation of CD8+ T cells with AZD4635 restored IFN-γ secretion back to baseline levels with an observed EC50 of 0.053 µM and 0.598 µM at 0.1 and 1 µM NECA, respectively (online supplementary figure 1C).
Supplemental material
In addition to its inhibitory effects on T cell function, adenosine has also been reported to suppress development and activity of antigen-presenting cells such as macrophages and DCs.11 13 40 To assess the effect of adenosine on macrophage differentiation, murine macrophages were matured ex vivo in the presence of NECA and AZD4635. Maturation of F4/80+ macrophages in the presence of NECA led to a dose-dependent decrease in MHCII expression, with 10 µM NECA leading to >90% suppression of MHCII expression relative to DMSO-treated controls (online supplementary figure 1B). AZD4635 fully restored MHCII expression at low NECA concentrations, and partially at higher concentrations.
AZD4635 reduces tumor growth and enhances the antitumor activity of checkpoint inhibitors in syngeneic tumor models
To evaluate the therapeutic benefit of AZD4635 in vivo, AZD4635 was administered as a monotherapy or in combination with checkpoint inhibitors in syngeneic mouse tumor models. Twice daily oral dosing of AZD4635 of 50 mg/kg was well tolerated and induced antitumor effects in several mouse syngeneic carcinoma models (figure 2A–D, online supplementary figure 2A‒C). Treatment of mice bearing B16F10-OVA carcinoma tumors with AZD4635 led to TGI alone (TGI=43%) and in combination with anti-PD-L1 (TGI=83%) (figure 2A). Similar responses were observed in mice bearing MC38 colorectal carcinoma tumors, with treatment of AZD4635 leading to statistically significant TGI alone (TGI=52%, p=0.03) and in combination with anti-PD-L1 antibody (TGI=91%, p=0.01) (figure 2D). In the murine MCA205 fibrosarcoma model, twice daily dosing with single agent AZD4635 led to significant TGI alone (TGI=68%, p=0.005) and completely suppressed tumor growth when combined with anti-PD-L1 antibody (TGI=100%, p=0.0001), including regressions in 2/10 animals (figure 2B, online supplementary figure 2C). The antitumor activity of AZD4635 was dose dependent in mice bearing MCA205 tumors with twice daily dosing of 50 mg/kg resulting in TGI=94%, 25 mg/kg resulting in TGI=70% and 10 mg/kg resulting in TGI=52%, when combined with anti-PD-L1 Ab (online supplementary figure 2D).

AZD4635 enhances the antitumor activity of checkpoint inhibitors in syngeneic tumors. (A) B16F10-OVA melanoma cells were implanted subcutaneously in C57Bl/6 mice and were treated with AZD4635 (50 mg/kg twice daily) alone or in combination with antiprogrammed death-ligand 1 (anti-PD-L1) (10 mg/kg twice daily). (B) C57Bl/6 mice bearing subcutaneous MCA205 fibrosarcoma tumors were treated with AZD4635 (50 mg/kg twice daily) alone or in combination with anti-PD-L1 (10 mg/kg twice daily). (C) Antitumor efficacy of AZD4635+anti-PD-L1 evaluated in MCA205 in NOD-scid IL2Rgnull (NSG) mice. (D) Mice bearing subcutaneous MC38 tumors were treated with AZD4635 (50 mg/kg twice daily) alone or in combination with anti-PD-L1 (10 mg/kg twice daily). (E) Mice bearing subcutaneous MC38-OVA tumors were injected with either isotype control or antimouse CD8 depleting antibody and treated with AZD4635 (50 mg/kg twice daily) in combination with anti-PD-L1 (10 mg/kg twice daily). Average tumor volume of 10–15 mice/group±SE are shown. *P<0.05 (two-tailed Student’s t-test).
To confirm the immune-mediated mechanism of TGI by AZD4635, immunodeficient NOD-scid IL2Rgnull (NSG) mice which lack mature T, B and functional NK cells were implanted with subcutaneous MCA205 tumors and treated with AZD4635 and anti-PD-L1 (D265A). The antitumor activity was completely abrogated in immunodeficient mice, confirming the immune-mediated mechanism of TGI (figure 2C). Similarly, depletion of CD8 T cells completely abrogated antitumor activity of AZD4635+PD-L1 treatments in MC38-OVA model, confirming a CD8 T cell-mediated mechanism of TGI (figure 2E).
Collectively, these studies demonstrate that AZD4635 reduces tumor growth and enhances the activity of anti-PD-L1 in several syngeneic models through a CD8+ T cell-mediated mechanism.
AZD4635 enhances immune activation in vivo
To study the molecular consequences of AZD4635 treatment on immune activation, changes in gene transcription in immune related pathways were evaluated in MC38 tumors harvested after being treated with AZD4635 for 14 days. Unsupervised clustering of gene expression changes revealed that AZD4635 treatment upregulated expression of a number of genes associated with immune activation, IFN-γ signaling (Gbp2b, Il12rb2), antigen processing (Cd1d2, H2-Ea-ps, Cd207), T cell function (Fasl), NK cell activity (Klrb1c) and macrophage and DC activity (Fcer1a, Cr2) relative to vehicle-treated animals (figure 3A). We did observe a somewhat unexpected increase in arginase gene expression which may be due to additional suppressive mechanisms being upregulated in response to A2AR inhibition however was not sufficient to ameliorate antitumor responses. Additionally, changes in TILs were analyzed in MC38 tumors following AZD4635 administration for 14 days. Consistent with in vitro findings, AZD4635 enhanced activation of antigen-presenting cells in the tumor microenvironment as demonstrated by increased expression MHCII and CD86 on CD11c+ DCs and F4/80+ macrophages (figure 3B). Interestingly, AZD4635 administration did not increase intratumoral frequencies of these cell populations suggesting AZD4635 treatment does not affect recruitment of these cells but restores antigen presentation function of DCs and macrophages. No significant changes in MHCII or CD86 expression were observed following anti-PD-L1 treatment (online supplementary figure 9). AZD4635 in combination with anti-PD-L1 significantly increased the frequency of infiltrating CD8+ cytotoxic lymphocytes into the tumor compared with vehicle-treated mice (figure 3C). Collectively, these results demonstrate that blockade of A2AR signaling by AZD4635 enhances priming activity of DCs and macrophages and increases infiltration of cytotoxic T cells in the tumor microenvironment.
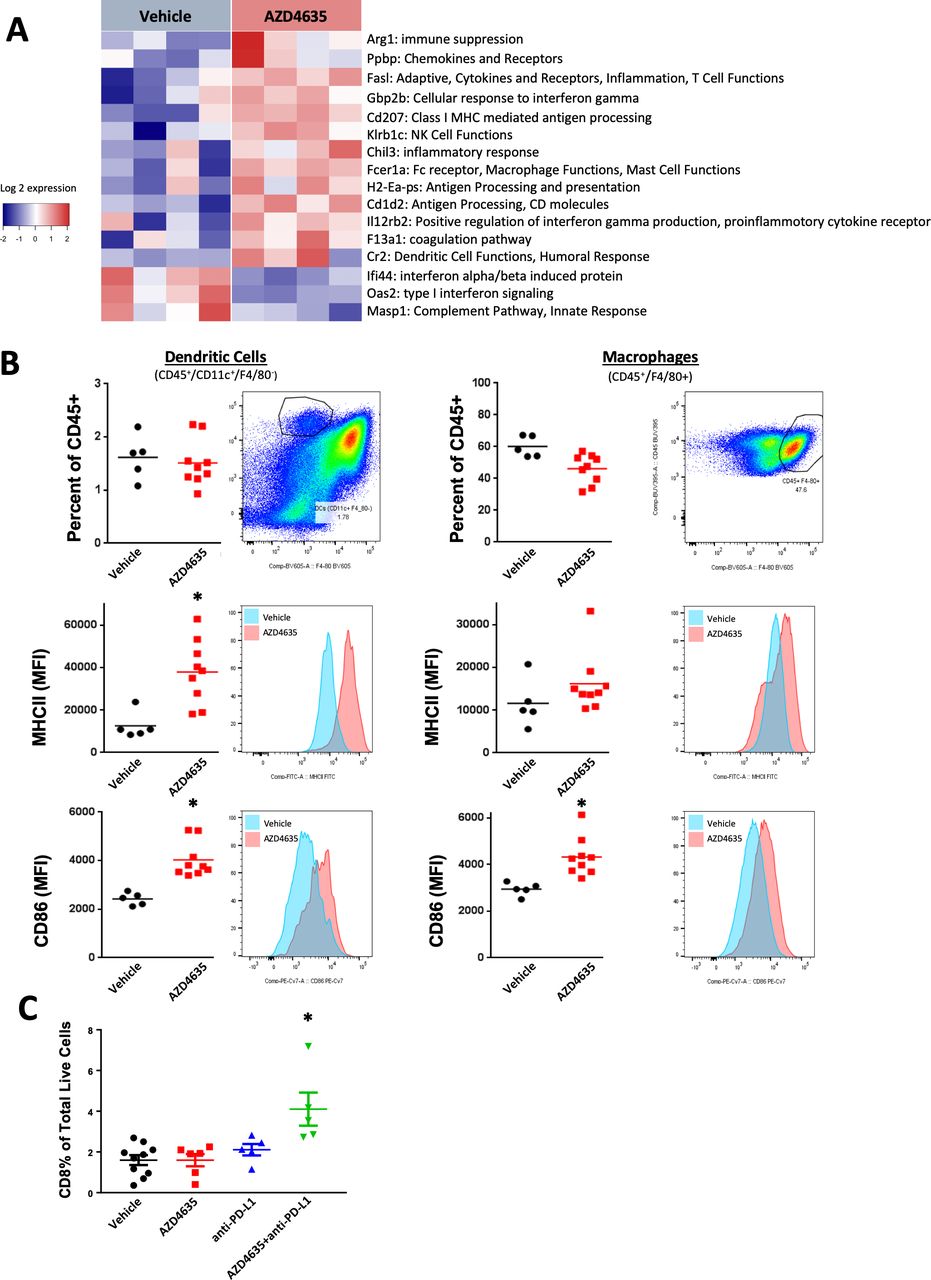
AZD4635 enhances immune activation and infiltration in vivo. (A) Mice bearing subcutaneous MC38 tumors were treated for 14 days with AZD4635 and gene expression changes were evaluated by the NanoString Mouse PanCancer Immune Profiling Panel. Unsupervised clustering of gene expression changes revealed upregulation of antigen processing and T cell function. Genes were filtered for significance (p≤0.05) and magnitude (fold change >2). (B) AZD4635 alone or in combination with antiprogrammed death-ligand 1 (anti-PD-L1) increased expression of markers of antigen presentation (MHCII) on dendritic cells (left panels) and macrophages (right panels) in MC38 tumors. (C) AZD4635 in combination with anti-PD-L1 increased intratumoral CD8+ infiltration in MC38 tumors. Data are representative of tumor-infiltrating lymphocyte (TIL) analysis from three repeated efficacy studies with similar results (n=6 mice per group), *p<0.05 two-tailed Student’s t-test.
Increases in antigen-specific immunity contributes to tumor efficacy with AZD4635 in combination with PD-L1 checkpoint blockade
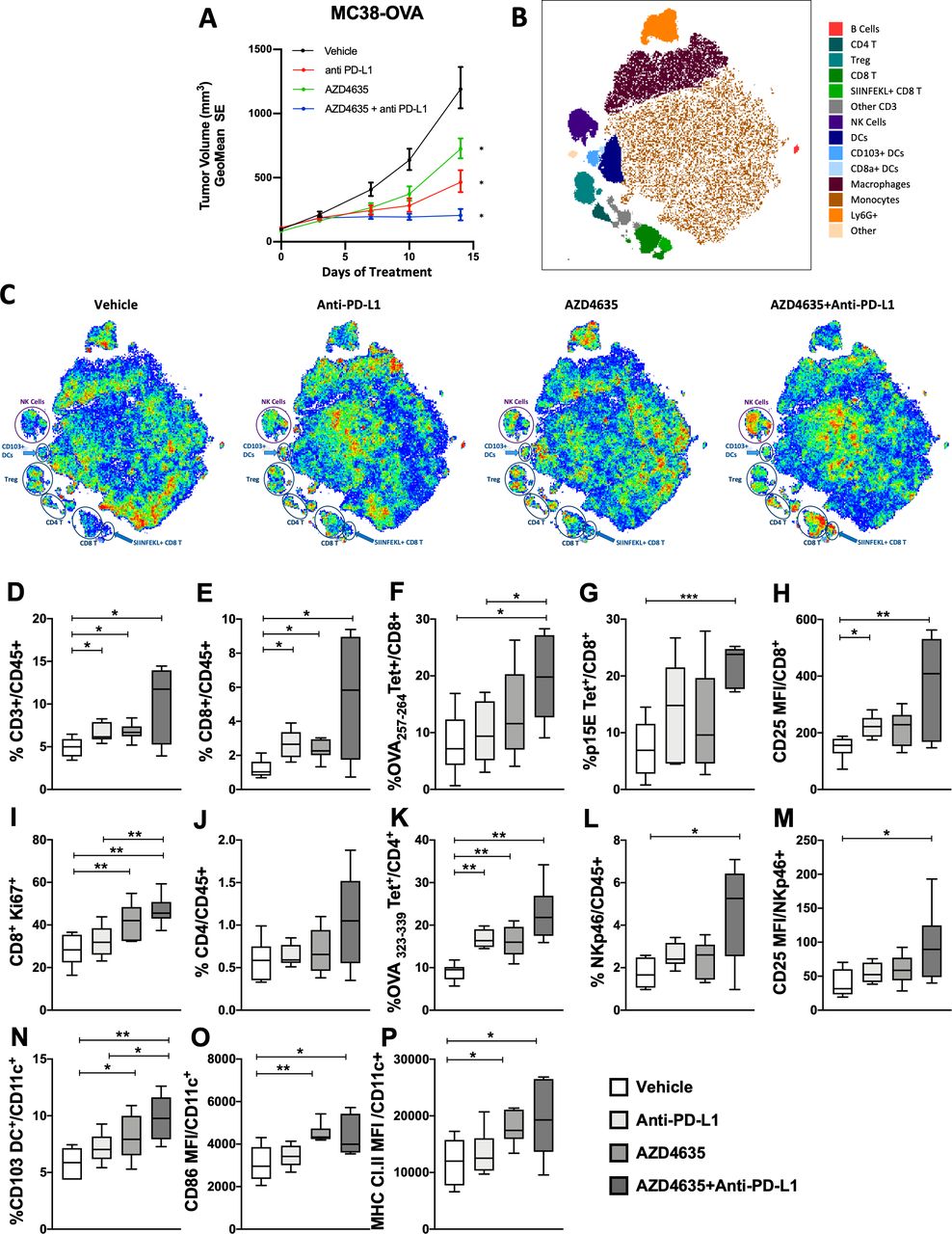
Given the promising efficacy seen in multiple syngeneic tumor models, including MC38, we next performed several in vivo studies using the MC38-OVA syngeneic model to explore antigen-specific responses. Repeated studies in this model demonstrated that 2 weeks of monotherapeutic regimen with αPD-L1 (10 mg/kg, twice weekly) or AD4635 (50 mg/kg, twice daily), resulted in significant tumor efficacy, with an appreciable beneficial combination effect (figure 4A,B). Changes in tumor infiltrating immune cell populations were analyzed from dissociated MC38-OVA tumors by flow cytometry, unbiased tSNE analysis of these data demonstrates broad immune cell changes with AZD4635 treatment and most significantly in combination with anti-PD-L1 (figure 4C). Innate and adaptive immune populations were increased in combination treated mice associated with efficacy (figure 4C,D). Quantification of the immune cell changes from the tSNE analysis showed an increase in T cells, importantly tumor antigen-specific CD8+and CD4+ were increased (figure 4E–K) as well as CD8+ T cell activation and proliferation were enhanced (figure 4H,I). Aligned with increased tumor antigen-specific T cell response was an increased presence of intratumoral CD103+ DCs, as well as CD86 and MHC expression, whose unique tumor antigen trafficking and T cells cross-priming activities are crucial for successful antigen-specific T cell-based tumor rejection (figure 4N-P).

Increased antigen-specific antitumor immune responses with AZD4635 combination treatment with antiprogrammed death-ligand 1 (anti-PD-L1) contributes to tumor efficacy in the MC38-OVA model. (A) Mice bearing subcutaneous MC38-OVA tumors (n=10) were administered AZD4635 (50 mg/kg twice daily) alone or in combination with anti-PD-L1 (10 mg/kg twice daily). Tumor volumes following 14 days of treatment with vehicles, anti-PD-L1 alone, AZ4635 alone and combination treatment with AZD4635 and anti-PD-L1 are shown (A), ±SEs are shown. Tumor efficacy data shown is from one representative study of three done with similar results. (B, C) Flow cytometric T-distributed stochastic neighbor embedding (tSNE) analysis of immune cells from dissociated tumors from the efficacy study shown in 4A, showing indicated lymphoid and myeloid populations associated with treatments. (D) CD3 tumor-infiltrating lymphocyte (TIL) as a percentage of CD45 infiltration in MC38-OVA tumors following treatments as indicated: vehicle, anti-PD-L1, AZD4635 and AZD4635 in combination with anti-PD-L1. Other CD8+ T cell populations, as a percentage of CD45% in TIL are shown as follows: (E) CD8+ T cells, (F) OVA 257-264-positive CD8+ T cells, (G) p15E-positive CD8 T cells, (H) activated CD8+ T cells indicated by CD25 mean fluorescent intensity (MFI) and (I) proliferating CD8+ T cells as indicated by Ki-67 proliferative antigen index. (J) CD4+ T cells in TIL as a percentage of CD45 infiltration in MC38-OVA tumors. (K) Antigen-specific OVA 323-339-positive CD4+ T cells. (L) Natural Killer (NK) cells determined as CD3−, NKp40+ as a percentage CD45+ TIL. (M) MFI of CD25 as a correlate of activated NK cells. (N) The percentage of CD103+ DC of CD11c bearing cells within TIL, (O) costimulatory molecule expression of CD86 and (P) MHC class II molecule expression, depicted as the MFI of expression in CD11c+ cells in TIL. Data are representative of TIL analysis from three repeated efficacy studies with similar results (n=6 mice per group), stained with antibodies to lymphoid and myeloid antigens as indicated in online supplementary methods. *P<0.05, **p<0.01, ***p<0.001 (Mann W-hitney U test).
To further understand the functional status of the T cells in the tumor microenvironment (TME), we performed an ex vivo stimulation to quantitate levels of cytokines, IFN-γ, tumor necrosis factor (TNF)-α, interleukin (IL)-2, granzyme B and surface expressed CD107a on CD8+ T cells were all enhanced (figure 5A–E) following single agent treatment of AZD4635 or anti-PD-L1 treatment, these changes were most significantly increased in the combination treatment. Cytokine production, IFN-γ, TNF-α, IL-2, was also strongly increased in CD4+ T cells, indicative of tumoricidal activity (figure 5F–H). There was also an increase in TIL NK cells (figure 4L) in the combination treatment correlating with increase activation marker CD25 (figure 4M) and a minimal increase in IFN-γ production (figure 5I). However, we observed a strong increase in TNF-α production and expression of CD107a—a marker of degranulation with combination treatment (figure 5J,K). To investigate tumor antigen-specific CD8 T cell activation in vivo, tumor draining LN cells were restimulated with either OVA peptide or MC38 endogenous tumor antigen p15E peptide in vitro. Strong activation of IFN-γ in response to both peptides confirm tumor antigen-specific CD8 T cell response in combination group (figure 5L,M). As expected on restimulation with foreign antigen OVA or tumor-specific antigen p15E, we observed loss of IFN-γ production in animals depleted of CD8 T cells in the combination group confirming the requirement of CD8 T cells in mounting an antitumor response. Taken together, AZD4635 with anti-PD-L1 treatment exhibited numerous pharmacodynamic effects in tumor infiltrating immune cell populations including enhanced recruitment and activity of antigen-specific T cells, increased CD103+ DC and NK populations, consistent with observed AZD4635-mediated TGI.

MC38-OVA tumor analysis demonstrates enhanced T cell function associated with tumor efficacy by AZD4635 in combination with anti-PD-L1. (A–I) Single cell suspension of MC38-OVA tumors were prepared and stimulated with PMA/ionomycin. CD8+ T cells as a percentage of CD3+, CD45+ cells were analyzed for (A). Interferon (IFN)-γ production (B). Tumor necrosis factor (TNF)-α (C). Interleukin (IL)-2 (D). Granzyme B (E). CD107a expression. CD4+ T cells were analyzed for (F). IFN-γ production (G). TNF-α (H). IL-2 (I). NK cells determined as CD3-, NKp46+ as a percentage CD45+ tumor-infiltrating lymphocyte (TIL) were analyzed for IFN-γ production (J, K). NK cells were analyzed for TNF-α production and CD107a expression, respectively (L, M). Tumor draining lymph node cells from MC38-OVA tumor-bearing mice were stimulated with OVA and p15E peptide (1 μg/mL) and IFN-γ production was analyzed using ELISpot assay. Data are representative of TIL analysis from three repeated efficacy studies with similar results (n=6), stained with antibodies to lymphoid and myeloid antigens as indicated in online supplementary figure 3. *P<0.05, **p<0.01, ***p<0.001 (Mann-Whitney U test).
The A2AR antagonist AZD4635 restores adenosine-mediated immunosuppression of CD103+ dendritic cells
Since we observed increased CD103+ DC in MC38-OVA tumors treated with AZD4635 and because the culture method for CD103+ DC has been recently published,36 we explored the impact of adenosine on the function of the cell populations in vitro. Previously, adenosine signaling has been shown to negatively impact the differentiation and function of granulocyte-macrophage colony-stimulating factor (GM-CSF)/IL-4 polarized DC populations.16 However, an adenosine receptor antagonist was not tested in this report and there is no report of adenosine effects on CD103+ cross-presenting DCs to our knowledge.
To evaluate the ability of AZD4635 to reverse adenosine-mediated suppression of CD103+ DC differentiation, C57BL/6 bone marrow cell suspensions were treated with FLT3L and GM-CSF for 16 days which resulted in 86% CD11c+ CD103+ CLEC9A+ cells by flow cytometry analysis (figure 6A) (online supplementary figure 3). The addition of NECA into immature myeloid cultures prevented differentiation into CD103+ DCs (p=0.0006), 57% CD103+ compared with 80% positive in the untreated cells. Cultures first treated with 3 µM AZD4635 and then exposed to NECA were able to differentiate into CD103+ DC efficiently. These data demonstrate that AZD4635 can rescue the immunosuppressive effects of adenosine signaling in DCs, thus promoted the generation of bone marrow-derived precursors into CD103+ DCs (figure 6A).To test if AZD4635 could restore adenosine-mediated suppression of pinocytosis in fully differentiated CD103+ DCs, cells were treated with NECA and/or AZD4635 then washed out and the uptake of fluorescent ovalbumin was evaluated. NECA (5 µM) reduced the surface expression of fluorescent OVA (figure 6B) by nearly 50%, whereas treatment with AZD4635 (3 µM) rescued OVA surface expression almost to levels in untreated control. Next, the ability of AZD4635 to rescue antigen presentation and processing was evaluated in vitro using SIINFEKL peptide or whole length ovalbumin peptide. In these assays, CD103+ DC cultures were pulsed with SIINFEKL peptide or with ovalbumin protein (figure 6C) in the presence of NECA (5 µM), AZD4635 (3 µM) or in combination. Using an antibody clone specific for Kb bound to SIINFEKL, the percentages of CD103+ DC positive for antigen presentation were determined. In SIINFEKL pulsed cultures, NECA reduced antigen presentation to less than a third compared with untreated CD103+ DC. AZD4635 rescued antigen presentation by twofold compared with NECA treatment. In cultures containing whole ovalbumin protein, surface antigen presentation of SIINFEKL in the context of Kb molecules generated in the presence of NECA was a log-fold reduced compared with untreated cells, suggesting adenosine signaling leads to combined deficiencies in antigen processing and presentation. In CD103+ DCs treated with AZD4635+NECA, antigen presentation was increased nearly fivefold compared with NECA and restored to a level comparable to untreated CD103+ DCs. Thus, AZD4635 was able to rescue CD103+ DC from adenosine-mediated suppression in differentiation and function in vitro.

AZD4635 mitigates adverse adenosine-mediated effects on mouse CD103+ DC generation, antigen presentation functions and T cell cross-priming. Bone marrow-derived CD103+ DC were generated and left untreated (UNTX), exposed to 5 µM 5′-(N-ethylcarboxamido) adenosine (NECA) or treated with 3 µM AZD4635 in addition to NECA (AZD4635+NECA) as indicated, and the effects on CD103+ DC were evaluated. (A) NECA reduced the differentiation of bone marrow precursors into CD11c+CD103+CLEC9A+ DCs but treatment of AZD4635 largely rescued the generation of CD103+ DCs. Results from two independent experiments from a total of seven individual animals are shown. (B) NECA reduced pinocytosis of fluorescent ovalbumin protein by CD103+ DC and AZD4635 pretreatment reduced the ingestion of this antigenic material. (C) Data show antigen presentation of Kb-SIINFEKL by flow cytometry on the surface of CD103+ DC with the indicated in vitro treatments during differentiation. CD103+ DC cultures were incubated with either SIINFEKL peptide (black bars) or ovalbumin (gray bars). (D) OT-1 T cell effector function, CD107a surface expression, black bars, intracellular interferon (IFN)-γ accumulation (gray bars), both with protein transporter inhibition, are shown following 24 hours co-culture with CD103+ DC exposed to SIINFEKL peptide shown in B. (E) CD107a surface accumulation, black bars, intracellular IFN-γ accumulation (gray bars), both with protein transporter inhibition, are shown following 24 co-culturing with CD103+ DCs left untreated, UNTX, exposed to 5 µM NECA, NECA or treated with 3 µM AZD4635+5 µM NECA, as indicated, incubated with dead MC38-OVA cells prior to addition of OT-1 CD8+ T cells. All results shown in A–E are combined results of two independent experiments done with bone marrow-derived CD103+ DC from seven individual mice. *P<0.05, **p<0.01 (Mann-Whitney U test).
AZD4635 rescues adenosine-mediated suppression of OVA antigen cross-priming of CD103+ to T cells
OT-1 CD8+ T cells were used to further explore the role of adenosine (NECA) on DC function and cross-priming ability. C57BL/6 BM-DC were differentiated in the presence or absence of 5 µM NECA in these assays, and then co-cultured with OT-1 CD8+ T cells in complete medium. When CD103+ DC were differentiated in the presence of 5 µM NECA, loaded with SIINFEKL peptide and then co-cultured with naïve OT-1 CD8+ T cells, a reduction in priming was observed. CD107a expression and IFN-γ secretion by OT-1 CD8+ T cells were reduced by 72% and 60%, respectively, compared with co-cultures containing DC differentiated without NECA (figure 6D). AZD4635 reversed this effect, restoring the expression of CD107a and IFN-γ comparable to untreated cells (figure 6D). Cross-priming of OT-1 CD8+ T cells to presented SIINFEKL peptide from dead MC38-OVA tumor cells was also evaluated in DC:OT-1 CD8+ T cells cultures. In these assays, both CD107a and IFN-γ responses were reduced by 75% in OT-1 CD8+ T cells that were co-cultured with CD103+ DCs that were differentiated in the presence of 5 μM NECA (figure 6E). AZD4635 improved CD107a responses suppressed by NECA-exposed CD103+ to within 87% of the responses elicited by untreated cells and the IFN-γ response was not statistically different compared with those elicited by untreated cells.
Next, the antigen cross-priming capability was evaluated in CD103+ DC sorted ex vivo from TdLN of mice bearing MC38-OVA tumors from the efficacy study shown in figure 4A (figure 7A). In this study, 5.8% of OT-1 CD8+ T cells showed proliferation on exposure to CD103+ DC sorted from TdLN isolated from vehicle-treated MC38-OVA tumor-bearing mice (figure 7A and online supplementary figure 6). All treatments produced statistically significant improvements in proliferation of OT-1 CD8+ T cells, compared with vehicle-treated controls: anti-PD-L1: 11%, AZD4635: 28%, with the anti-PD-L1+AZD4635 combination treatment showing an even bigger improvement to 53% (figure 7B). Both AZD4635 groups, as monotherapy and in combination with anti-PD-L1, enhanced OT-1 CD8+ T cell proliferation as a result of enhanced T cell cross-priming, with the combination statistically higher than single agent treatment.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
AZD4635 reverses tolerogenic dendritic cells (DCs) and promotes TAA-specific T cell priming ex vivo and in human in vitro assays. (A) Ex vivo CD103+ DC cross-priming was determined by flow sorting CD103+ DCs from the TdLN of MC38-OVA tumor-bearing mice treated for 14 days with vehicles, antiprogrammed death-ligand 1 (anti-PD-L1) alone, AZ4635 alone and combination treatment with AZD4635 and anti-PD-L1. Treatment with AZD4635 improved ability to stimulate proliferation of CFSE-labeled OT-1 CD8+ T cells after 4 days of co-culture. Data shown are representative of two independent experiments with similar results with n=12 per group. *P<0.05, **p<0.01, ***p<0.001 (Mann-Whitney U test). (B) Human monocytes were treated with granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin (IL)-4 to generation DCs which were untreated, UNTX, exposed to 5 µM 5′-(N-ethylcarboxamido) adenosine (NECA), or treated with 3 µM AZD4635 in addition to NECA. Cultures were washed and the effects on the expansion of HLA-A*0201-restricted Melan-A CD8+ T cells, in the presence of ELAGIGILTV cognate peptide, from a pool of naïve CD8+ T cells was tested. Representative flow cytometry dot plot histogram from one donor. (C) Tabulated data of the percentage of Melan-A tetramer-positive CD8+ T cells from two independent assays performed with a total of 4 individual HLA-A*0201 positive donors, *p<0.05, **p<0.01 (Mann-Whitney U test).
This study underscores the mechanism of action of AZD4635 in improving CD103+ DC antigen presentation and cross-priming of antigen-specific T cells to tumor-associated antigens, both in vitro and in vivo.
AZD4635 reverses DC tolerogenesis and promotes T cell priming in vitro
Few human in vitro culture systems are suitable for testing the effects of DC priming on the generation of tumor antigen-specific CD8+ T cells. However, based on the detectable frequency of HLA-A*0201 within the T cell repertoire of healthy individuals, a protocol was described38 in which these responses can be studied under conditions where autologous GM-CSF polarized monocyte-derived DCs are co-cultured with naïve CD8+ T cells in the presence of the ELAGIGILTV peptide and exogenous cytokines. Similar to our murine cultures, we studied the effects of NECA and AZD4635 treatment on antigen presentation of Melan-A TAA by DCs. Since the T cells were added after NECA and AZD4635 treatment were removed, any change in cross-priming to HLA-A*0201 Melan-A tetramer positive CD8+ T cells would be a direct effect on the DCs (see online supplementary figure 7 for assay scheme). Priming and expansion of naïve Melan-A26-35-specific CD8+ T cells with DCs differentiated in the absence of NECA resulted in the detection of 32% tetramer-positive antigen-specific T cells (figure 7B,C), in agreement with published literature.38 41 In contrast, NECA suppressed the cross-priming function of DCs to a greater than 90% reduction. Treatment of AZD4635 in the presence of NECA resulted in 16% Melan-A26-35 tetramer positive CD8+ T cells compared with 2.5% with NECA, thus a 52% recovery was obtained with A2AR inhibition. Thus, both murine and human antigen-specific models of T cell priming show that adenosine antagonism using AZD4635 in DCs rescues many features of DC function required for a productive antitumor response.
Discussion
Evasion of immune response is a hallmark of cancer and seminal experiments by Sitkovsky et al identified accumulation of adenosine in the tumor microenvironment as a critical mechanism in immune evasion.1–3 42 43 To promote tumorigenesis, cancers induce a hypoxic environment leading to the accumulation of extracellular ATP and subsequently accumulation of adenosine via ectoenzymes CD39 and CD73 in the tumor microenvironment.44 Intratumoral adenosine signals through the high-affinity A2AR receptor, expressed on the surface of a wide range of immune populations, leading to suppression in immune effector activity and contributing to evasion of immune surveillance. Herein, we characterize AZD4635, a novel, orally bioavailable and selective A2AR inhibitor which reverses adenosine-mediated immune suppression. In this study, we demonstrate that AZD4635 exhibits antitumor activity as a monotherapy and enhances activity of αPD-L1 across a range of syngeneic mouse carcinoma models. AZD4635 restores effector function in a number of immune populations, importantly we show CD8+ T cells are required for combination efficacy. Treatment of AZD4635 restores adenosine-mediated suppression of IFN-γ secretion in CD8+ T cells both in vitro and in vivo. Effects on T cells recruitment were also observed in vivo, where combined AZD4635 and anti-PD-L1 treatment induced significant intratumoral infiltration of CD8+ T cells in MC38 tumors. Importantly, these T cells were functionally more active when restimulated with endogenous tumor antigen ex vivo. These data are consistent with previous reports demonstrating that antitumor efficacy is enhanced by A2AR antagonism by restoring T cell function.19 In addition to its effects on cytotoxic lymphocytes, AZD4635 exhibited robust effects on antigen-presenting cells. Efficient antigen presentation by antigen-presenting cells requires expression of MHC (signal 1) and a co-stimulatory receptor (CD80 or CD86, signal 2) providing the essential signals to T cells to induce activation and survival.45 In mice bearing MC38 tumors, treatment with AZD4635 significantly increased surface expression of MHCII and CD86 on both macrophages and DCs and was not observed with anti-PD-L1 treatment. Therefore, inhibition of A2AR signaling by AZD4635 represents a mechanism of immune activation distinct from, and complementary to, classical checkpoint inhibition by simultaneously reversing suppression of both lymphocytes and antigen-presenting cells.
In this report, we provide evidence of a novel mechanism of A2AR antagonism in rescuing the cross-priming function of CD103+ DCs. This cell population has been shown to be essential in recruitment of, and cross-presentation of tumor-associated antigens to cytotoxic lymphocytes. Abundance of CD103+ DCs in tumors has also been shown to correlate with immune-mediated control and favorable outcome in both mouse models and in human.20 46–48 We provide mechanistic data demonstrating adenosine prevents CD103+ DC differentiation and its cross-priming function in mature DCs, both of which can be restored with AZD4635 treatment. Our data provide evidence that inhibition of A2AR signaling is critical for enhancing CD103+ function and accumulation in syngeneic tumor models. CD103+ DCs sorted from the draining LNs of tumor-bearing mice treated with AZD4635 were able to more efficiently present antigen to CTLs, an effect which was further enhanced in combination with αPD-L1 treatment. This suggests that AZD4635 and αPD-L1 combined therapy can promote the priming and expansion of tumor-specific CD8+ T cells. This concept is consistent with the study by Salmon et al, which used administration of the FLT3L and poly I:C to expand and activate CD103+ DCs in combination with PD-L1 blockade to produce robust tumor regressions, providing rational for combination therapies that can boost priming and T cell activity. Importantly, we demonstrate the role of A2AR signaling blockade with AZD4635 is conserved in human DCs, this is in line with the recent report showing mouse CD103+ and the analogous human subset are highly conserved.49 Although we were not able to test AZD4635 directly on human CD141+ DCs due to the scarcity of this cell type, we hypothesize A2AR inhibition will have a similar effect of reversing adenosine-mediated suppression, further work is required to explore the role of adenosine on the CD141+ subset of DCs.
The broad immune cell changes we observe with AZD4635 treatment suggest activation of a NK-DC cell axis, which is a recently emerging concept shown to be critical in orchestration of a productive antitumor response.50 51 Stimulatory DCs and NK cell levels in the TME have been shown to predict responses to checkpoint blockade.51 Consistent with this concept, A2AR engagement has been shown to act as a checkpoint negatively impacting NK cell maturation contributing to tumor control.5 We show A2AR inhibition boosts both NK cells and CD103+ DCs in the TME pointing to AZD4635 treatment as a potential therapy to enhance the NK-DC axis. Additional studies are needed to work out the interplay of NK cells and DCs in adenosine-mediated TME.
All together, our data support an emerging field of small molecule immunotherapy that can be used to treat a variety of solid tumors, which rely on adenosine as the main mechanism of immunosuppression. We provide rationale for combining AZD4635 treatment to rescue CD103+ DC function with checkpoint blockade to enhance tumor eradication. We believe enhancement of CD103+ DCs by AZD4635 contributes to the clinical responses that are being explored in the clinic and support emerging literature showing the requirement of stimulatory DC for enhanced T cell responses.
Acknowledgments
The authors would like to thank Susan Cantin, Maryann San Martin, Kelly Goodwin, Petar Pop-Damkov, Adrian Fretland, India (Lynne) Neveras, Victor Ospina, Michelle Lamb, Ruth Illingworth and Simon Barry for their help with this work and critical review of the manuscript.
References
Footnotes
AB and CMB contributed equally.
Contributors Conceptualization: AB, CMB, AGS, DAM. Data curation and flow analysis: LP and DC. Investigation: AGS, RW, DAM. Performed in vitro assays: MY, JS, CB. Performed in vivo studies: YW, ND, AB, AGS. Transcriptional analysis: BL, KS. Project administration: WS. PK analysis of AZD4635: JDC Resources: CR, LD, SF. Chemical design and synthesis of AZD4635: GAB, JDC, JB, EH, ASD, RKYC, MC. Writing manuscript: AB, CMB and DAM. All authors reviewed and approved the final manuscript.
Funding The work described herein was funded in its entirety by AstraZeneca Pharmaceuticals.
Competing interests A.B., C.M.B., Y.W., N.D., M.Y., J.S., K.S., D.C., L.P., J.D.C., B.L., W.S., R.W., C.R., L.D., S.F., A.G.S. and D.A.M. are employees of AstraZeneca Pharmaceuticals. J.D.C., G.A.B., J.B., M.C., R.K.Y.C., A.S.D., E.H. are employees of Heptares Therapeutics.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. Deanna.mele@AstraZeneca.com.