Article Text

Abstract
Background Current immune checkpoint blockade strategies have been successful in treating certain types of solid cancer. However, checkpoint blockade monotherapies have not been successful against most hematological malignancies including multiple myeloma and leukemia. There is an urgent need to identify new targets for development of cancer immunotherapy. LILRB1, an immunoreceptor tyrosine-based inhibitory motif-containing receptor, is widely expressed on human immune cells, including B cells, monocytes and macrophages, dendritic cells and subsets of natural killer (NK) cells and T cells. The ligands of LILRB1, such as major histocompatibility complex (MHC) class I molecules, activate LILRB1 and transduce a suppressive signal, which inhibits the immune responses. However, it is not clear whether LILRB1 blockade can be effectively used for cancer treatment.
Methods First, we measured the LILRB1 expression on NK cells from cancer patients to determine whether LILRB1 upregulated on NK cells from patients with cancer, compared with NK cells from healthy donors. Then, we developed specific antagonistic anti-LILRB1 monoclonal antibodies and studied the effects of LILRB1 blockade on the antitumor immune function of NK cells, especially in multiple myeloma models, in vitro and in vivo xenograft model using non-obese diabetic (NOD)-SCID interleukin-2Rγ-null mice.
Results We demonstrate that percentage of LILRB1+ NK cells is significantly higher in patients with persistent multiple myeloma after treatment than that in healthy donors. Further, the percentage of LILRB1+ NK cells is also significantly higher in patients with late-stage prostate cancer than that in healthy donors. Significantly, we showed that LILRB1 blockade by our antagonistic LILRB1 antibody increased the tumoricidal activity of NK cells against several types of cancer cells, including multiple myeloma, leukemia, lymphoma and solid tumors, in vitro and in vivo.
Conclusions Our results indicate that blocking LILRB1 signaling on immune effector cells such as NK cells may represent a novel strategy for the development of anticancer immunotherapy.
- antibodies, neoplasm
- cytotoxicity, immunologic
- receptors, immunologic
- killer cells, natural
- immunotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- antibodies, neoplasm
- cytotoxicity, immunologic
- receptors, immunologic
- killer cells, natural
- immunotherapy
Background
NK cells play critical roles in anticancer immunity and their functions against cancer cells can be enhanced by engaging activating receptors or blocking inhibitory receptors.1 Among activating receptors, FcγRIII (CD16) plays critical roles in antibody-dependent cellular cytotoxicity (ADCC) induced by therapeutic monoclonal antibodies (mAbs) to treat both hematological malignancies (rituximab for lymphoma and daratumumab for multiple myeloma (MM)) and metastatic solid cancer (cetuximab and trastuzumab).2 3 Antibodies engaging activating receptors such as NKG2D and NKp46 also showed natural killer (NK)-dependent tumor immunity in preclinical studies.4 5 Importantly, it has also been demonstrated that antibodies that block inhibitory receptors on NK cells, such as KIR and NKG2A, enhance NK cell function against cancer cells.6 7 Thus, developing antibodies to target immune receptors on NK cells can provide novel cancer immunotherapeutic strategies.
LILRB1, an immunoreceptor tyrosine-based inhibitory motif-containing receptor, is expressed on a variety of human immune cells. This includes all B cells, monocytes and macrophages, dendritic cells and subsets of NK cells and T cells. LILRB1 ligands, including MHC class I molecules, activate LILRB1 and transduce a negative signal that downregulates the immune response.8 The percentage of LILRB1+ NK cells is significantly higher in patients with advanced stage prostate and breast cancer than in healthy donors or patients with localized cancer.9–11 Blockade of LILRB1 signaling in immune cells was capable of activating the activity of NK cells against solid tumor and leukemia,10 12 and activating T cells or macrophages against solid tumors,13–15 using in vitro models. However, it is unknown whether LILRB1 can be targeted to turn ‘on’ immune cells in vivo for cancer treatment. In addition, it is also reported that LILRB1 is expressed on some tumor cells and stimulates immune response.16 17 Thus, it is not clear whether the net outcome of blockade of LILRB1 signaling on both tumor cells and immune cells is to activate or suppress antitumor immune response.
In this study, we found that the percentage of LILRB1+ NK cells in the peripheral blood from patients with persistent MM after treatment is significantly higher than that in peripheral blood from health donors or from patients with minimal disease or complete response. We also found that the percentage of LILRB1 expressing NK cells in peripheral blood from patients with late-stage (3B+3C) prostate cancer is significantly higher than that from healthy donors. We generated a novel anti-LILRB1 mAb (B1-176) that blocks the activation of LILRB1 on NK cells by MHC class I ligands and is able to stimulate cytotoxic activity of NK cells against MM, leukemia and solid tumor cells. Our in vitro and in vivo results suggest that blockade of LILRB1 signaling in NK cells is a promising strategy for treatment of patients with MM and other malignancies.
Materials and methods
Mice
Female NOD-SCID interleukin (IL)2Rγ-null (NSG) mice, aged 6–8 weeks (weight about 20 g), were purchased from the animal core facility of UT Southwestern. Mice were kept in a specific pathogen free room with a 12 hours light/dark cycle, controlled room temperature and ab libitum food and water. Mice were randomly allocated to each treatment group for experiments.
Cell lines and primary samples
Expi293F (Cat#A14528) was obtained from Life Technologies (Carlsbad). Hematological cancer cell lines 697, MHH-CALL-2, and OPM2 were purchased from DSMZ (Braunschweig, Germany). KMS27, KMS26, KMS12PE and KMS20 were purchased from Health Sciences Research Resources Bank, Japan Health Sciences Foundation. LILRB1 reporter cells were described previously.18 All other cell lines were purchased from ATCC except as noted. Hematological cancer cell lines were maintained in Roswell Park Memorial Institute (RPMI) 1640 supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Sigma Aldrich) (R10). Solid tumor cell lines were maintained in Dulbecco's Modified Eagle Medium (DMEM) with 10% heat-inactivated FBS except for H460 and H1299, which were maintained in R10. NKL cells were cultured as previously described.19 All cell culture medium were supplemented with 1% penicillin and streptomycin.
Peripheral blood mononuclear cells (PBMC) were separated from the buffy coats of healthy donors (Interstate Blood Bank) by gradient centrifugation using Ficoll media (GE Lifesciences). To isolate LILRB1 positive NK cells, PBMC were incubated with antihuman CD56 microbeads (Miltenyi Biotech) and separated using an AutoMACS Pro Separation System. Isolated CD56+ cells were then stained with anti-CD3-PE (clone: HIT3a, BioLegend)), anti-CD56-FITC (clone: TULY56, eBioscience) and anti-LILRB1-APC (clone: HP-F1, eBioscience) or Mouse IgG1 kappa Isotype Control-APC (eBioscience). LILRB1+ NK cells (CD56+CD3-) were sorted using a fluorescence-activated cell sorting (FACSAria) I system. Sorted LILRB1+ NK cells were maintained in the same medium as NKL cells for 2–3 days. Cancer patient samples were obtained from UT Southwestern Medical Center (UTSW) Tissue Bank and Hematologic Malignancies Tissue Bank of UTSW. Patient NK cells were isolated using the same protocol except that AutoMACS-separated NK cells were cultured, activated, and used for cytotoxicity assays without FACS.
Generation of anti-LILRB1 mAbs
Two New Zealand white rabbits were immunized subcutaneously (sc.) with 0.5 mg recombinant human LILRB1 protein (untagged ECD, Sino Biological). Four boosters were given after the initial immunization in a 3-week interval. Serum anti-LILRB1 titers were evaluated by indirect ELISA. Single LILRB1 positive memory B cells were cultured in 96-well plates for 14 days (RevMAb Biosciences USA). The culture supernatants were analyzed by ELISA for antibody binding to LILRB1 protein (untagged ECD, Sino Biological). Antibody variable region genes were cloned from those positive B cells using methods described previously.20
Expression and purification of mAbs
Full length IgG was produced in human embryonic kidney Expi293F cells using an ExpiFectamine 293 Transfection Kit (Catalog #: A14528, Gibco) by cotransfection HEK293 cells with both heavy and light chain expression constructs.20 After 7 days of fed-batch culture, the culture supernatants were harvested and antibodies were purified by affinity chromatography using protein A resin (Catalog #: 10–2001-XM, Repligen) as described previously.20
Epitope binning and affinity measurement with bio-layer interferometry
Classical sandwich epitope binning experiments were performed on an 8-channel Octet RED96 System as described.20 First, a baseline was established in kinetics buffer (Cat#18–1105, ForteBio) for 3 min. Then antibodies (30 µg/mL) were captured by protein A biosensors (Cat#18–5010, ForteBio) for 4 min. The remaining Fc-binding sites on the biosensors were then blocked with an irrelevant rabbit antibody (200 µg/mL) for 4 min, followed by soaking the biosensors in kinetics buffer for 10 s. The biosensors were exposed to recombinant LILRB1 (25 µg/mL) for 4 min, and then exposed to the secondary antibodies (40 µg/mL). The surfaces were regenerated for 45 s in 100 mM glycine (pH 2.6). The antibody pairs were assessed for competitive binding. Additional binding signal observed by the secondary antibody indicates an unoccupied epitope (non-competitor). A lack of additional binding by the secondary antibody indicates epitope blocking (competitor).
For antibody affinity measurement, antibodies (analyte, 30 µg/mL) were loaded onto the protein G biosensors (Cat#18–5082, ForteBio) for 4 min. Following a short baseline in kinetics buffer for 20 s, the loaded biosensors were exposed to solutions of recombinant LILRB1 in a range of concentrations (0.1–133 nM). After background was subtracted, data were fit to a 1:1 binding model to extract an association rate (kon) and dissociation rate (koff). The KD was calculated using the ratio koff/kon. Kinetic constant ranges for Octet RED96 systems is between 1 mM to 10 pM. Thus, affinity higher than 10 pM will be shown as KD <10 pM. All experiments were performed with shaking at 1000 rpm. All raw data were processed using ForteBio Octet Data Analysis Software V.9.0.
Generation of LILRB1-Fc, mutants-Fc and LILRBs-Fc fusion proteins
Ig-like C2-type domains, which including D1, D2, D3, D4 of LILRB1, were defined as described previously.21 The motif between D4 and transmembrane domain is considered as the stalk region or attached region. Different mutants of LILRB1 were generated using QuikChange Lightning Site-Directed Mutagenesis Kits (Agilent, Cat #210519) with LILRB1 wild type DNA construct as the template. LILRBs ECD, LILRB1 mutants and different domains were cloned into the Fc fusions expression vectors in frame with an antibody heavy chain signal sequence and the Fc portion of human IgG1 (hIgG1). Expression, purification and quantification of the Fc fusion proteins are the same as those for the antibodies described above.
ELISA
LILRB1 recombinant proteins (50 ng/well, 100 µL per well) were coated on Corning 96-well ELISA plates at 37°C for 4 hours. Plates were blocked for 2 hours at 37°C with 5% non-fat milk. After washing with phosphate buffered saline with tween (PBST), 100 µL of serial diluted anti-LILRB1 antibodies were added and incubated for 60 min at 37°C. Subsequently, the plates were washed with PBST for five times and incubated for 30 min with anti-rabbit or anti-human F(ab')2 HRP-conjugated antibody (Jackson ImmunoResearch, Cat #111-036-003 and #109-036-003). Plates were washed again with PBST for another five times, then the immunoreactions were developed with 3,3',5,5'-Tetramethylbenzidine (TMB) substrates (Sigma, Cat #T0440) and stopped by the addition of 2 M H2SO4 before the plate was read at 450 nm.
Humanization of rabbit mAb
Anti-LILRB1 rabbit antibodies were humanized by CDR-grafting as described previously.20 Briefly, CDRs in the heavy and light chains of the rabbit antibody were defined by a combination of three methods: Kabat, IMGT and Paratome. The parental rabbit mAb and the most homologous human germline sequences were aligned and residues that are known not to be structurally critical or subjected to change during the in vivo maturation process were identified in the mutational lineage guided analysis and humanized. DNA sequences encoding humanized light chain variable (VL) and heavy chain variable (VH) were synthesized (GENEWIZ) and a hIgG signal peptide and a Kozak sequence were engineered at the 5′ ends of the VL and VH sequences.22 The humanized VL and VH fragments were then cloned into expression vector in fusion with human constant regions for mammalian expression.
Lentivirus/retrovirus infection
HLA-G-1 cDNA with a signal peptide mutant23 was cloned into pLentiLox3.7-PuroR plasmid. Lentivirus plasmids pLentiLox3.7-luciferase-PuroR, pLentiLox3.7-HLA-G-PuroR and pLVX-MICA-ZsGreen plasmids were used to overexpress target proteins in cell lines as described previously.24 The infected cells were enriched by selection with 1 µg/mL puromycin for luciferase or HLA-G overexpression. MICA-positive cells were enriched by FACS for three times (clone: 6D4, BioLegend). LILRB2-5 reporter cells and LILRA1-6 reporter cells were made by retrovirus infection, according to previous reported.18 25
Flow cytometry-based in vitro killing assay
Carboxyfluorescein succinimidyl ester (CFSE, Thermo Fisher Scientific) labeling of target cells combined with propidium iodide (PI) staining of dead cells is a reliable method to measure in vitro cell killing by flow cytometry.26–28 Briefly, NK cells were cocultured with CFSE-labeled leukemia cells in U-bottom 96-well plates for 4 hours. Then, each sample was mixed with PI and analyzed by FACS Calibur. Cell lysis was calculated by the percentage of PI-positive leukemia cells among total leukemia cells. Spontaneous cancer cell death, in the absence of NK cells, was less than 5% and subtracted from total killing in the presence of NK cells.
Cytokine measurement
A total of 5×104 NKL cells were cocultured with 5×104 cancer cells in U-bottom 96-well plates for 24 hours. Interferon-γ (IFN-γ) release was detected in culture supernatants by ELISA (BioLegend) following the manual provided by the vendor.
In vivo killing assay
A total of 5×106 (CFSE)-labeled 697 (NKL resistant) and 697-MICA (NKL sensitive) cells were mixed and injected in combination with 5×107 NKL into NSG mice intraperitoneally. Anti-LILRB1 antibody (10 mg/kg) or control hIgG was administrated retro-orbitally. After 24 hours, cells from mice peritoneal cavities were harvested and stained with anti-MICA antibody and analyzed by FACS Calibur1 (BD Biosciences). In vivo cytotoxic activities of NKL were calculated as:
NK=Ratio of 697-MICA/697 in mice receiving 697, 697-MICA cells and NKL cells.
CN=Ratio of 697-MICA/697 in mice receiving 697 and 697-MICA cells.
% in vivo cytotoxic activity= (1- NK /CN)*100.
In vivo cytotoxic activity of NKL cells against 697-MICA was also tested in NSG mice sc. A total of 1×106 luciferase-expressing 697-MICA (697 MICA-luci) cell was mixed with 5×106 NKL cells and injected into mice sc. Anti-LILRB1 antibody (10 mg/kg) or control hIgG was administrated to each mouse retro-orbitally. Bioluminescence imaging (BLI) was conducted 48 hours later to monitor the remaining 697-MICA cells in mice.
MM xenograft
Age 6–8 weeks NSG mice were sublethally irradiated with 200 cGy X-ray on day −1. On day 0, each mouse was given 5×105 KMS27-luci together with 5×106 NKL cells via tail vein injection. Anti-LILRB1 antibody (10 mg/kg) or control hIgG was administrated retro-orbitally on day 0, day 3 and day 7, then once a week for 1 month. Another 5×106 NKL cells were injected on day 14. A total of 10 000 IU human IL2 were administrated to each mouse through IP injection every other day. Tumor burden was measured by BLI on day 28 and day 35.
Statistical analysis
Data are presented as mean±SE. Statistical significance between two groups is calculated by two-tailed unpaired t-test except as noted. Kaplan-Meier survival curves were analyzed using a log-rank test. Differences are considered statistically significant if p<0.05.
Results
LILRB1 is highly expressed on NK cells from the peripheral blood of patients with MM and prostate cancer
We have characterized the function of immune inhibitory receptors of the LILRB family and immune activating receptor NKG2D in cancer development.8 19 20 24 25 29–31 To investigate if LILRB1 can be a molecular target for cancer immunotherapy, we determined LILRB1 expression on NK cell from patients with cancer in comparison with that from health donors. Results showed that LILRB1 is mainly expressed on CD56dim NK cells, rather than CD56bright NK cells, from both healthy donors and patients with cancer (figure 1A). We then evaluated the percentage of LILRB1 expressing NK cells among CD56dim NK cells from peripheral blood of patients with prostate cancer or MM. We found that percentages of LILRB1+ NK cells (among CD56dim NK) were significantly higher in blood from patients with late-stage prostate cancer (3b and 3 c) than in blood from healthy donors (figure 1B, online supplementary table 1). Moreover, NK cells in peripheral blood from patients with MM who had persistent disease while on treatment had greater percentage of LILRB1+ NK cells (among CD56dim NK) than that from health donors or patients with minimal disease to complete response (figure 1C, online supplementary table 2). These results suggest that the percentage of LILRB1+ NK cells is significantly higher in patients with late-stage cancer or poor prognosis, and LILRB1 may be a molecular target for immunotherapy for these patients.
Supplemental material
Supplemental material

The expression of LILRB1 on natural killer (NK) cells from human peripheral blood. (A) PBMC were isolated from healthy donors’ buffy coats or peripheral blood of patients with multiple myeloma (MM) or prostate cancer by density gradient centrifugation using Ficoll-Paque plus medium, and then stained with anti-CD3-PE, anti-CD56-FITC, anti-LILRB1-APC or isotype-APC antibodies. NK cells were gated as SSClowFSClowCD56+CD3-. The LILRB1 expression on NK cell was gated according to the isotype-APC antibody. representative flow cytometry plots showed that LILRB1 were mainly expressed on CD56dim, instead of CD56bright NK cells. (B) The percentage of LILRB1+ NK cells among CD56dim NK cells from healthy donors (n=12), patients with stage 2B or 2C prostate cancer (n=16), and stage 3B or 3C prostate cancer (n=17) were determined by flow cytometry. (C) The percentage of LILRB1+ NK cells from healthy donors (same as figure 1.B), patients with newly diagnosed MM (n=10), and patients with MM who had partial response or refractory disease (n=8) and good response to treatment (CR+VGPR, n=9) were determined by flow cytometry. *P<0.05; ***P<0.001. SSC, side scatter, FSC, forward scatter, FITC, fluorescein isothiocyanate, PE, phycoerythrin, APC, allophycocyanin, CR, complete remission, VGPR, very good partial remission.
Generation and characterization of anti-LILRB1 antagonistic mAbs
To evaluate therapeutic potential of anti-LILRB1 mAbs in activating NK cells, we generated a panel of rabbit anti-LILRB1 mAbs by screening single memory B clones, cloning antibody genes and evaluating the blocking activity of recombinant mAbs (online supplementary figure S1A,B).32–34 In total, 229 anti-LILRB1 specific single B cell clones were generated from 384 LILRB1 B cell culture wells and assessed by ELISA (online supplementary table 3). Among them, we identified 174 single B cell clones can produce mAbs binding to LILRB1 expressed on cell surface of LILRB1 NFAT-green fluorescent protein (GFP) reporter cell (online supplementary table 4), which express GFP when the cell surface LILRB1 is cross-linked.18 We selected 60 binders for further cloning and expression, finding 44 antibodies with EC50 of low nanomolar range (0.05–3 nM) (online supplementary figure S1C). To group the 44 mAbs by their binding epitopes, classic sandwich epitope binning assay was performed by using Octet RED96, and twelve epitope bins were identified for these 44 high binders (online supplementary figure S1D).
Supplemental material
Supplemental material
Supplemental material
To screen for antagonistic anti-LILRB1 mAbs, LILRB1 receptor cells were cocultured with K562 cells overexpressing HLA-G1 (K562-HLA-G), the MHC class I molecule with the highest affinity for LILRB1 (figure 2A). The LILRB1 reporter cells were activated by K562-HLA-G cells, but not by parental K562 cells (MHC class I negative) (figure 2A). We screened all 44 antibodies using this reporter assay. Antibodies in bin 1, bin 7, bin 8 and bin 9 showed antagonistic effects (online supplementary figure S1E). Antibodies from bin 1 showed the strongest blocking efficacy (figure 2B). B1-176 in bin 1 showed specific binding to LILRB1 as assessed by flow cytometry (figure 2C), ELISA (figure 2D) and Octet RED96 (online supplementary figure 1F). B1-176 showed high affinity to LILRB1 with a KD value of about 10 pM (figure 2E), significantly higher than that of two commercial anti-LILRB1 mAbs HP-F1 and GHI/75 (online supplementary figure 1G). Therefore, we chose B1-176 to further evaluate its binding epitope and function in modulation of NK cell activation.

Generation and characterization of rabbit antagonistic anti-LILRB1 mAbs. (A) Left panel: the expression of HLA-G on cell surface of K562 and K562 cells overexpressed with HLA-G1 (K562-HLA-G) were stained with anti-HLA-G antibody (clone: MEM-G/9, Abcam) and analyzed by flow cytometry. Right panel: K562 or K562-HLA-G were cocultured with LILRB1 reporter cells. K562/K562-HLA-G cells were stained with DDAOSE before coculturing with LILRB1 reporter cells. The percentage of activated LILRB1 reporter cells (GFP) was analyzed by flow cytometry, 24 hours after coculture. (B) Titration of antagonistic anti-LILRB1 mAbs in blocking the activation of LILRB1 reporter cells stimulated by K562-HLA-G. LILRB1 reporter cells were cocultured with K562-HLA-G and incubated with different concentration of antagonistic anti-LILRB1 mAbs. (C) Representative flow cytometry plots showed that B1-176 binds to LILRB1 specifically. The LILR reporter cells were incubated with 0.5 µg/mL rabbit B1-176 at 4°C for 30 min. Then, the reporter cells were incubated with anti-Rabbit-IgG PE antibody (Jackson laboratory) and analyzed with flow cytometry. (D) Binding specificity of B1-176 rabbit antibody to LILRBs and LILRAs, determined by ELISA. (E) Affinity of B1-176 performed by Octet RED96. (F) Screening of B1-176 binding to LILRB1-Fc fusion proteins with different domain deletion, determined by ELISA. (G) Binding ability of B1-176 to LILRB1-D1D2 Fc fusion proteins with different mutant was determined by ELISA (top panel). two amino acids (Y76 and R84) in LILRB1 were identified to be critical for the binding of B1-176 (bottom panel). (H) Detailed binding surface of LILRB1 by rabbit mAb B1-176 was generated by molecular docking using discovery studio. mAbs, monoclonal antibodies; PBS, phosphate buffered saline.
To understand mechanism of B1-176 in blocking LILRB1 activation, we determined its binding epitope. B1-176 showed specific binding to the D1D2 region of LILRB1 (online supplementary figure 2A) not to the other domains, as assessed by ELISA (figure 2F). We made a series of amino acid mutations in the D1D2 region of LILRB1 to determine key amino acids for B1-176 binding (online supplementary figure 2B). Two amino acids (R84 and Y76) were found to contribute to LILRB1 interacting with B1-176 (figure 2G). Molecular docking of B1-176 was performed using Discovery Studio 2017. Based on the PDB structure of LILRB1 D1D2 region (PDBID:1UFU) and the modeling structure of B1-176 using the two key amino acids R84 and Y76, we identified the most possible binding model. In this model, the long HCDR3 of B1-176 inserts into the narrow ligand-binding pocket on LILRB1 D1 domain. Arg-84 of LILRB1 may interact with Tyr-102, Glu-103 and Asp-109 from HCDR3 loop by hydrogen bonds and electrostatic interactions. Tyr-76 of LILRB1 may be involved in hydrogen bonds with Asp-104 from HCDR3 (figure 2H).
Supplemental material
Humanization of rabbit anti-LILRB1 mAb
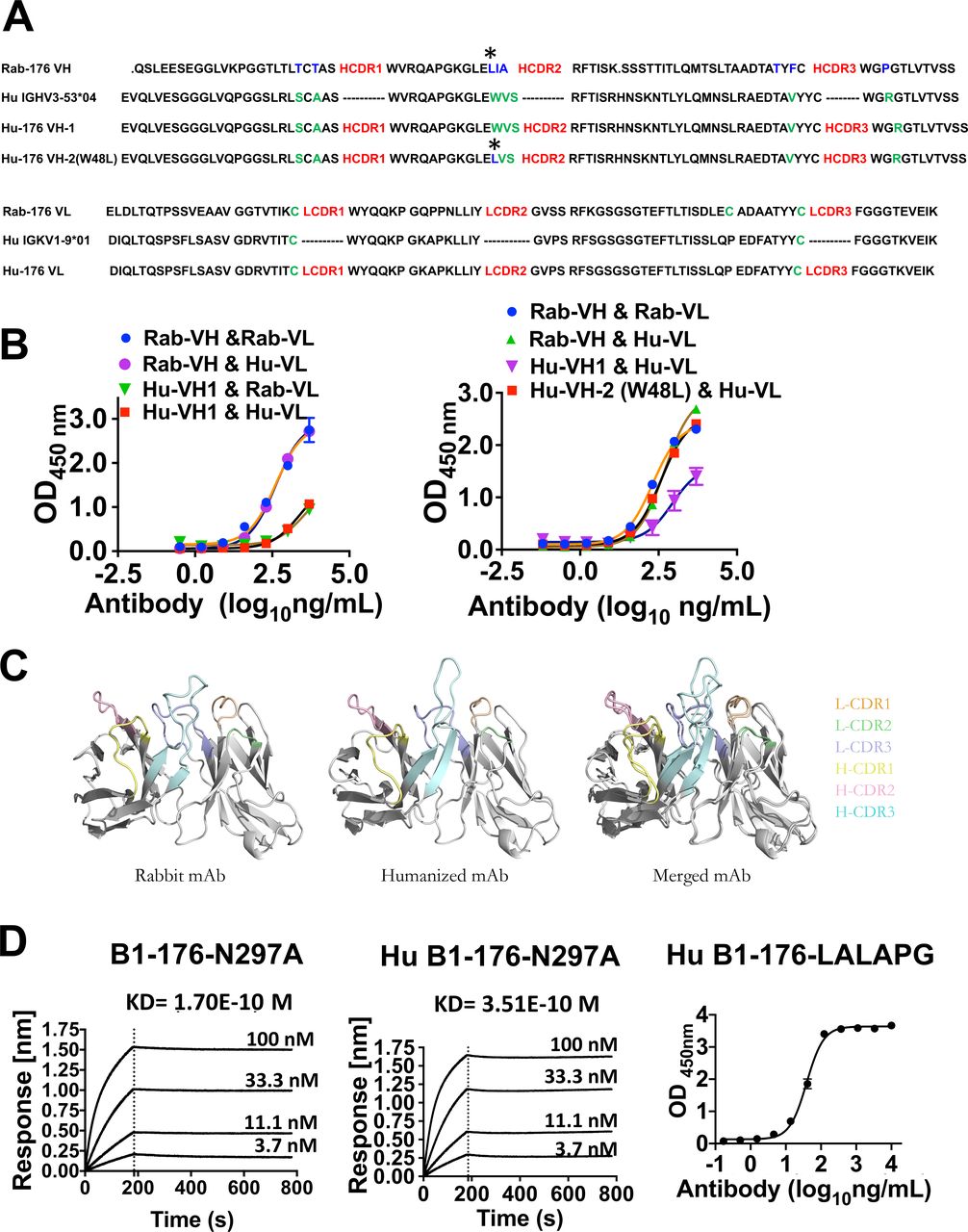
To minimize the immunogenicity of B1-176 for potential clinical applications, we humanized B1-176 and conducted molecular engineering for optimization of the antibody sequences. The Kabat/IMGR/Paratome combined-CDRs in the heavy and light chain of B1-176 were identified as described previously.20 The CDR sequences here are showed as heavy chain complementarity-determining regions (HCDR) and light chain CDR (LCDR). Human germline IGHV3-53*04 and IGKV1-9*01 were selected as the backbones. Heavy chain Hu-176 VH-1 and light chain Hu-176 VL were generated from the CDR grafting (figure 3A). However, antibodies with humanized heavy chain sequence VH-1 showed reduced binding to LILRB1 (figure 3B). After multiple rounds of single back-mutations in VH-1, mutation evaluation showed that amino acid L48 in FR2 was important to restore the binding activity (figure 3A,B). Molecular docking showed that Fabs of humanized and rabbit B1-176 shared similar superposition structures (figure 3C).

Humanization of anti-LILRB1 blocking antibody 176. (A) A combined Kabat/IMGT/Chothia CDR-grafting strategy was used to humanize rabbit antibody 176. The heavy chain and light chain of rabbit antibody B1-176 were showed as Rab-176 VH, Rab-176 VL. the heavy chain and light chain of human antibody framework were showed as Hu IGHV3-53*04 and HU IGKV1-9*01. Amino acids different from rabbit original ones (shown in blue), which were near HCDR, were shown in green. The heavy chain of B1-176 was humanized based on the human antibody framework, showed as Hu-176 VH-1 or Hu-176 VH-2 (W48L), which has a mutation noted with asterisk. The light chain of B1-176 was humanized based on the human antibody framework, showed as Hu-176 VL. (B) The ELISA results showed that rabbit amino acid L48 (noted with asterisk) in FR2 need to be remained at the same position to keep the binding activity of humanized B1-176 as same as rabbit B1-176. (C) Molecular docking data showed that the structures of rabbit B1-176 and humanized Hu B1-176–N297A are similar. (D) The affinities of rabbit human chimeric anti-LILRB1 antibody B1-176 N297A and humanized anti-LILRB1 antibody expressed in human IgG1 with N297A or LALAPG mutations were determined by Octet RED96 or ELISA. mAbs, monoclonal antibodies.
Considering the wide expression of LILRB1 on normal immune cells, rabbit B1-176 was expressed as hIgG1 with N297A mutation (B1-176-N297A) and humanized B1-176 was expressed as hIgG1 with N297A mutation (Hu B1-176-N297A) or with L234A, L235A, and P329G mutations (Hu B1-176-LALAPG) to abolish Fc mediated immune effector function.35 36 All B1-176 with Fc mutants maintained strong affinity to LILRB1 (figure 3D).
Anti-LILRB1 mAbs block activation of LILRB1 reporter cells by cancer cells
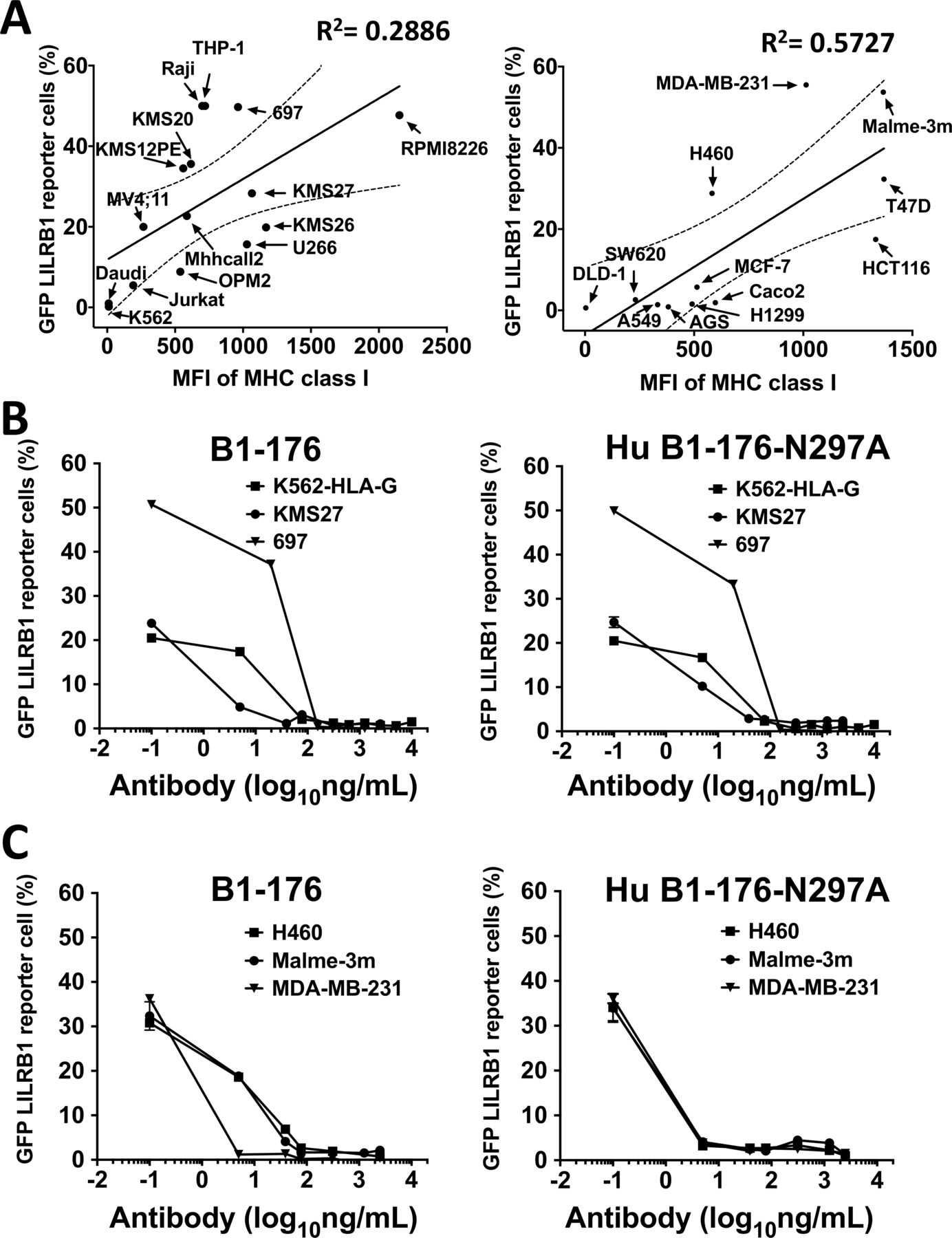
We screened for cancer cell lines that could activate LILRB1 by coculturing cancer cells with LILRB1 reporter cells (figure 4A). Flow cytometry was used to assess the expression of MHC class I molecules (antibody clone:HP-1F7, Santa Cruz), the ligands of LILRB1, on the cell surface of hematological and solid cancer cell lines (figure 4A). We found that the majority of cancer cell lines expressed MHC class I molecules and stimulated the activation of LILRB1 reporter cells. Exceptions were K562, DLD1 and Daudi cells, which did not express MHC class I molecules on cell surface. These results suggest that the ability of cancer cells to activate the LILRB1 reporter cells is positively correlated with their MHC class I expression levels. B1-176 and Hu B1-176-N297A blocked the activation of LILRB1 reporter cells stimulated by hematological cancer cell lines (figure 4B) or solid tumor cell lines (figure 4C). These results suggest that rabbit and humanized versions of B1-176 can block LILRB1 activation induced by HLA-G and other MHC class I molecules. It is noteworthy that LILRB1 binds to the conserved alpha3 domain of MHC class I.37 This may explain why our antagonistic anti-LILRB1 antibody screened from K562-HLA-G cells can block LILRB1 activation by other cancer cell lines.

Antagonistic LILRB1 mAbs can block the activation of LILRB1 receptor cells stimulated by both hematological cancer and solid tumor cell lines. (A) The levels of MHC class I on hematological cancer cell lines (left panel) or solid cancer cell lines (right) was detected by flow cytometry, using anti-pan MHC class I antibody (clone: HP-1F7, SANTA cruz biotechnology). Cancer cell lines (prestained with 9H-(1,3-Dichloro-9,9-Dimethylacridin-2-One-7-yl)-succinimidyl ester, DDAOSE) were cocultured with LILRB1 reporter cells to test cancer cells’ ability to stimulate LILRB1. The percentage of activated LILRB1 reporter cells (GFP+) were analyzed by flow cytometry. (B) Rabbit B1-176 (left panel) and humanized Hu B1-176-N297A (right panel) can block the activation of LILRB1 receptor cells induced by hematological cancer cell lines in a dose-dependent manner. LILRB1 reporter cells were cocultured with K562-HLA-G, multiple myeloma cell line KMS27 or pre-B leukemia cell line 697, then incubated with anti-LILRB1 mAbs. The activation of LILRB1 reporter cells was analyzed by flow cytometry 24 hours after treatment. (C) Rabbit B1-176 (left panel) and humanized HU B1-176-N297A (right panel) were titrated to block the activation of LILRB1 receptor cells by solid tumor cell lines. LILRB1 reporter cells were cocultured with lung carcinoma cell line H460, melanoma cell line MALME-3M or triple-negative breast cancer cell line MDA-MB-231, then incubated with anti-LILRB1 mAbs. The activation of LILRB1 reporter cells was analyzed by flow cytometry 24 hours after treatment. mAbs, monoclonal antibodies.
In vitro function of anti-LILRB1 mAb on NK cells
We assessed the in vitro efficacy of anti-LILRB1 mAbs using the NK cell line NKL. NKL cells express high levels of LILRB1 on surface (online supplementary figure 3A). Treatment with different versions of anti-LILRB1 mAb: B1-176-N297A, Hu B1-176-N297A, and Hu B1-176-LALAPG, increased the cytotoxic activities of NKL cells against the MM cell line KMS27 at similar levels (figure 5A). Anti-LILRB1 antibody also increased the cytotoxic activity of NKL against other MM cell lines OPM2 and RPMI8226, and the T cell leukemia line Jurkat (figure 5B). By flow cytometry, we found that LILRB1 was expressed on the cell surface of certain leukemia and lymphoma cell lines, such as 697 and Raji cells, but not on MM cell lines, Jurkat cells, or the majority of malignant plasma cells from a patient with MM (online supplementary figure 3B-D). Since the N297A mutation or LALAPG on Fc would abolish ADCC function of the antibody, our results indicate that the antagonistic anti-LILRB1 antibodies increased the natural cytotoxic activity of NKL cells against MM cell lines. Interestingly, we found that pre-B leukemia cell line 697 and Burkitt lymphoma cell line Raji were resistant to NKL cytotoxicity even when treated with anti-LILRB1 antibody (figure 5C). A previous study reported that MHC class I chain-related gene AB (MICA/B), a ligand for the NK cell activating receptor NKG2D, was expressed on NK-sensitive acute lymphoblastic leukemia (ALL) cells but was absent on NK-resistant ALL cells.38 To sensitize the 697 cells and Raji, we overexpressed MICA on 697 and Raji cells. As expected, MICA on the cancer cell surface stimulated the cytotoxic activities of NKL cells against these cells and anti-LILRB1 antibody further increased the cytotoxic activities of NKL cells (figure 5C,D). These results suggest that LILRB1 blockade and NKG2D activation on NKL cells synergistically increase their cytotoxic activity. Besides hematologic cancer cells, anti-LILRB1 antibody stimulated NKL cells to kill solid tumor cancer cell lines, such as breast cancer cell line MDA-MB-231 and melanoma cell line MALME-3M (figure 5E).
Supplemental material

Antagonistic LILRB1 mAbs can regulate functions of NKL cell lines against cancer cells in vitro. (A) Multiple myeloma cell line KMS27 was cocultured with NKL and incubated with 10 µg/mL control human IgG (hIgG) or B1-176-N297A (left panel, n=3), Hu B1-176-N297A (middle panel, n=3), and Hu B1-176-LALAPG (right panel, n=2). Four hours later, cytotoxic activity of NKL against cancer cells was analyzed by flow cytometry. (B) Multiple myeloma cell lines OPM2, RPMI8226 or acute T cell leukemia Jurkat cells were cocultured with NKL and incubated with 10 µg/mL B1-176-N297A or hIgG. n=3. (C) Pre-B leukemia 697 cells (left panel) or Burkitt lymphoma Raji cells (right) were overexpressed with MICA (697-MICA, Raji-MICA) and cocultured with NKL. 10 µg/mL B1-176-N297A or hIgG was added into the culture. (D) 697-MICA was cocultured with NKL (E: T=20) and incubated with different concentrations of B1-176-N297A. (E) Solid tumor cell lines MDA-MB-231 or MALME-3M were cocultured with NKL and incubated with 10 µg/mL B1-176-N297A or hIgG. n=3. (F) KMS27, OPM2, RPMI8226, Jurkat cells or 697/697MICA cells were cocultured with NKL (E:T ratio=1:1). Ten µg/mL control hIgG, B1-176-N297A (for RPMI8226, Jurkat and 697/697 MICA) or Hu B1-176-N297A (for KMS27 and OPM2 treatment) was added into cell culture. IFN-γ levels in cell culture supernatants were determined by ELISA 24 hours later. n=3–4, statistical significance was analyzed between anti-LILRB1 antibody and hIgG. *P<0.05; **P<0.01; ***P<0.001. IFN-γ, interferon-γ; mAbs, monoclonal antibodies.
NK cells can also improve the response of other immune cells against cancer cells by secreting cytokines. Anti-LILRB1 antibody increased IFN-γ secretion from NKL cells stimulated by T cell leukemia Jurkat cells and MM cell lines RPMI8226, OPM2 cells and KMS27 (figure 5F). We observed that anti-LILRB1 antibody increased the IFN-γ secretion from NKL cells induced by 697-MICA but not 697. This result suggests that LILRB1 blockade and NKG2D activation synergistically stimulated IFN-γ from NKL cells (figure 5F).
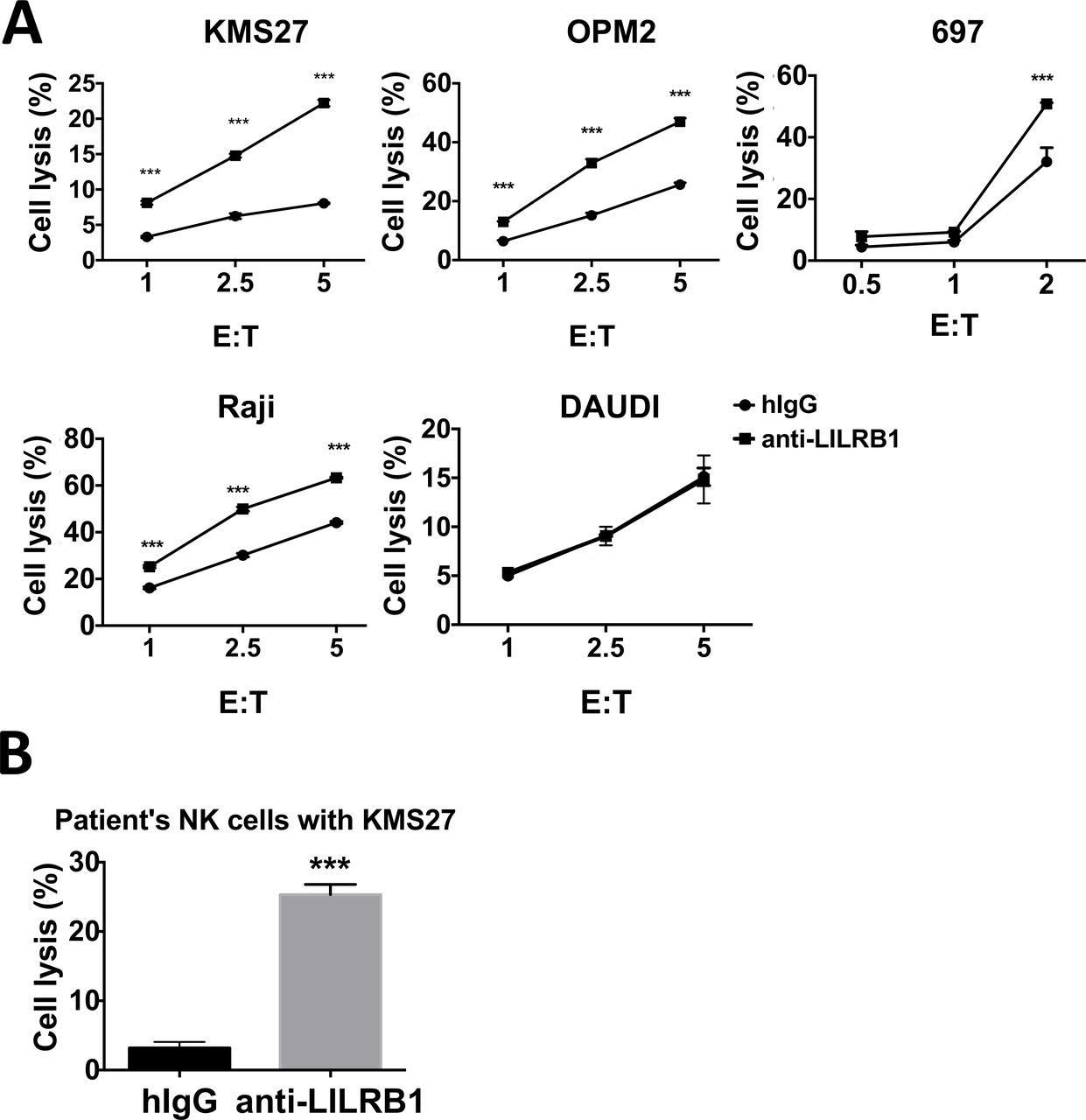
Moreover, we confirmed the function of anti-LILRB1 antibodies using primary NK cells. Primary NK cells from healthy donors’ buffy coats were isolated and LILRB1+ NK cells were enriched by FACS. Anti-LILRB1 antibody increased the cytotoxic activities of primary LILRB1+ NK cells from healthy donor against MM cell lines (KMS27 and OPM2), 697 cells or Raji cells, but not against Daudi cells, which do not express the LILRB1 ligand MHC class I (figure 6A). Since NK cells also express MHC class I, this result suggests that MHC class I on neighboring NK cells or cis-MHC class I does not inhibit the cytotoxic activity of NK cells against cancer cells. Here, 697 and Raji cells were sensitive to the cytotoxic activity of primary NK cells, likely due to the more potent cytotoxic activity of primary NK cells than that of NKL cells. One previous publication also reported that activated primary NK cells showed stronger cytotoxic activity than NKL cells, possibly due to the relative higher level of cytotoxicity receptors and NK94 on primary NK cells.39 We also isolated NK cells from the peripheral blood of one patient with MM whose NK cells was about 80% LILRB1 positive, and used the patient’s NK cells for cytotoxic assay. Anti-LILRB1 mAb increased cytotoxic activity of patient’s NK cells against MM cell line KMS27 (figure 6B).
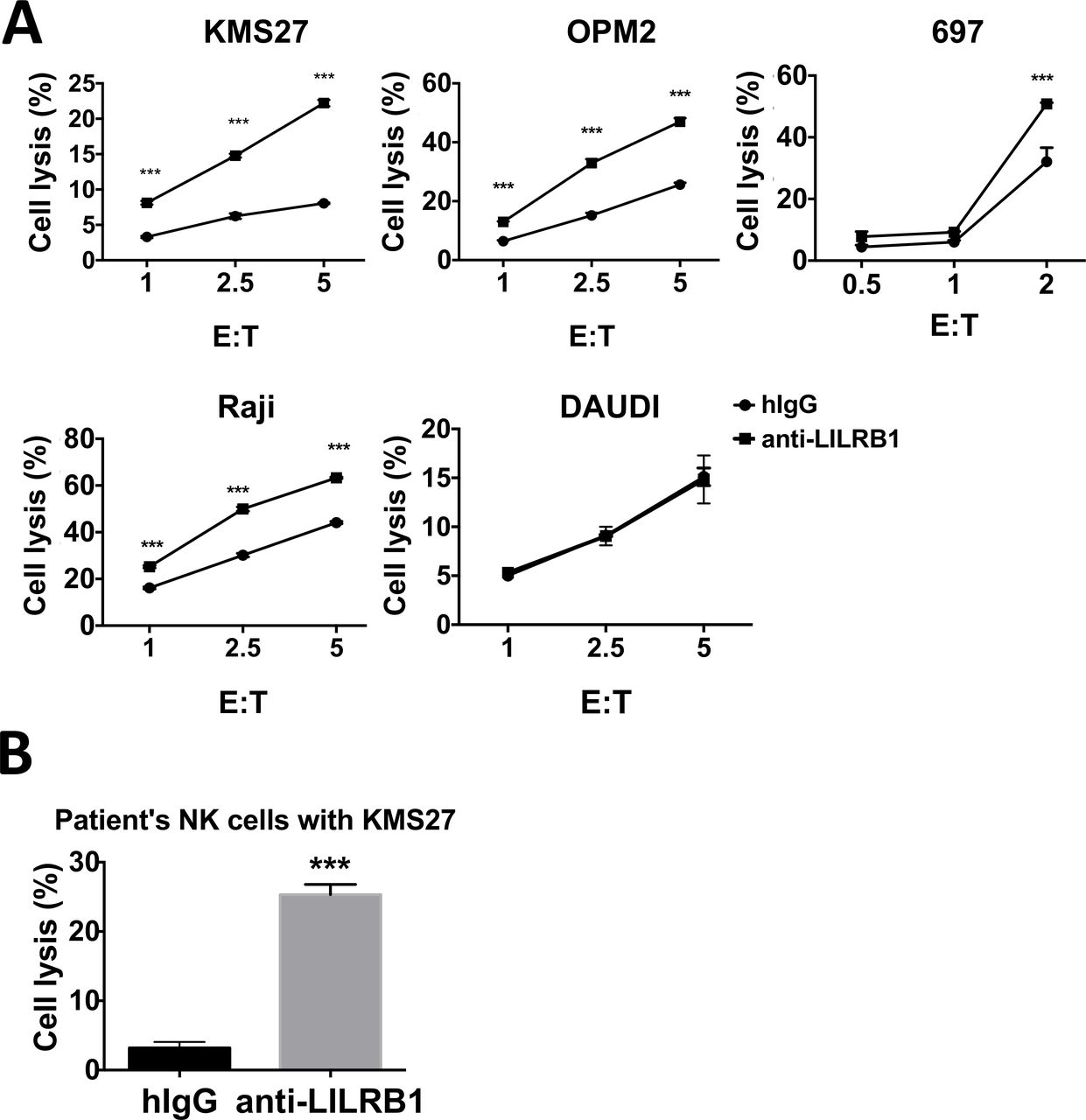
Antagonistic LILRB1 mAbs can increase cytotoxic activities of primary NK cells against cancer cells in vitro. (A) Multiple myeloma cell lines KMS27 and OPM2, pre-B leukemia cell line 697 or lymphoma cell line Raji and Daudi cells were cocultured with FACS-sorted LILRB1+ NK cells isolated from healthy donors’ PBMC. 10 µg/mL control hIgG or B1-176-N297A was added into cell culture. n=3, except Daudi cells n=2. (B) KMS27 cells were cocultured with NK cells from one multiple myeloma patient (E:T ratio=2.5:1) and incubated with 10 µg/mL control hIgG or Hu B1-176-N297A. Cytotoxic activity of primary NK cells was analyzed by flow cytometry 4 hours after coculture. n=3, ***P<0.001. hIgG, human IgG; mAbs, monoclonal antibodies; NK, natural killer.
In vivo function of anti-LILRB1 mAb on NK cells
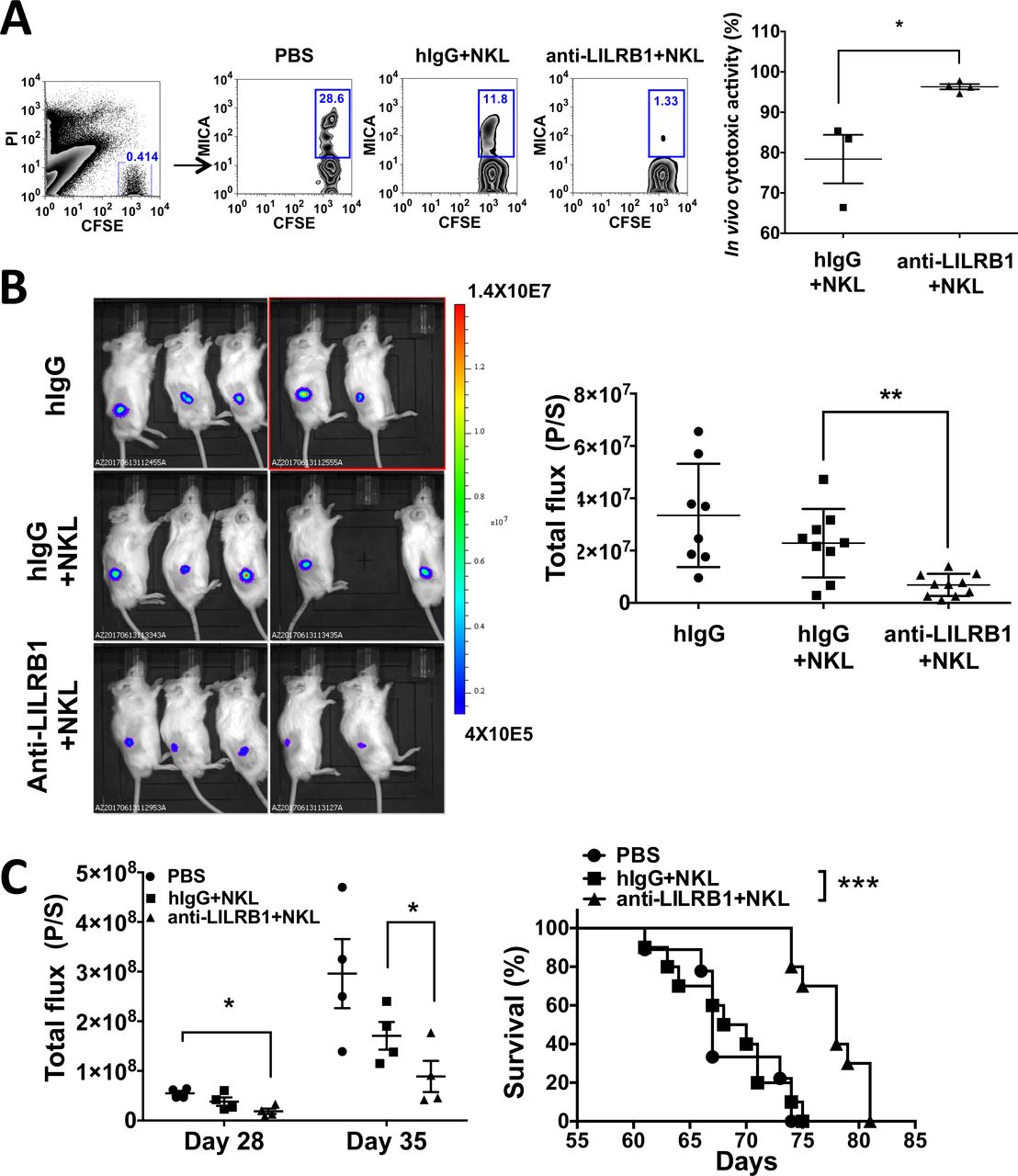
We tested the in vivo function of anti-LILRB1 antibody using NSG mice. NKL-resistant 697 cell and NKL-sensitive 697 MICA mixtures were injected together with NKL cells into the peritoneal cavity of NSG mice. After 24 hours, the ratio of 697-MICA/697 was analyzed and calculated as the cytotoxic activity of NKL cells in vivo (figure 7A). Our results showed that anti-LILRB1 antibody increased the in vivo cytotoxic activity of NKL cells against 697-MICA cells. The cytotoxic activity of NKL cells was also tested in vivo by injecting the NKL and luciferase-overexpressed 697-MICA cell mixtures into NSG mice sc. After 48 hours, BLI showed that mice receiving anti-LILRB1 antibody had significantly lower tumor burdens compared with control hIgG-treated mice (figure 7B). Together, the anti-LILRB1 antibody can improve the in vivo cytotoxic function of NKL cells.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Antagonistic LILRB1 mAbs can increase cytotoxicity activities of NKL cells in vivo. (A) 5×106 CFSE-stained 697 (NKL resistant) and 697 MICA (NKL sensitive) cell mixtures were injected together with 5×107 NKL cells into the peritoneal cavity of NSG mice. Ten mg/kg of control hIgG or B1-176-N297A was injected into mice through retro-orbitally. Twenty-four hours later, peritoneal cells were harvested and stained with anti-MICA-APC antibody. The ratio of 697-MICA to 697 cells was quantified by flow cytometry. Left panel: the representative flow plot. Right panel: the ratio of 697-MICA to 697 was used to calculate the cytotoxic activity of NKL cells in vivo. n=3–4, *P<0.05. (B) 1×106 697 MICA luciferase-overexpressing cells (697 MICA-luci) were mixed with 5×106 NKL cells and injected into NSG mice subcutaneously. 10 mg/kg of control hIgG or B1-176-N297A was administrated retro-orbitally. Bioluminescence imaging (BLI) was conducted 48 hours later. quantification of BLI data (total flux (P/S)) from two independent experiments demonstrates that mice received B1-176-N297 showed significantly lower tumor burden compared with control hIgG-treated mice, suggesting that B1-176-N297 improves the cytotoxic function of NKL cells in vivo. n=8–10, **P<0.01. (C) NSG mice were irradiated sublethally and injected with 5×105 luciferase-overexpressing KMS27 and 5×106 NKL cells via tail vein injection on day 0. Another 5×106 NKL cells were injected into NSG mice via tail vein injection on day 14. Mice received 10 mg/kg of control hIgG or B1-176-N297A retro-orbitally on day 0, day 3 and day 7, and then once a week for 1 month further. BLI was conducted on day 28 and day 35. Summary BLI data showed that B1-176-N297 significantly decreased tumor development in mice (left panel). n=4, data from one representative experiment. Statistical significance was calculated by one-tailed unpaired t-test. survival analysis of multiple myeloma mouse xenograft showed that B1-176-N297 treated mice significantly improved survival compared with control hIgG-treated mice (right panel). n=9–10, pooled data from two representative experiments. ***P<0.001. CFSE, carboxyfluorescein succinimidyl ester; hIgG, human IgG; mAbs, monoclonal antibodies; PBS, phosphate buffered saline.
We further assessed the efficacy of anti-LILRB1 antibody to prevent MM development in a xenograft mouse model. NSG mice were engrafted with KMS27 cells and NKL cells through their tail vein and were administrated with control IgG or anti-LILRB1 antibody, with cancer development monitored by BLI. The mice that received NKL cells and anti-LILRB1 antibody had significantly lower disease burden and longer survival than did mice that received NKL cells and control IgG or mice that received only PBS (figure 7C). Taken together, our results suggest the antagonistic anti-LILRB1 antibody can improve the function of NKL cells against MM development in vivo.
Discussion
LILRB1 is expressed on subsets of T cells and NK cells. We demonstrated that LILRB1 is mainly expressed on CD56dim NK cells from healthy donors and patients with different malignancies. In addition, we found that the percentage of LILRB1+ NK cells from peripheral blood of patients with MM with persistent disease while on treatment is significantly higher than that in NK cells from health donors or from patients with minimal disease or complete response. The percentage of LILRB1+ NK cells is also significantly higher in the peripheral blood of patients with late stage prostate cancer (stage 3B and 3C) than in that of healthy donors. These results are in accordance with previous studies showing that LILRB1 level is higher on peripheral blood CD56dim NK cells than CD56bright NK cells.40 Other prior studies also reported that the percentage of LILRB1+ NK cells was significantly higher for NK cells in the peripheral blood of patients with metastatic prostate and breast cancers than for NK cells from healthy donors or patients with localized cancers.9 10 Previous studies also reported that there is a strong association between the percentage of circulating CD8+LILRB1 T cells and the recurrence risk of non-muscle-invasive bladder cancer.41 Taken together, our results and previous reports suggest that LILRB1 expression on NK cells and T cells has prognostic value. Although the precise mechanism of the LILRB1 polymorphism in human NK cells is unknown, the expression of LILRB1 on NK cells is linked to particular haplotypes and a polymorphic regulatory region.42 The increase of LILRB1+ NK cells from certain patients with cancer was well documented. NK cells upregulated LILRB1 expression when cultured with cancer cells in vitro.9 HLA-G and soluble HLA-G were reported to upregulate LILRB1 expression on NK cells, T cells and antigen presenting cells.43 Patients with elevated plasmatic soluble HLA-G may have high LILRB1 expression on NK cells.10 Terminally differentiated NK cells, marked by the expression of CD57 or multiple KIRs, have high expression of LILRB1 and poor proliferative capacity.44 The high number of circulating CD57+ NK cells was associated with resistance to HER2-specific therapeutic antibodies in patient with primary breast cancer,45 which may explain the increased LILRB1+ NK cells in patients with cancer that have poor response to treatment.
Importantly, we demonstrated here that blockade of LILRB1 with antagonistic mAbs increase the immune functions of NK cells against MM cells both in vitro and in vivo. In an earlier study, Heidenreich and colleagues reported that blockade of LILRB1 on NK92 cell line did not increase their cytotoxic activity against MM cell lines.46 We speculate that this discrepancy is related to the use of the NK92 cell line, which may have more potent cytotoxic activity47 than the NKL cell line and primary NK cells we used in this study. MHC class I, ligands of LILRB1, is strongly expressed on late-stage MM cell lines, with a direct correlation between expression levels and clinical stage of disease.48 We showed that RPMI8226, the cell line used in the study of,46 and other MM cell lines, express high levels of MHC class I molecules at the cell surface and have a strong capacity to activate the LILRB1 reporter cells. These data indicate that MM cells may become NK cell-resistant by activating LILRB1 on NK cells through engagement of the MHC class I molecules expressed by MM cells.
NK cell function is determined by the balance of various activating and inhibitory signals in the cell. Targeting multiple immune receptors may optimize the function of NK cells against cancer cells. We demonstrated that combinatorial blockade of LILRB1 and activation of NKG2D receptor (by its ligand MICA), acts synergistically to increase the cytotoxic function of NK cells. Our results are concordant with a previous report showing that overexpression of HLA-G on MICA-expressing M8 melanoma cell line blocked NKL cells’ cytotoxic activity against the cancer cell line by activating LILRB1.49 Several methods are being developed to increase the surface abundance of MICA on cancer cells.4 19 Other studies have reported combinatorial effects of LILRB1 blockade with KIR blockade or ADCC-inducing mAbs in vitro.10 12 More studies are needed to test the in vitro and in vivo effects of the combination of LILRB1 blockade with antibodies targeting other immune receptors such as KIR, NKG2D and CD16, on the activity of NK cells against cancer. Adoptively transferred allogeneic haploidentical NK cells are considered to have improved function because of the mismatch of MHC class I molecules and KIRs.1 50 Since LILRB1 may still be activated on these allogeneic NK cells, it will be interesting to study the combinatorial effect of LILRB1 blockade and allogeneic NK cell adoptive transfer.
Interestingly, there are reports that LILRB1 on cancer cells, such as transformed B lymphoid cancer cells and Monoclonal gammopathy of undetermined significance (MGUS) cell, may activate immune responses.16 17 We confirmed that LILRB1 is expressed on pre-B leukemia cells and Burkitt lymphoma Raji cells. However, we observed that administration of anti-LILRB1 blocking antibodies increased the NK cell function against the LILRB1 positive cell line 697 cells and Raji cells, in agreement with previous report that LILRB1 blockade increased the cytotoxic activity of NK cells against pre–B-ALL.12 These results suggest that immune stimulating functions of LILRB1 may be context dependent. As we observed that LILRB1 expression on NK cells is significantly higher in patients with persistent disease following treatment, we believe that LILRB1 blockade may increase the function of NK cells in patients with persistent MM. However, LILRB1 expression level on both myeloma cells and NK cells should be monitored before and during anti-LILRB1 antibody treatment. To minimize the risk of modulating LILRB1 receptor on myeloma cells, the anti-LILRB1 antibody treatment should prioritize those patients with high expression level of LILRB1 on NK cells and no LILRB1 expression on myeloma cells.
In conclusion, our results suggest that blockade of LILRB1 by antagonistic antibodies has potential as an immunotherapy approach for treatment of patients with various types of cancer with high levels of expression of LILRB1 on NK cells.
Supplemental material
Acknowledgments
We thank Dr. Georgina T. Salazar for editing the manuscript. We thank Dr. Maria Costa for critical review of the manuscript. We thank Simmons Cancer Center’s Tissue Management Shared Resource for collecting blood samples from patients with prostate cancer and Meagan Woodard for collecting blood samples from patients with MM.
References
Footnotes
HC and YC contributed equally.
Contributors HC and YC performed experiments, analyzed data and drafted the manuscript. MD, SJ, XG, JK, GW, JX and LZ performed the experiments. AK, WC and CL reviewed and interpreted patient data. HA provided material. YH and HY conducted molecular docking experiments. RH, XL, WL and NX reviewed and proofread the manuscript. NZ, ZA and CCZ supervised the study and draft the manuscript. All authors read and approved the final manuscript.
Funding This work was supported by National Cancer Institute (1R01 CA248736), the Cancer Prevention and Research Institute of Texas (RP180435, RP150551 and RP190561), the Welch Foundation (AU-0042–20030616), Immune-Onc Therapeutics (Sponsored Research Grant #111077), and National Cancer Institute of the National Institutes of Health (P30 CA142543).
Competing interests ZA, and CCZ. are Scientific Advisory Board members with Immune-Onc Therapeutics. A.K. is an advisory Board member of BMS, Takeda, Pfizer, Janssen, Sanofi, Karyopharm and GSK. ZA, and CCZ, NZ, MD, XG, and JK hold equity in Immune-Onc Therapeutics. ZA, CCZ, NZ, HC, YC are inventors of LILRB1-related patent.
Patient consent for publication Not required.
Ethics approval The UT Southwestern Human Research Protection Program (HRPP) has reviewed the project and determined that it does not meet the definition of human subjects’ research under 45 CFR46.102 and therefore does not required IRB approval or oversight.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as online supplementary information.