Article Text

Abstract
Background The mitogen-activated protein kinases (MAPKs) are important for T cell survival and their effector function. Mixed lineage kinase 3 (MLK3) (MAP3K11) is an upstream regulator of MAP kinases and emerging as a potential candidate for targeted cancer therapy; yet, its role in T cell survival and effector function is not known.
Methods T cell phenotypes, apoptosis and intracellular cytokine expressions were analyzed by flow cytometry. The apoptosis-associated gene expressions in CD8+CD38+ T cells were measured using RT2 PCR array. In vivo effect of combined blockade of MLK3 and CD70 was analyzed in 4T1 tumor model in immunocompetent mice. The serum level of tumor necrosis factor-α (TNFα) was quantified by enzyme-linked immunosorbent assay.
Results We report that genetic loss or pharmacological inhibition of MLK3 induces CD70-TNFα-TNFRSF1a axis-mediated apoptosis in CD8+ T cells. The genetic loss of MLK3 decreases CD8+ T cell population, whereas CD4+ T cells are partially increased under basal condition. Moreover, the loss of MLK3 induces CD70-mediated apoptosis in CD8+ T cells but not in CD4+ T cells. Among the activated CD8+ T cell phenotypes, CD8+CD38+ T cell population shows more than five fold increase in apoptosis due to loss of MLK3, and the expression of TNFRSF1a is significantly higher in CD8+CD38+ T cells. In addition, we observed that CD70 is an upstream regulator of TNFα-TNFRSF1a axis and necessary for induction of apoptosis in CD8+ T cells. Importantly, blockade of CD70 attenuates apoptosis and enhances effector function of CD8+ T cells from MLK3−/− mice. In immune-competent breast cancer mouse model, pharmacological inhibition of MLK3 along with CD70 increased tumor infiltration of cytotoxic CD8+ T cells, leading to reduction in tumor burden largely via mitochondrial apoptosis.
Conclusion Together, these results demonstrate that MLK3 plays an important role in CD8+ T cell survival and effector function and MLK3-CD70 axis could serve as a potential target in cancer.
- adaptive immunity
- CD8-positive T-lymphocytes
- cytotoxicity, immunologic
- lymphocytes, tumor-infiltrating
- tumor microenvironment
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- adaptive immunity
- CD8-positive T-lymphocytes
- cytotoxicity, immunologic
- lymphocytes, tumor-infiltrating
- tumor microenvironment
Background
The members of mitogen-activated protein kinases (MAPKs) regulate key cellular function, such as growth, survival, proliferation, metabolism and differentiation in a variety of cell types, including immune cells.1 The three principal MAPK signal transduction pathways in mammalian cells include the extracellular signal-related kinases (ERKs), c-Jun N-terminal kinases (JNKs) and p38 MAP kinases. All three MAP kinase pathways relay their membrane-originated signals through sequential phosphorylation and activation of downstream kinases and finally targeting specific transcription factors in the nucleus, culminating to a specific function.
A broad range of T cell functions is associated with MAPK activation, including early thymocyte development; positive and negative selection of thymocytes; activation, differentiation and pro-inflammatory T cell responses.2–6 The ERK1/2 differentially regulates positive selection and maturation of CD4+ and CD8+ T cells in thymus.7 In T cell receptor (TCR)-mediated T cell activation, the ERK1/2 get activated by sequential activation of Ras-Raf-1-MEK1/2 cascade.8 The role of ERK has also been reported in interleukin 2 (IL-2) production and in proliferation of T cells.9 The ERK1/2 signaling regulates differentiation and effector function of activated CD8+ T cells.10 Similarly, the p38 MAPK has also been reported to get activated as one of the downstream kinase in TCR signaling.3 The p38 MAPKs regulate T cell activation and CD8+ T cell effector function against bacteria and tumor cells via selective activation of nuclear factor of activated T-cells, cytoplasmic (NFATc).11 12 Likewise, the stress-activated MAPK member, JNK, has been reported to regulate IL-2 production in T cells, although IL-2 differentially regulates CD4+ and CD8+ T cells.13–15 The JNK signaling is reported to use distinct substrates to regulate specific T cell function. For apoptosis of thymocytes, the c-Jun is substrate for JNK2, while in T cell proliferation, the DNA binding activity of NFAT transcription factor serves as a substrate for JNK2.16 These reports suggest that members of MAPKs play important roles in T cell function and specific targets in these pathways can be harnessed for immunotherapy.
Earlier we reported that a member of mixed lineage kinase (MLK) family, MLK3, acts as a potent activator of JNK; however, its detail function in immune compartment is not known.17 The MLK group of kinases is clinically important because the pan-MLK inhibitor, CEP-1347, has gone through clinical trial for Parkinson’s diseases and MLK3-specific inhibitor URMC-099 has been proposed for HIV-associated dementia.18 It is also stated that MLK3/MLK inhibitors could play important roles in cancer; yet, its mechanism of action on cancer and immune cells is not elucidated.19–21
Here, we report that the genetic loss or pharmacological inhibition of MLK3 induces CD70-mediated apoptosis in CD8+ T cells but not in CD4+ T cells. In activated CD8+ T cell phenotypes, the CD8+CD38+ T cells are most sensitive to cell death due to loss of MLK3. Additionally, the protein expression of TNFRSF1a is significantly higher in activated CD8+ T cells compared with CD4+ T cells isolated from MLK3−/− mice. Moreover, CD70 regulates TNFα-TNFRSF1a axis to induce apoptosis in CD8+ T cells. The blockade of CD70 in MLK3−/− mice attenuates CD8+ T cell apoptosis and increases recruitment and cytotoxic function. We also demonstrate that combined inhibition of MLK3 and CD70 leads to increased infiltration of cytotoxic CD8+ T cell in breast tumors and reduces the tumor burden mainly via intrinsic pathway of apoptosis.
Methods
Mouse and cell lines
The MLK3−/+ breeding pairs in C57BL/6 background were initially obtained from Dr Roger Davis (UMass Medical Center, Worcester, Massachusetts, USA) and bred in-house to obtain wild type (WT) and homozygous MLK3−/− mice. Gender-matched and age-matched WT and MLK3−/− mice (5–10 weeks old) were considered for each experiment. The age-matched WT mice were treated (intraperitoneal injection) with MLK3 inhibitor, URMC-099 (dose 7.5 mg/kg body weight; Selleck Chemicals) or vehicle control (control) for 3 weeks daily to inhibit MLK3 activity in vivo. Female BALB/c mice were obtained from Charles River Laboratories. The mice were housed in a BRL facility on commercial diet and water. All animal experiments were performed under a protocol approved by IACUC. Jurkat cells (clone E6-1), E.G7-OVA cells and 4T1 cells (from ATCC) were grown in the RPMI-1640 medium (Gibco) supplemented with 10% heat-inactivated fetal bovine serum and 100 IU/mL penicillin/streptomycin (Gibco).
Reagents and antibodies
Zombie aqua fixable viability kit, Annexin-V/7AAD kit, fixation buffer, permeabilization/wash buffer, recombinant mouse IL-2 (mIL-2) and Brefeldin-A solution were purchased from BioLegend. Antibodies for NF-kB, Bid and vinculin were procured from Cell Signaling Technology; antibodies for phospho-NF-kB p65, FAS, Bcl-2 and Bax were purchased from Santa Cruz Biotechnology; M2-flag antibody was purchased from Sigma; tumor necrosis factor-α (TNFα) and GAPDH antibodies were purchased from Proteintech; Granzyme B and COX IV antibodies were purchased from Abcam; secondary antibodies were procured from either Jackson ImmunoResearch Laboratories or Life Technologies.
Antibodies for flow cytometry
The antibodies for flow cytometry experiments: anti-mouse CD3; anti-mouse CD4; anti-mouse CD8a; anti-mouse CD25; anti-mouse CD38; anti-mouse CD69; anti-mouse CD70; anti-mouse CD11b; anti-mouse CD11c; anti-mouse CD19; anti-CD62L; anti-mouse CD80; anti-mouse CD86; anti-mouse CD68; anti-mouse F4/80; anti-mouse TNFα; anti-Granzyme B; anti-mouse/human CD44; anti-mouse CD183 (CXCR3); anti-mouse CD184 (CXCR4); anti-mouse CD185 (CXCR5); anti-mouse CD186 (CXCR6); anti-mouse CD1d; anti-mouse CD120a (TNFRSF1a); anti-mouse CD152 (CTLA-4); anti-mouse CD279 (PD-1); anti-mouse CD274 (B7-H1 or PD-L1); anti-mouse CD273 (B7-DC or PD-L2); anti-mouse CD117 (c-Kit); anti-mouse Ly-6A/E (SCA-1); anti-mouse Lineage Cocktail with Isotype Ctrl; mouse IgG1, κ Isotype; mouse IgG2b, κ Isotype Ctrl; mouse IgG2a, κ Isotype; rat IgG2b, κ Isotype; Armenian Hamster IgG Isotype; Syrian Hamster IgG Isotype; rat IgG2a, κ Isotype and rat IgG1, κ Isotype Ctrl were procured from BioLegend. The anti-mouse CD34, anti-perforin 1 antibodies and regulatory T cells (Treg) staining kit were purchased from eBioscience.
Isolation of splenocytes, thymocytes, lymph nodes, bone marrow cells and tumor cell suspension
The thymus, spleen, lymph nodes, bone marrow and tumors from mice were harvested immediately after euthanization. Single cell suspensions from thymus, spleen, lymph nodes, bone marrow and tumors were prepared using standard protocols.22 23
Purification of pan, CD4+ and CD8+ T cells
In brief, mice were sacrificed, and spleens were excised under sterile condition. The splenic pan T cells from single cell suspension were isolated on LS Columns (Miltenyi Biotec) in QuadroMACS Separator system (Miltenyi Biotec) using mouse Pan T Cell Isolation Kit II (Miltenyi Biotec). The CD8+ T cells were isolated by CD8a+ T cell isolation kit, mouse (Miltenyi Biotec) through negative selection and CD4+ T cells were isolated by positive selection from purified pan T cells, respectively.
Flow cytometry
For surface staining, cells were counted, washed with 1× phosphate-buffered saline (PBS) (pH 7.4) and non-specific sites were blocked by using TruStainfcX (anti-mouse CD16/32) antibody (BioLegend) for 5 min. Cells were stained with indicated antibodies for 20 min at room temperature (RT) in dark. For intracellular staining, cells were fixed in fixation buffer (200 µL/sample) for 20 min, washed twice in 1 mL of perm/wash buffer (5 min each) and indicated antibodies were added for 30 min at RT in dark. For nuclear staining, True-Nuclear Transcription Factor Buffer Set (BioLegend) was used. The cells were stained with Annexin-V and 7AAD in Annexin binding buffer (BioLegend) for cell death assays. The multi-color flow cytometry compensation was achieved using UltraComp eBeads (Thermo Fisher Scientific) and events were captured using flow cytometer (BD LSR Fortessa). The unstained and isotype control antibodies were used for gating. Data were analyzed by using FlowJo V.10 software (FlowJo, LLC).
In vitro and in vivo T cell activation
The pan T cells (2×106/well) were activated using 20 µL of anti-CD3ε and anti-CD28 antibodies loaded MACSiBead particles (Miltenyi Biotec) in the presence of 50 U/mL of rec mIL-2. The T cells were also activated with PMA/ionomycin cocktail (2 µL/mL; this cocktail contains phorbol-12-myristate 13-acetate (40.5 µM) and ionomycin (669.3 µM) in DMSO; BioLegend) for various time points. For blocking of soluble TNFα released from T cells, 1 µM Enbrel (Immunex Corporation) was used. The in vivo blocking of CD70 was achieved using anti-mouse CD70 monoclonal antibody (mAb; 100 µg/mouse; BioXcell). The rat IgG2b isotype control, anti-keyhole limpet hemocyanin (BioXcell), was used as an isotype control. For in vivo activation of T cells, mice were treated with single dose of LEAF purified anti-mouse CD3ε mAb (50 µg/mouse; BioLegend). Jurkat cell lines were activated with ImmunoCult Human CD3/CD28/CD2 T Cell Activator (Stem Cell Technology).
Effector function of CD8+ T cells
The chemotaxis assay of CD8+ T cells was performed using CytoSelect 96-well cell migration assay kit (Cell Biolabs) in the presence of CCL3 (BioLegend) and CCL4 (BioLegend), following manufacturer protocol. The reading was recorded at excitation 485 and emission 538 nm. For cytotoxicity assay using E.G7-OVA cells, WT and MLK3−/− mice were sensitized with OVA as described earlier.24 The CD8+ T cells termed as ‘effector cells’ were isolated from OVA-sensitized mice. Dendritic cells (DCs) from splenocytes of naive WT mice were purified (purity <60%) by CD11c micro beads (Miltenyi Biotec) and pulsed with OVA (2 µg/mL; InvivoGen) and matured with LPS (0.5 µg/mL; Sigma-Aldrich) for 24 hours. The effector and OVA-pulsed DCs were co-cultured for 24 hours and subsequently ‘target cells’ (eg,E.G7-OVA) were co-cultured for 6 hours. The intracellular GZMB in CD8+ T cells was estimated by flow cytometry.
Caspase-3 activity
The caspase-3 activity was determined using colorimetric method as described earlier.25 DEVD-AFC (Enzo Chemicals) was used as a substrate for determining caspase-3 activity. The fluorescence was recorded in plate reader (BioTek) at excitation 400/30 nm and emission 508/20 nm.
Immunofluorescence and immunohistochemistry
In brief, cells were fixed in 4% paraformaldehyde in PBS (pH 7.4) for 10 min at RT and permeabilized with 0.25% Triton X-100 in PBS for 10 min. Blocking was done by using 10% goat serum containing 0.25% Triton X-100 in PBS and incubated overnight at 4°C in primary antibody. The cells were subsequently incubated in secondary antibody for 3 hours and mounted with ProLong Gold Antifade Mountant with DAPI (Thermo Fisher). The images were captured by an inverted laser-scanning confocal microscope, equipped with an oil immersion lens (Carl Zeiss). The fluorescence signals were quantified using Image J software (NIH).
For immunohistochemistry, briefly, tumor sections were rehydrated and antigen retrieved in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) in Decloaking Chamber (Biocare Medical). The tissue were peroxide quenched, and non-specific staining was blocked by bovine serum albumin along with goat serum. The tissue sections were incubated overnight at 4°C with primary antibodies, subsequently incubated with secondary antibodies for 1 hour at RT and developed by DAB. The images were captured by Nikon Eclipse Ti microscope (Nikon).
Overexpression of MLK3 in Jurkat cells
The MLK3 (WT) lentivirus expression vectors were generated by the Gateway Technology. Briefly, MLK3 WT was amplified from their cDNAs and recombined into the donor vector (pDONR221) using BP clonase. The expression clone was created by recombining donor clone with the lentivirus destination vector using LR clonase. Lentivirus expression vectors were confirmed in transient transfection for MLK3 protein expression and packaged into the 293FT cells (Thermo Fisher) for viral particle production. Lentivirus titer was determined by the qRT-PCR kit (Clontech), and Jurkat cells were transduced with viral particles to overexpress either MLK3 (WT) or empty vector (Lac Z). The Jurkat cells stably expressing either MLK3 (WT) or empty vector were selected using blasticidin (5 µg/mL; Gold Bio).
The RT2 PCR array
The mouse CD8+CD38+ T cells from activated pan T cells were isolated using FACS (MoFlo Astrios—Beckman Coulter) and cDNA was prepared from RNA, following manufacturer protocol (Qiagen). The cDNA was used for RT² Profiler PCR array mouse apoptosis (Qiagen) using real-time PCR (ABI StepOnePlus). The RT² Profiler PCR array data were analyzed by using SA Biosciences software.
MLK3 inhibition and CD70 blockade in breast cancer xenograft model
Mouse breast cancer cell line, 4T1, was used to generate orthotopic xenograft in BALB/c mice. The 4T1 cell line was authenticated by short tandem repeat (STR) profiling and tested for any microbial or bacterial contamination before using in mice. The 4T1 cells (10,000/mouse) were implanted orthotopically in mammary fat pads of female Balb/c mice (10–12 weeks). The tumor-bearing mice with average tumor diameter of 5 mm were randomized into five groups and treated as follow: group 1—control; group 2—isotype control (first two dose 100 µg/mouse; next two dose of 50 µg/mouse); group 3—anti-mouse CD70 mAb (first two dose 100 µg/mouse; next two dose of 50 µg/mouse), group 4—URMC-099 (dose 7.5 mg/kg body weight daily) and group 5—URMC-099 (dose 7.5 mg/kg body weight daily) and anti-mouse CD70 mAb (first two dose 100 µg/mouse; next two dose of 50 µg/mouse). All treated and control mice were sacrificed on day 14. To deplete CD8+ T cells in tumor-bearing mice, anti-CD8 antibody (10 mg/kg, clone YTS 169.4, Bio X Cell) was administered,26 1 day prior to treatment with URMC-099. The second and third dose of anti-CD8 antibody was given on day 6 and day 11 from first day of URMC-099 administration. The serum and organs were used for enzyme-linked immunosorbent assay (ELISA), flow cytometry and immunohistochemistry (IHC) analyses.
Enzyme-linked immunosorbent assay
The serum TNFα concentration in 4T1 tumor-bearing mice was determined by using mouse TNF-alpha Quantikine HS ELISA kit (R&D Systems) following manufacturer’s protocol. The absorbance was recorded at 450 nm using a plate reader (BioTek).
Statistical analysis
To compare two groups, data were analyzed by Student’s t-test (unpaired, two-tailed). For more than two groups, the data were analyzed by one-way analysis of variance followed by Bonferroni’s multiple comparisons test. Data are presented as mean±SEM or mean±SD. p<0.05 was considered statistically significant. *p<0.05; **p<0.001 and ***p<0.0001.
Results
Genetic loss of MLK3 decreases murine CD8+ T cell population
The role of MLK3 in immune system is poorly understood. The size, shape and mass of immune organs like spleen and thymus are greatly influenced by immune modulation27 28; therefore, we examined the weights of spleen and thymus, and compared with body weights in MLK3−/− and WT mice. We noted a slight increase in spleen versus body weight ratio and thymus versus body weight ratio in MLK3−/− mice compared with WT (figure 1A and online supplementary figure S1A). Furthermore, estimation of splenic B and T cell populations indicated a significant decrease in T cell population in the absence of MLK3 (figure 1B). On further examination of T cell subsets, there was significant decrease in splenic CD8+ T cell population in MLK3−/− mice, whereas the CD4+ T cells were partially increased (figure 1C). However, there was no significant change in the regulatory T cell (Treg) population due to loss of MLK3 (online supplementary figure S1B). The absolute counts of splenic pan T cells and CD8+ T cells were also decreased in MLK3−/− mice compared with WT (figure 1D,E). Since there was relative increase in CD4+ T cell population due to loss of MLK3 and NF-kB activity is associated with T cell survival,29 we therefore estimated total and phospho-NF-kB expressions in T cells. The expression of total and phospho-NF-kB was higher in CD4+ compared with CD8+ T cells from MLK3−/− mice (online supplementary figure S2A–C). To explore how loss of MLK3 decreases CD8+ T cell population, various parameters that regulate T cell development and maturation were determined. The hematopoietic progenitor (c-Kit+, Lin− and SCA-1+ cells aka KLS) and stem (c-Kit+Lin−SCA-1+CD34dim aka CD34−/dimKSL cells) cells from bone marrow of WT and MLK3−/− mice were isolated, and flow cytometry showed partial increase in progenitor and hematopoietic stem cells in MLK3−/− mice (online supplementary figure S3). The T cells go through progressive developmental stages in thymus and since we observed a significant decrease in splenic CD8+ T cell population in MLK3−/− mice, therefore, we estimated various stages of thymic T cell development, including DN1 (CD44+CD25−), DN2 (CD44+CD25+), DN3 (CD44−CD25+), DN4 (CD44−CD25−) and DP (CD4+CD8+). There was no effect of MLK3 loss on thymic T cell development (figure 1F) and loss of MLK3 had no significant effect on either CD4 or CD8 thymic single positive (CD4 SP and CD8 SP) populations (figure 1G). Since we did not observe any defect in thymic T cell development due to loss of MLK3, we therefore analyzed status of various surface molecules on splenocytes. The expression of several splenic surface molecules was increased including CD70 in MLK3−/− mice compared with WT (online supplementary table S1). Taken together, these results demonstrate that genetic loss of MLK3 decreases CD8+ T cell population.
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material

MLK3 depletion induces decrease in CD8+ T cell population. (A) Representative images of spleen (left) and spleen versus body weight ratio (right) in WT and MLK3−/− mice (n=5 mice/group). (B) Representative contour plots (left) and quantification (right) for expression of CD19 and CD3 gated on live splenocytes, analyzed by flow cytometer (n=4 mice/group). (C) Representative contour plots (left) and quantification (right) of splenic CD8+ and CD4+ populations, gated on T cells (n=5 mice/group). (D and E) Representative contour plots (left) and quantification (right) for precision count of T cells and CD8+ T cells in splenocytes by flow cytometer (n=3 mice/group). (F) Representative pseudo color plots (left) and quantification (right) of T cell developmental phases in thymocytes isolated from WT and MLK3−/− mice. (n=3 mice/group). (G) Representative pseudo color plots (left) and quantification (right) of double positive (DP), CD4+ single population (CD4 sp) and CD8+ single population (CD8 sp) in thymus isolated from WT and MLK3−/− mice (n=3). For figure A, B, C, D, E and G, values are the mean±SEM and for figure F, values are the mean±SD. P values (*p<0.05) for knockout versus WT by unpaired t test. MLK3, mixed lineage kinase 3; WT, wild type.
Loss of MLK3 is associated with CD70 upregulation and apoptosis in CD8+ T cells
Since we observed decline in CD8+ T cell population in MLK3−/−, we wanted to know why and how loss of MLK3 regulates CD8+ T cell population. We observed that several surface molecules were altered on splenocytes due to loss of MLK3, including significant upregulation of CD70 expression (online supplementary table S1). Further analyses of splenic T cell populations showed significant increase in CD70 expression on splenic CD4+ and CD8+ T cells (figure 2A,B). To further understand any functional relation between MLK3 and CD70, the expression of CD70 was measured in Jurkat cells, stably expressing either MLK3 (WT) or vector control (figure 2C). The basal expression of CD70 was higher in vector control compared with MLK3 (WT) Jurkat cells (figure 2D). The CD70 has been implicated in apoptosis of immune effector cells30 and to directly prove involvement of MLK3-regulated CD70 in CD8+ T cell apoptosis; the CD70 was blocked in vivo using anti-CD70 mAb (clone FR70) (figure 2E). On loss of MLK3, CD70 blockade did not decrease apoptosis in CD4+ T cells (figure 2F); however, blockade of CD70 attenuated apoptosis in CD8+ T cells derived from MLK3−/− mice (figure 2G). Collectively, these results suggest that loss of MLK3 induces CD70 expression and cell death in CD8+ T cells, and thus blockade of CD70 should increase the life span of CD8+ T cells.
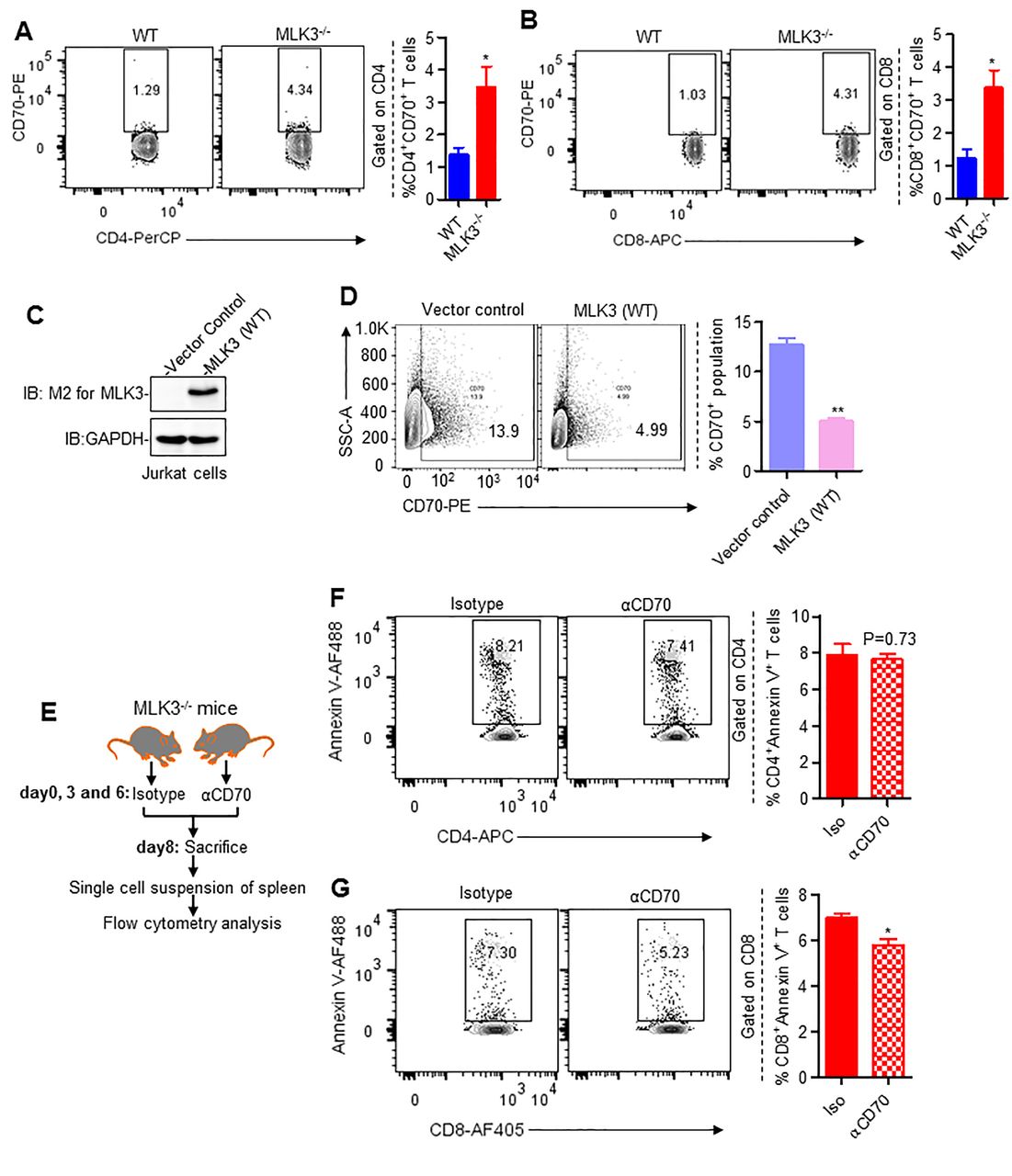
Loss of MLK3 promotes CD70-mediated apoptosis in CD8+ T cells. (A and B) Representative contour plots (left) and quantification (right) of CD4+CD70+ and CD8+CD70+ T cell populations, gated on splenic CD4+ and CD8+ T cells, respectively. Values are mean±SEM, *p<0.05 for knockout versus WT by unpaired t test (n=4 mice/group). (C) Protein expression of M2-MLK3 in lysates of vector control and MLK3 (WT) overexpressing Jurkat cells. GAPDH was taken as loading control. (D) Representative contour plots (left) and quantification (right) of CD70 expression on vector control and MLK3 (WT) overexpressing Jurkat cells. Values are the mean±SEM, **p<0.001 for MLK3 (WT) versus vector control by unpaired t test (n=3). (E) Experimental plan for figure F and G. (F and G) Representative contour plots (left) and quantification (right) of splenic CD4+Annexin V+ T cells and CD8+Annexin V+ T cell population. Values are the mean±SEM, p values (*p<0.05) for αCD70 mAb versus isotype control by unpaired t test (n=5 mice/group). mAb, monoclonal antibody; MLK3, mixed lineage kinase 3; WT, wild type.
MLK3 deficiency promotes apoptosis in activated CD8+ T cells
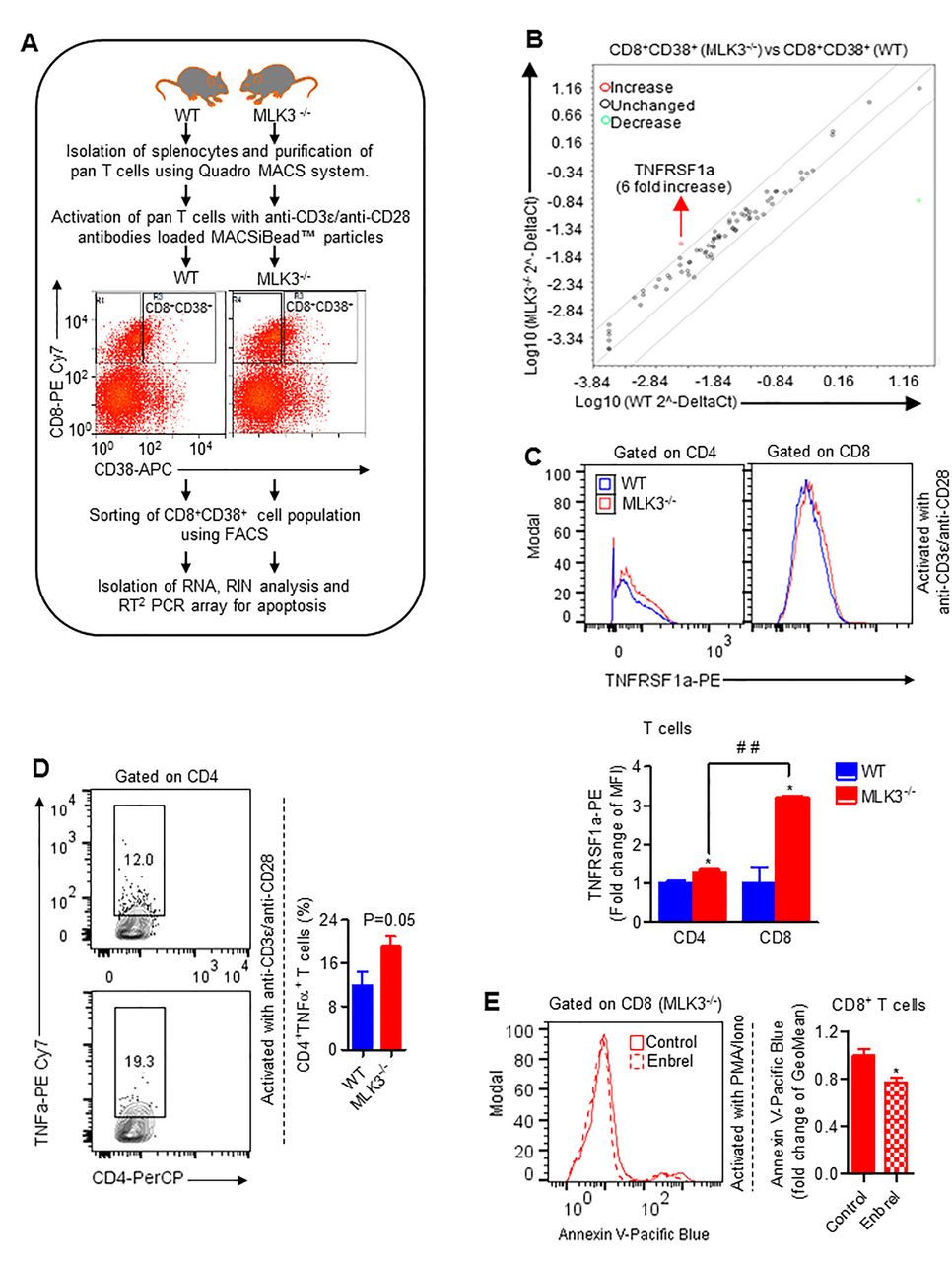
The activated T cells after achieving its effector function undergo a process of cell death.31 To explore the role of MLK3 in activation-induced apoptosis, we first used Jurkat cells, expressing MLK3 (WT) and estimated cell death on activation with ImmunoCult (ie, anti-CD3/CD28/CD2 antibodies). There was significant diminished apoptosis and caspase-3 activity in MLK3 (WT) Jurkat cells compared with vector control (figure 3A,B). Importantly, the expression of CD70 was lower in activated Jurkat cells expressing MLK3 (WT) compared with vector control (figure 3C). In a complimentary experiment, the pan T cells were isolated from MLK3−/− and WT mice and activated with anti-CD3ε and anti-CD28 antibodies loaded MACSiBead particles and markers for different T cell phenotypes were determined. There were no major changes in CD44lowCD62Lhigh (ie, naive), CD44highCD62Lhigh (ie, central memory) and CD44highCD62Llow (ie, effector memory) phenotypes due to loss of MLK3. However, CD8+ T cell activation was significantly increased due to loss of MLK3 (online supplementary figure S4A,B). The flow cytometry analyses indicated that loss of MLK3 induces T cell apoptosis (figure 3D) and caspase-3 activity on activation (figure 3E). Since CD8+ T cell population was diminished at basal level due to loss of MLK3, we planned to determine the effect of MLK3 on cell death/survival of T cell subsets on activation. The result showed that CD8+ T cells, but not CD4+ T cells, were more sensitive to cell death due to loss of MLK3 (figure 3F; online supplementary figure S5A). Moreover, the activated phenotypes of CD8+ T cells, including CD8+CD25+, CD8+CD38+ and CD8+CD69+ T cells from MLK3−/− mice, had higher apoptosis compared with WT (figure 3G). The apoptosis among all the activated phenotypes of CD8+ T cells, the CD8+CD38+ T cells from MLK3−/− had the maximum apoptosis compared with WT mice (figure 3G). To understand the mechanism of apoptosis in activated CD8+ T cells due to loss of MLK3, the apoptosis-associated genes in CD8+CD38+ T cells were determined by RT2 PCR array (figure 4A). Several genes were altered due to loss of MLK3, including six fold increase in TNFRSF1a in CD8+CD38+ T cells from MLK3−/− mice (figure 4B; online supplementary table S2). The TNFRSF1a protein expression was also increased on CD8+ T cells from MLK3−/− mice (figure 4C). The soluble TNFα is a ligand of TNFRSF1a and CD4+ T cells are known to produce TNFα.32 Therefore, we estimated intracellular TNFα in activated CD4+ T cells from WT and MLK3−/− mice; the TNFα was elevated in CD4+ T cells derived from MLK3−/− mice (figure 4D). To understand the contribution of TNFα-TNFRSF1a axis in MLK3-mediated apoptosis in T cells, pan T cells derived from MLK3−/− mice were activated in absence and presence of Enbrel, an antagonist of TNFα,33 and T cell apoptosis was determined. The blocking of TNFα by Enbrel decreased apoptosis in CD8+ T cells but not in CD4+ T cells isolated from MLK3−/− mice (figure 4E and online supplementary figure S5B). Together our results demonstrate that loss of MLK3 upregulates TNFRSF1a to promote apoptosis in activated CD8+ T cells.
Supplemental material
Supplemental material
Supplemental material
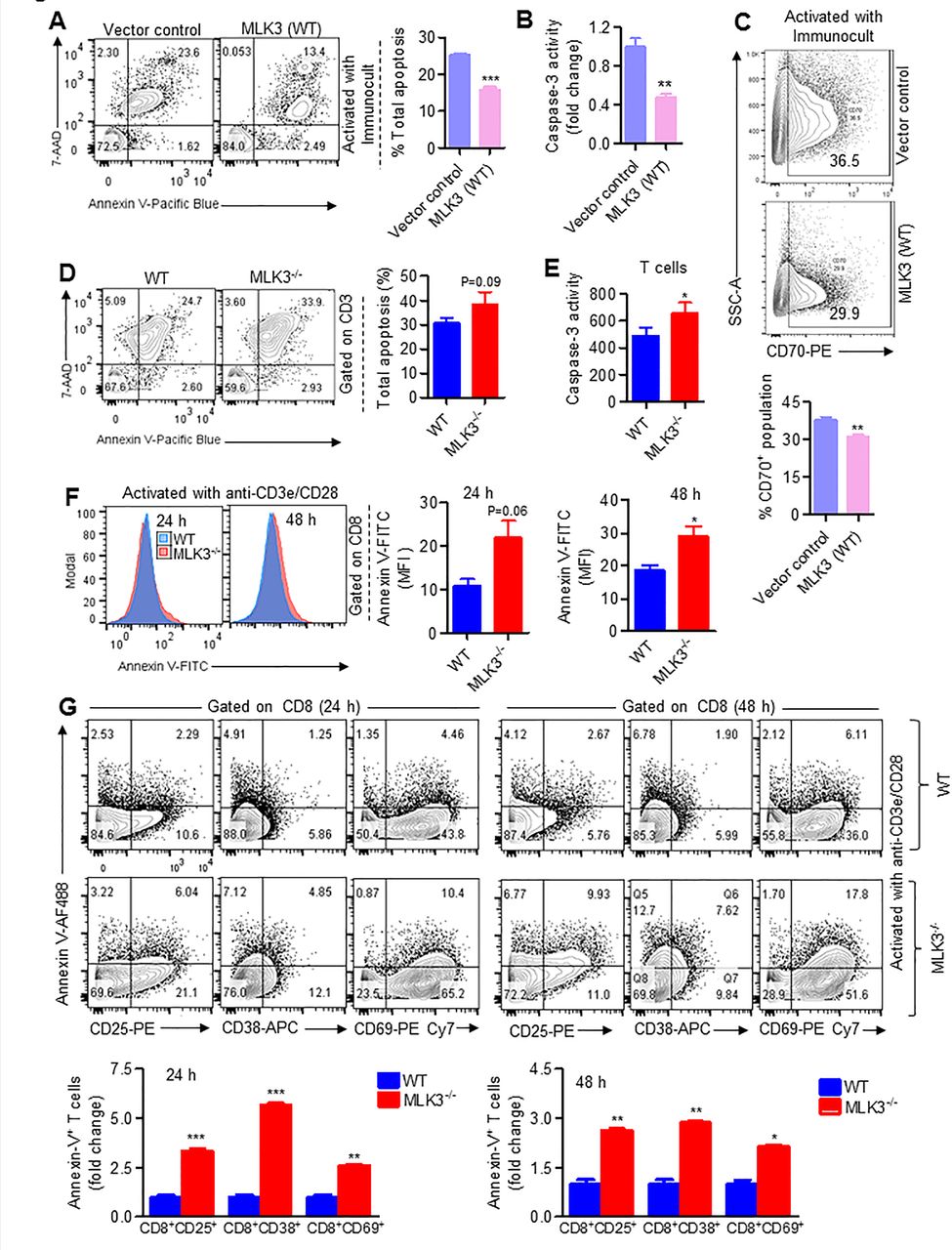
Loss of MLK3 promotes activation-induced apoptosis in CD8+ T cells. (A) Representative contour plots (left) and quantification (right) of cell death and apoptosis in activated vector control and MLK3 (WT) overexpressing Jurkat cells. Values are the mean±SEM, ***p<0.0001 for MLK3 (WT) versus vector control by unpaired t test (n=3). (B) Caspase-3 activity in cell lysates of vector control and MLK3 (WT) overexpressing Jurkat cells on activation with ImmunoCult for 72 hours. DEVD-AFC was taken as substrate for caspase-3 activity. Values are the mean±SD, **p<0.001 for MLK3 (WT) versus vector control by unpaired t test (n=3). (C) Representative contour plots (upper panel) and quantification (lower panel) of CD70 expression on activated vector control and MLK3 (WT) overexpressing Jurkat cells. Values are the mean±SEM, **p<0.001 for MLK3 (WT) versus vector control by unpaired t test (n=3). (D) Representative contour plots (left) and quantification (right) of cell death and apoptosis, gated on activated T cells from WT and MLK3−/− mice. Values are the mean±SEM, p value for knockout versus WT by unpaired t test (n=6 mice/group). (E) Caspase-3 activity in cell lysates of activated pan T cells from WT and MLK3−/− mice. Values are the mean±SEM, *p<0.05 for knockout versus WT by unpaired t test (n=4 mice/group). (F) Representative histogram plots (left) and quantification (right) of annexin V expression, gated on CD8+ T cells isolated from WT and MLK3−/− mice. Values are the mean±SEM, p values (*p<0.05) for knockout versus WT by unpaired t test (n=3 mice/group). (G) Representative contour plots (upper panel) and quantification (lower panel) of CD25+Annexin V+, CD38+Annexin V+ and CD69+Annexin V+ population, gated on CD8+ T cells from WT and MLK3−/− mice. Values are the mean±SD, *p<0.05, **p<0.001 and ***p<0.0001 for knockout versus WT by unpaired t test (n=3 mice/group). MLK3, mixed lineage kinase 3; WT, wild type.

Loss of MLK3 induces TNFα-TNFRSF1a axis-mediated apoptosis in CD8+ T cells. (A) Experimental approach for RT2 PCR array for apoptosis-associated genes . (B) RT2 PCR array for apoptosis-associated genes in purified CD8+CD38+ T cells derived from WT and MLK3−/− mice (pooled, n=3 mice/group). (C) Representative histogram plots (upper panel) and quantification (lower panel) of TNFRSF1a surface expression, gated on CD4+ and CD8+ T cells, respectively. Values are the mean±SD, *p<0.05 for knockout versus WT by unpaired t test and # #p<0.001 for CD8+ T cells (from knockout) versus CD4+ T cells (from knockout) by unpaired t test (n=2 mice/group). (D) Representative contour plots (left) and quantification (right) of intracellular TNFα expression, gated on CD4+ T cells. Brefeldin-A (1 µg/mL) was added in culture medium 12 hours before harvesting the cells. Values are the mean±SEM, p value for knockout versus WT by unpaired t test (n=4 mice/group). (E) Representative histogram plots (left) and quantification (right) of Annexin V expression, gated on CD8+ T cells in presence and absence of Enbrel. Values are the mean±SD, *p<0.05 for Enbrel versus control by unpaired t test (n=7 mice/group). FACS, fluorescence-activated cell sorting; MLK3, mixed lineage kinase 3; RIN, RNA integrity number; TNFα, tumor necrosis factor-α; WT, wild type.
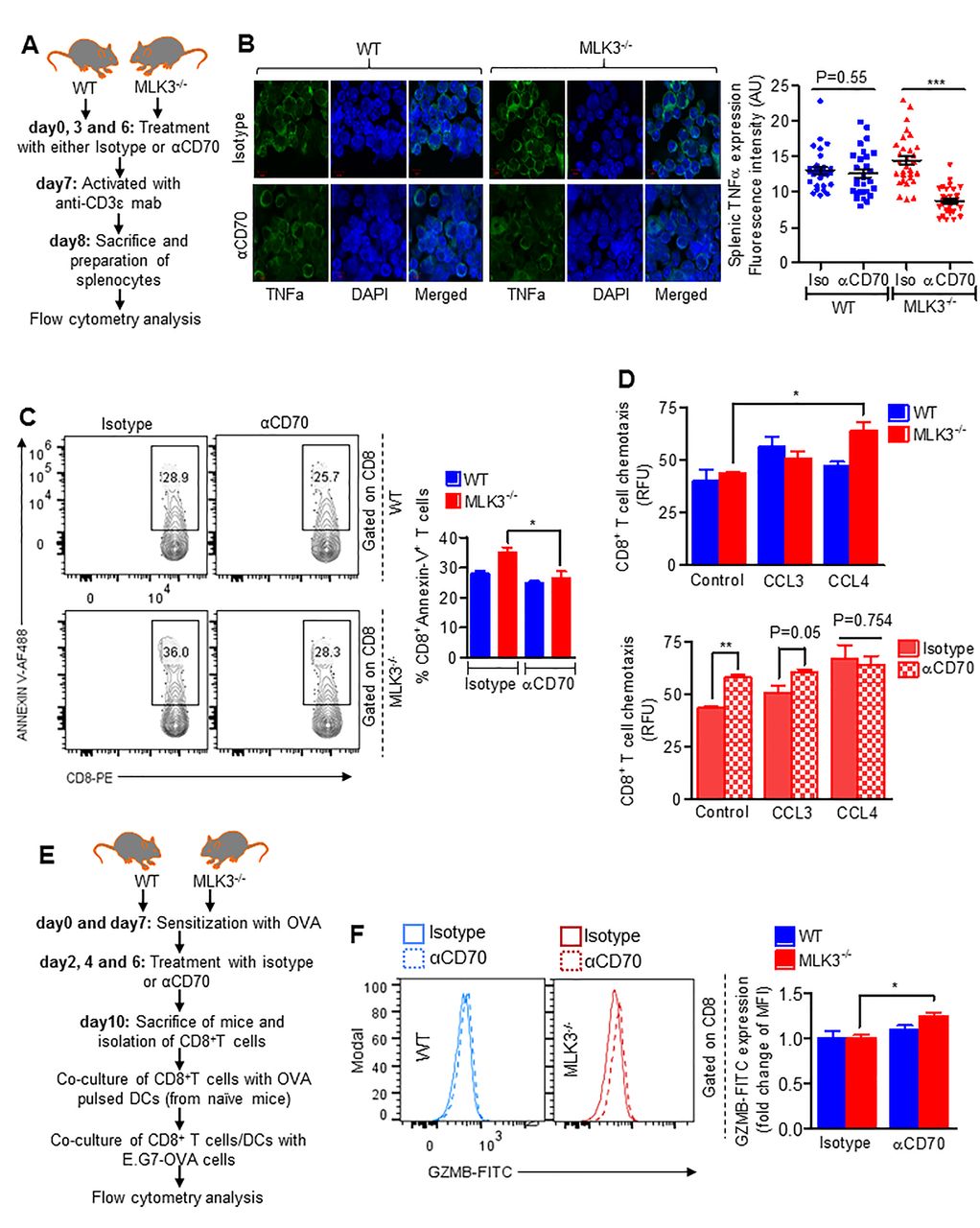
Blockade of CD70 induces survival and effector function of MLK3-deficient CD8+ T cells
Since TNFα and CD70 both were elevated in T cells derived from MLK3−/− mice and induced apoptosis in CD8+ T cells, we wanted to determine any physiological relation between CD70 and TNFα in mediating CD8+ T cell apoptosis on activation. The CD70 was first blocked and then T cells were activated in vivo (figure 5A); the blockade of CD70 decreased TNFα level in splenocytes of MLK3−/− mice compared with isotype-treated mice (figure 5B) and CD8+ T cell apoptosis was attenuated in MLK3−/− mice treated with anti-CD70 (figure 5C). We next sought to determine how loss of MLK3 and blockade of CD70 affect CD8+ T cell effector function. First, we determined the release of cytokines by activated CD8+ T cells (from MLK3−/− and WT mice) using proteome profiler array. The array analyses showed that loss of MLK3 promotes release of CCL3 and CCL4 (online supplementary figure S6A). We also determined the expressions of different chemokine receptors under activated conditions in CD8+ T cells from WT and MLK3−/− mice. Interestingly, under activated conditions, the expression of CXCR3 on CD8+ T cell was upregulated in the absence of MLK3 (online supplementary figure S6B). Both CCL3 and CCL4 are reported to induce CD8+ T cell migration,34 and therefore, we determined CCL3-induced and CCL4-induced chemotaxis of CD8+ T cells, isolated from WT and MLK3−/− mice on CD70 blockade and in vivo activation (figure 5A). The chemotaxis assay indicated an increased migration of CD8+ T cells derived from MLK3−/− mice on CD70 blockade (figure 5D). To understand the role of MLK3 and CD70 in CD8+ T cell cytotoxicity, OVA-sensitized WT and MLK3−/− mice were treated either with anti-CD70 or isotype control, and CD8+ T cells were purified. These CD8+ T cells were co-cultured with OVA peptide-pulsed DCs from naive WT mice, along with E.G7-OVA cells (figure 5E). The flow cytometry analyses indicated a significantly increased Granzyme B (GZMB) expression in CD8+ T cells derived from MLK3−/− mice treated with anti-CD70 (figure 5F). Taken together, these results suggest that blockade of CD70 promotes life and effector function of CD8+ T cells in the absence of MLK3.
Supplemental material

CD70 blockade increases life and effector function of CD8+ T cells deficient in MLK3. (A) Experimental plan of CD70 blockade and activation of T cells in vivo (for figure (B–D)). (B) Representative images for protein expression of TNFα (left) and quantification (right) in splenocytes isolated from WT and MLK3−/− mice. For quantification of TNFα, fluorescence in single cell from three different images was quantified by Image J software. Values are the mean±SEM, p values (***p<0.0001) for αCD70 mAb treatment versus isotype control by unpaired t test (n=30 cells/group). (C) Representative contour plots (left) and quantification (right) of splenic Annexin V+CD8+ T cells on in vivo CD70 blockade and T cell activation. Values are the mean±SEM, *p<0.05 for αCD70 mAb versus isotype control by unpaired t test (n=3 mice/group). (D) CD8+ T cell chemotaxis in the presence of CCL3 or CCL4 (each, 100 ng/mL) for 3 hours. Values are the mean±SEM, p values (*p<0.05, **p<0.001) for CCL3 or CCL4 versus control by one-way analysis of variance, Bonferroni's multiple comparison test (upper panel) or αCD70 mAb versus isotype control by unpaired t test (lower panel) (n=3 mice/group). (E) Experimental approach for figure F. (F) Histogram plot of intracellular GZMB expression (left) and quantification (right), gated on CD8+ T cells. Values are the mean±SD, *p<0.05 values for αCD70 mAb treatment versus isotype control by unpaired t test (n=4 mice/group). DAPI, 4′, 6-diamidino-2-phenylindole; DCs, dendritic cells; FITC, fluorescein isothiocyanate; MLK3, mixed lineage kinase 3; OVA, ovalbumin; RFU,relative fluorescence units; WT, wild type.
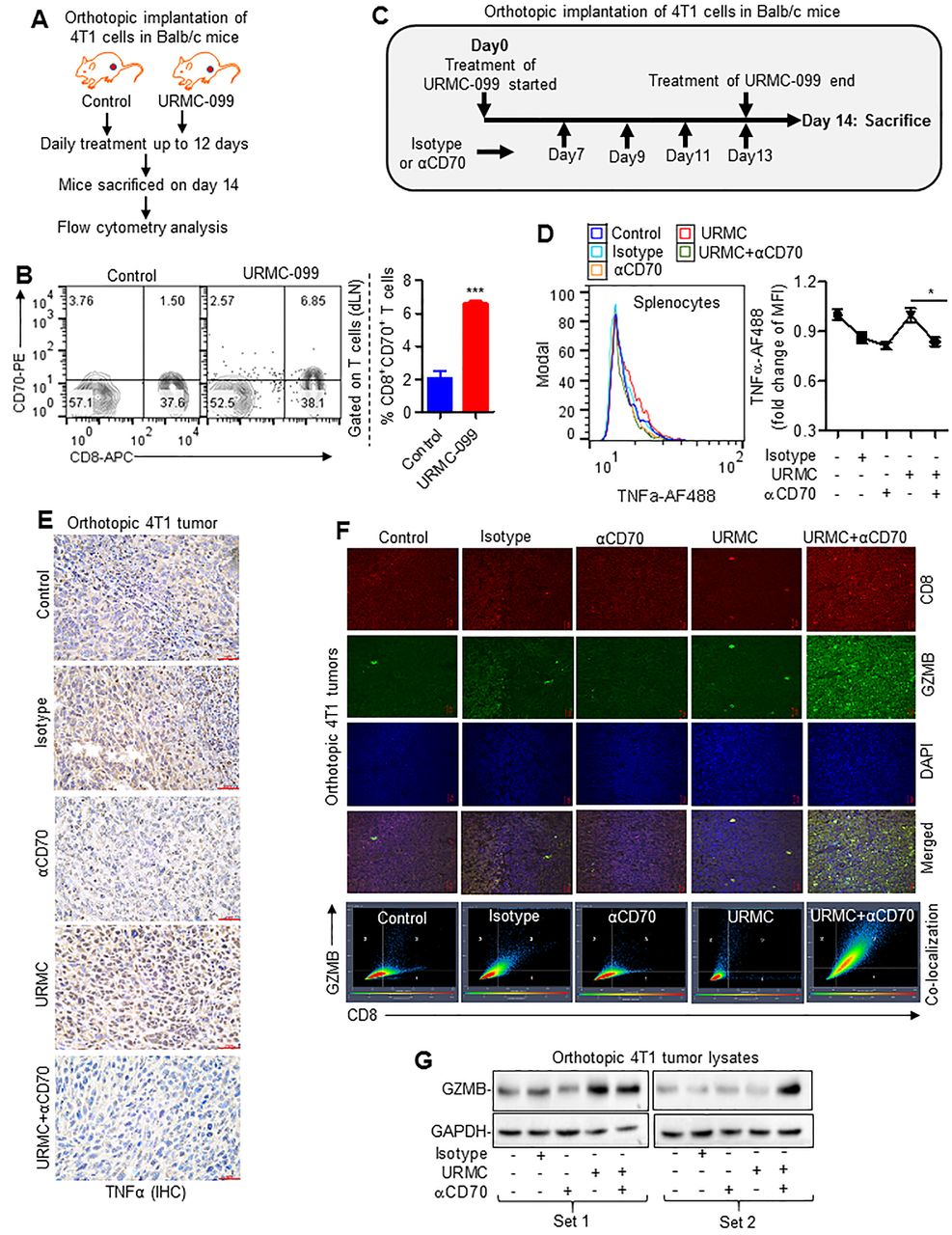
The combined inhibition of MLK3 and CD70 increases cytotoxic CD8+ T cell infiltration and reduces breast tumor burden
The small molecule URMC-099 is reported as a specific inhibitor of MLK3.35 To determine the in vivo efficacy of URMC-099 on T cell function, similar to genetic loss of MLK3, the C57BL/6 mice were treated with MLK3 inhibitor (online supplementary figure S7A). The hematopoietic stem cell population (ie, c-Kit+Lin−SCA-1+CD34dim) in bone marrow was increased in treated mice compared with non-treated group (online supplementary figure S7B), as seen in MLK3−/− mice (online supplementary figure S3). To determine that URMC-099 also affects activation-associated T cell death, similar to MLK3 loss, the pan T cells were isolated from splenocytes of control and URMC-099-treated mice and subjected to activation using anti-CD3ε and anti-CD28 antibodies loaded MACSiBead particles. The result showed increased expression of CD70 (online supplementary figure S7C) associated with higher apoptosis in CD8+ T cells from mice pretreated with URMC-099 (online supplementary figure S7D).
Supplemental material
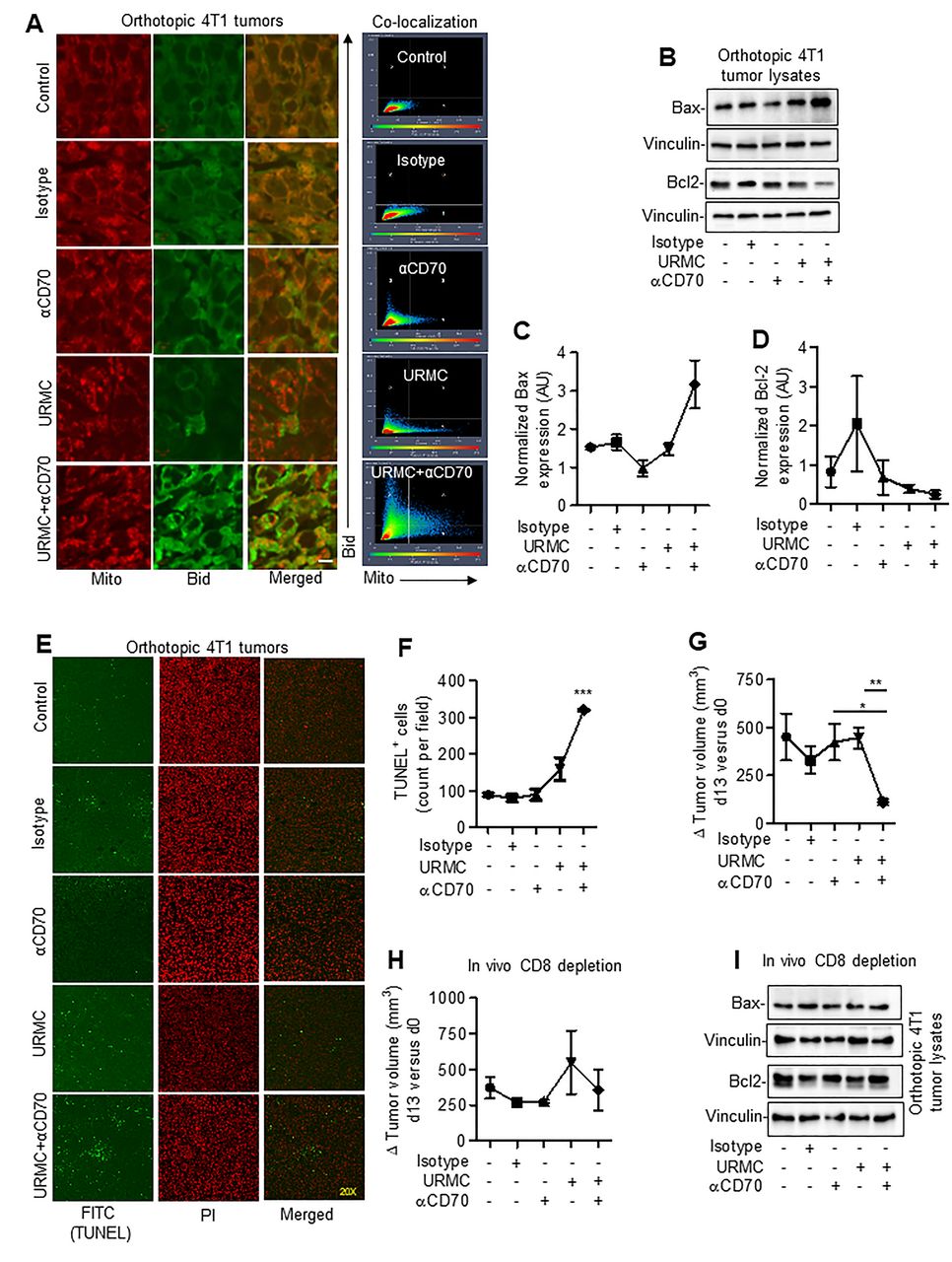
To understand the physiological significance of MLK3-regulated CD70 expression in CD8+ T cells and its impact on tumor immunity, expression of CD70 on CD8+ T cells derived from draining lymph node (dLN) of 4T1 breast tumor-bearing mice treated with MLK3 inhibitor (ie, URMC-099) was determined (figure 6A). The URMC-099 treatment increased the CD8+CD70+ T cell population in dLN compared with control mice (figure 6B). Since we observed that increase in CD70 due to loss/inhibition of MLK3 was associated with TNFα-TNFRSF1a-mediated apoptosis in CD8+ T cells, therefore we determined TNFα in splenocytes. Interestingly, combined blockade of MLK3 and CD70 significantly decreased TNFα level in comparison with MLK3 inhibition alone (figure 6C,D). Further analysis of peripheral CD4+ T cells indicated a partial increase in CD4+TNFα+ T cell population on MLK3 inhibition, which was reduced on blocking of CD70 (online supplementary figure S8A). The tumor infiltrating CD4+TNFα+ T cell population was similar in both control and URMC-099-treated mice. However, the combined inhibition of MLK3 and CD70 significantly decreased the CD4+TNFα+ T cell population in tumors (online supplementary figure S8B). Similar to results with splenocytes, TNFα protein expression was also significantly decreased in breast tumors in mice treated with MLK3 and CD70 inhibitors (figure 6E). Interestingly, circulating TNFα level was below detection limit (less than 0.80 pg/mL) in serum of tumor-bearing mice treated with combination of MLK3 and CD70 inhibitors (online supplementary table S3). Remarkably, combined blockade of MLK3 and CD70 significantly increased the numbers of tumor infiltrating CD8+ T cells and increased the GZMB expressing tumor infiltrating CD8+ T cells (figure 6F). We also estimated the GZMB protein expression in tumor lysates and observed an overall increase, especially in mouse tumors treated with URMC-099 and anti-CD70 mAb together (figure 6G). We also estimated perforin 1 (PRF) expression in peripheral and tumor infiltrating CD8+ T cells. There was slight increase in perforin expression in peripheral CD8+ T cells; however, its expression was increased almost two folds in tumor infiltrating CD8+ T cells, in mice treated with MLK3 and CD70 inhibitors together (online supplementary figure S8C,D). Since GZMB expressing tumor infiltrating CD8+ T cells were increased on combined inhibition of MLK3 and CD70, and CD8+ T cells release GZMB that ultimately activates Bid,36 therefore, we determined expression and mitochondrial localization of Bid in mouse tumors. The IHC results showed overall increased expression of Bid (online supplementary figure S9A) and mitochondrial localization in tumor sections treated with URMC-099 and anti-CD70 mAb together (figure 7A). We also examined protein expression of additional proteins involved in intrinsic cell death pathway, like Bax and Bcl-2 in 4T1 breast tumor lysates. There was significant increase in the expression of proapoptotic protein Bax, whereas the expression of prosurvival Bcl2 was decreased on blockade with URMC-099 and anti-CD70 mAb together (figure 7B–D). Since Bid, Bax and Bcl2 proteins mediate intrinsic cell death pathway, we therefore also measured protein expression of Fas that participate in extrinsic cell death pathway. The expression of Fas was equally increased by URMC and anti-CD70 alone or in combination (online supplementary figure S9B,C). Furthermore, we also measured the effector caspase-3 activity, which was increased on combined blockade of MLK3 and CD70 (online supplementary figure S9D). The in situ terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay with tumor sections also showed increased apoptosis on combined blockade of MLK3 and CD70 (figure 7E,F). These results collectively suggest that combined blockade of MLK3 and CD70 increases infiltration of cytotoxic CD8+ T cells within tumors and that finally promote breast cancer cell death and decrease in overall tumor burden (figure 7G and online supplementary figure S9E).
Supplemental material
Supplemental material
Supplemental material

Combined blockade of MLK3 and CD70 increases tumor infiltration of cytotoxic CD8+ T cells. (A) Experimental approach for figure B. (B) Representative contour plots (left) and quantification (right) of CD8+CD70+ T cells, gated on T cells derived from dLN of tumor-bearing mice treated either with control or URMC-099. Values are the mean±SEM, ***p<0.0001 for URMC-099 versus control by unpaired t test (n=4 mice/group). (C) Experimental plan for combination therapy in 4T1 breast cancer model. (D) Representative contour plots (left) and quantification (right) of intracellular TNFα expression in splenocytes from control and treated tumor-bearing mice. Values are the mean±SD, *p<0.05 for combination of URMC-099 and αCD70 mAb versus URMC-099 alone treatment by unpaired t test (n=3 mice/group). (E) Representative IHC images of TNFα expressions in tumor sections from control, isotype, anti-CD70 mAb, URMC-099 and combination of URMC-099 and anti-CD70 mAb-treated mice (n=3 mice/group). Size bar=20 µm. (F) Representative confocal microscopy images of CD8 and GZMB expressions in tumor sections from control and treated mice (n=3 mice/group). Co-localization data were generated from confocal microscopy images. Size bar=20 µm. (G) Protein expressions of GZMB in tumor lysates isolated from control and treated mice. GAPDH was taken as loading controls. dLN, draining lymph node; IHC, immunohistochemistry; MLK3, mixed lineage kinase 3; TNFα, tumor necrosis factor-α.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Combined blockade of MLK3 and CD70 induces mitochondrial apoptosis in tumor cells. (A) Representative confocal microscopy images of Bid protein expression in tumor sections from control, isotype, anti-CD70 mAb, URMC-099 and combination of URMC-099 and anti-CD70 mAb-treated mice (n=3 mice/group). Co-localization data are generated from confocal microscopy images. Size bar=5 µm. (B) Protein expressions of Bax and Bcl-2 in tumor lysates isolated from control and treated mice and (C and D) quantification by densitometry using Image Lab software. Vinculin was taken as loading controls. Values are the mean±SEM (n=2 mice/group). (E) In situ terminal deoxynucleotidyl transferase dUTP nick endlabeling (TUNEL) assay for determination of apoptosis in tumor sections from control and treated mice (n=3 mice/group). (F) For quantification of FITC positive (TUNEL) cells, Image J software was used. Magnification=20×. Values are the mean±SEM, ***p<0.0001 values treatment versus control by one-way analysis of variance, Bonferroni's multiple comparison test (n=3 mice/group). (G) Change in tumor volume (day 13 to day 0) in control and treated mice. Values are the mean±SEM, p value (*p<0.05 and **p<0.001) for combination of URMC-099 and αCD70 mAb versus URMC-099 alone or anti-CD70 alone treatment by unpaired t test (n=5 mice/group, except isotype treatment where n=4 mice/group). (H) Change in tumor volume (ie, day 13 to day 0) in CD8+ T cell depleted mice, control or treated with: isotype, anti-CD70, URMC-099 and combination of anti-CD70+URMC−099. Values are the mean±SEM (n=3 mice/group). (I) Protein expressions of Bax and Bcl-2 in tumor lysates, isolated from control, and treated (as indicated in figure 7H above) mice. Vinculin was taken as loading controls (n=3 mice/group). MLK3, mixed lineage kinase 3.
We next sought to determine the direct role of CD8+ T cells in antitumor efficacy of combined inhibition of MLK3 and CD70. The CD8+ T cells were depleted in tumor-bearing mice using mAb. The tumor volume and protein expressions of Granzyme B, Bid, Bax, Bcl-2 and cleaved caspase-3 were determined in tumors at the conclusion of the experiment. The depletion of CD8+ T cells almost completely blocked the antitumor efficacy of combined MLK3 and CD70 inhibitors (figure 7H; online supplementary figure S10A). The protein analyses of tumor lysates did not show any significant changes in Bax and Bcl-2 expression between control and treated mice (figure 7I and online supplementary figure S10B,C). There was no change in expression of GZMB, Bid and c-caspase-3, either due to the combined (MLK3 and CD70 inhibitors) or single inhibitor treatments (online supplementary figure S10D). Collectively, these results suggest that combined inhibition of MLK3 and CD70 induces CD8+ T cell-mediated mitochondrial apoptosis in tumor cells.
Supplemental material
Discussion
The immune-modulating effects of targeted therapies against the MAPKs along with immunomodulatory antibodies, targeting checkpoint inhibitors like PD1/PDL1 and CTLA4, have shown efficacy in various malignancies.37 The MLKs/MLK3 are reported to have either prosurvival or prodeath functions in solid malignancies; however, its impact on immune cells is not established.19–21 For the first time, our results demonstrate that MLK3 plays a prosurvival role in CD8+ T cells and its loss/inhibition promotes CD70-mediated apoptosis in CD8+ T cells. Interestingly, blockade of CD70 in the absence of MLK3 was able to increase life span as well as effector function of CD8+ T cells. While loss of MLK3 induces CD70-mediated apoptosis in CD8+ T cells, however in CD4+ T cells, the MLK3 loss increases CD4+ T cell population, suggesting that MLK3 plays a paradoxical role in CD4+ and CD8+ T cell survival. The transcription factor, NF-kB, has been implicated in MAPK-dependent survival/death of different cell types, and therefore, to delineate the differential function of MLK3 in CD8+ and CD4+ T cells, we determined the total and activated (phospho-NF-kB) NF-kB expressions. Even though the expression of total NF-kB was comparatively lower in CD4+ T cells under basal condition, however, it was significantly increased on loss of MLK3. Moreover, the relative expression of activated NF-kB was significantly upregulated in CD4+ compared with CD8+ T cells. These results partly support that perhaps this differential regulation of NF-kB by MLK3 is the basis of disparity in CD4+ and CD8+ T cell survival. Interestingly, the downstream target of MLK3, JNK has also been reported to play a differential role in CD8+ and CD4+ T cell survival.13 14
It is known that activated T cells undergo cell death on performing their effector function. Our phenotypic assessment for activated CD8+ T cells that is more prone to cell death due to loss of MLK3 identified that CD8+CD38+ T cells were dependent on MLK3 expression. Furthermore, the loss of MLK3 upregulated TNFRSF1a on CD8+ T cells, which was much higher compared with CD4+ T cells, leading to TNFRSF1a-dependent apoptosis in activated CD8+ T cells. The TNFRSF1a is an established receptor for soluble TNFα, and it has also been reported that mutation in TNFRSF1a decreases TNFα-induced apoptosis.32 38 39 TNFα is regulated by the transcription factor, NF-kB,40 and we observed that loss of MLK3 was able to induce NF-kB in CD4+ T cells, and therefore, we can speculate that induction of NF-kB can increase TNFα production due to loss of MLK3. We also observed that loss of MLK3 induced CD70 expression on T cells, and it is reported that treatment of human peripheral blood mononuclear cells with CD70 induces rapid decrease of inhibitor of kappa B (IκBα), indicating that CD70-CD27 axis can trigger activation of the canonical NF-kB pathway and this could result in TNFα induction/synthesis.41 Interestingly, the blockade of CD70 decreased TNFα protein expression in splenocytes of MLK3−/− mice. Since the ligand of TNFRSF1a (ie, TNFα) was decreased upon blockade of CD70, this observation supports our result of improved survival of activated CD8+ T cells in the absence of MLK3.
Interestingly, the blockade of CD70 in the absence of MLK3 also increased the effector functions of CD8+ T cells, including migration and cytotoxicity. T cell migration is a key process that allows activated T cells to traffic to inflammatory or non-inflammatory sites.42 Chemokines and their receptors play essential roles in T cell migration and homing.43 Our results showing that loss of MLK3 upregulates the chemokine receptor, CXCR3 and chemokines, CCL3 and CCL4 to promote T cell migration and homing are in agreement with earlier reports.34 44 Cytotoxicity is an important effector function of CD8+ T cells that is important for its tumoricidal effect. The combined inhibition of MLK3 and CD70 showed an enhanced intracellular expression of GZMB in CD8+ T cells, indicating that inhibition of both MLK3 and CD70 increases cytotoxic T cells.
The small molecule URMC-099 was synthesized as a specific inhibitor of MLK3.35 Our in vivo results suggest that pharmacological inhibition of MLK3 using URMC-099 affects T cell survival and effector function similar to MLK3 loss in mice. These findings have opened up a possible clinical use of URMC-099 for immunotherapy to manage T cell functions. Concurrent with the possibility of URMC-099 for immunotherapy, the pharmacological inhibition of MLK3 and CD70 induced tumor infiltration and cytotoxicity of CD8+ T cells. We further observed that combined inhibition of MLK3 and CD70 increases expression of proapoptotic proteins, Bid, Fas and Bax in tumor cells. Therefore, one of the mechanisms of apoptosis in tumor cells could be through GZMB released by cytotoxic CD8+ T cells and subsequent activation of caspase-8 and Bid in tumor cells. 36 It is reported that caspase-8 activates effector caspase-3 that leads to extrinsic apoptosis in cancer cells.45 Our results also suggest that combined inhibition of MLK3 and CD70 was independent of Fas-mediated extrinsic cell death because single or combined agents did not affect Fas expression. Furthermore, it is reported that Bid induces mitochondrial apoptosis by oligomerization of Bax and/or Bak, causing the release of cytochrome c to activate caspase-9.46 The activated caspase-9 binds to apaf-1 and forms apoptosome to activate caspase-3 and induces intrinsic apoptosis in cancer cells.47 Our results clearly showed that combined inhibition of MLK3 and CD70 increased mitochondrial localization of Bid, Bax expression and decreased Bcl2 expression, suggesting that combined inhibition induces predominantly intrinsic apoptotic pathways in tumor cells.
Conclusion
These results define the function of MLK3 in T cell survival, effector function and tumoricidal efficacy. The increased CD70-mediated apoptosis in CD8+ T cells due to loss/inhibition of MLK3 is a major shortcoming of targeting MLK3 that can be rectified by blocking CD70. The MLK3/MLKs inhibitor has gone through clinical trial for Parkinson’s disease, and several specific monoclonal anti-CD70 biologics are under clinical trials to manage cancers expressing CD70. Therefore, a combination of MLK3 inhibitor and anti-CD70 biologics can easily be repurposed for cancer immunotherapy. Based on our current data and published results, it is tempting to propose that combined inhibition of MLK3 and CD70 could serve as a novel therapeutic approach in breast and perhaps other cancers.
Acknowledgments
We acknowledge Dr Enrico Benedetti, Chair Department of Surgery, for providing access to departmental resources and financial support. The authors also acknowledge Ms Janet York for her administrative support and Dr Balaji Ganesh, Director of UIC Flow Cytometer Core facility for his technical advice.
References
Footnotes
Collaborators NA.
Contributors SK and AR designed and performed experiments, analyzed and interpreted the data. SK drafted and AR edited the manuscript. SKS, NV, PS, MD, GS and RSN contributed in reagent preparation and completing some of the experiments. SCS helped in URMC-099 synthesis and characterization for some of the initial experiments. KH and GT participated in scientific discussion, analyzed and interpreted the data. BR and KH contributed to scientific suggestions, manuscript correction and data analyses. AR and BR funded the study.
Funding We acknowledge financial supports from National Cancer Institute (NCI) to AR (CA 176846 and CA 216410) and BR (CA 178063). Additional supports from Veteran Administration Merit Awards to AR (BX002703) and BR (BX003296) are also acknowledged.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All experiments were performed in accordance with the guidelines of Ethics Committee of University of Illinois at Chicago. Human data are not used.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information. Materials described in this manuscript may be made available to qualified, academic, non-commercial researchers through a material transfer agreement upon request.