Article Text

Abstract
The recent success of multiple immunomodulating drugs in oncology highlights the potential of relieving immunosuppression by directly engaging the immune system in the tumor bed to target cancer cells. Durable responses to immune checkpoint inhibitors experienced by some patients may be indicative of the formation of a T cell memory response. This has prompted the search for preclinical evidence of therapy-induced long-term immunity as part of the evaluation of novel therapeutics. A common preclinical method used to document long-term immunity is the use of tumor rechallenge experiments in which tumor growth is assessed in mice that have previously rejected tumors in response to therapy. Failure of rechallenge engraftment, typically alongside successful engraftment of the same tumor in naive animals as a control, is often presented as evidence of therapy-induced tumor immunity. Here, we present evidence that formation of tumor immunity often develops independent of therapy. We observed elevated rates of rechallenge rejection following surgical resection of primary tumors for four of five commonly used models and that such postexcision immunity could be adoptively transferred to treatment-naïve mice. We also show that tumor-specific cytolytic T cells are induced on primary tumor challenge independent of therapeutic intervention. Taken together these data call into question the utility of tumor rechallenge studies and the use of naïve animals as controls to demonstrate therapy-induced formation of long-term tumor immunity.
- drug evaluation, preclinical
- immunologic memory
- immunotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
The potential for cures in late stage cancer through the use of immune checkpoint inhibitors has profoundly altered cancer treatment paradigms over the past decade.1 2 The pursuit of additional therapeutics to extend the activity of these inhibitors into additional indications and patients continues at an ever-aggressive pace. One hallmark of the response to checkpoint inhibitors is durability of response suggestive of long-lived antitumor immunity, or a T-cell memory response. As this is considered a key feature of cancer immunotherapies, the induction of T-cell memory is often assessed in preclinical studies. Following treatment with an experimental regimen that leads to complete tumor eradication, surviving mice are subsequently challenged a second time with either the same tumor inoculum, and/or, with a second unrelated syngeneic tumor cell line. Whereas tumor formation after inoculation of different cells into surviving mice occurs just as well as after the inoculation of the same cells into naïve control mice, the rechallenge of the same cells into surviving mice typically fails to result in tumor growth. Since the first immune checkpoint inhibitor to reach market, ipilimumab, was approved, this very study design continues to be implemented frequently to support claims that efficacious experimental immunotherapy promoted the induction of antitumor immunity.3
We employed surgical tumor resection (STR) to test whether growth of a primary tumor in mice is sufficient to lead to the rejection of a tumor rechallenge in the absence of therapy in five syngeneic models selected based on their response to checkpoint inhibitor therapies (MC-38, CT26, EMT6-Luc, TC-1, JC) (figure 1A). MC-38, CT26 and EMT6-Luc are all sensitive to treatment with checkpoint inhibition whereas we have not observed responses in either TC-1 or JC. With exception of TC-1, tumor rejection rates following STR were higher than observed in response to a primary inoculation. We also demonstrate that tumor antigen-specific T cells can be detected in spleen and lymph nodes in response to primary inoculation of MC-38 tumor cells and that immunity observed post STR can be adoptively transferred to naïve animals in three models tested (MC-38, CT26 and EMT6-Luc). Together, such findings bring attention to the fact that many rechallenge studies designed to show induction of long-lived antitumor immunity published today are flawed and fail to consider the inherent immunogenicity of the tumors themselves which may be sufficient to induce immunity regardless of therapeutic intervention.

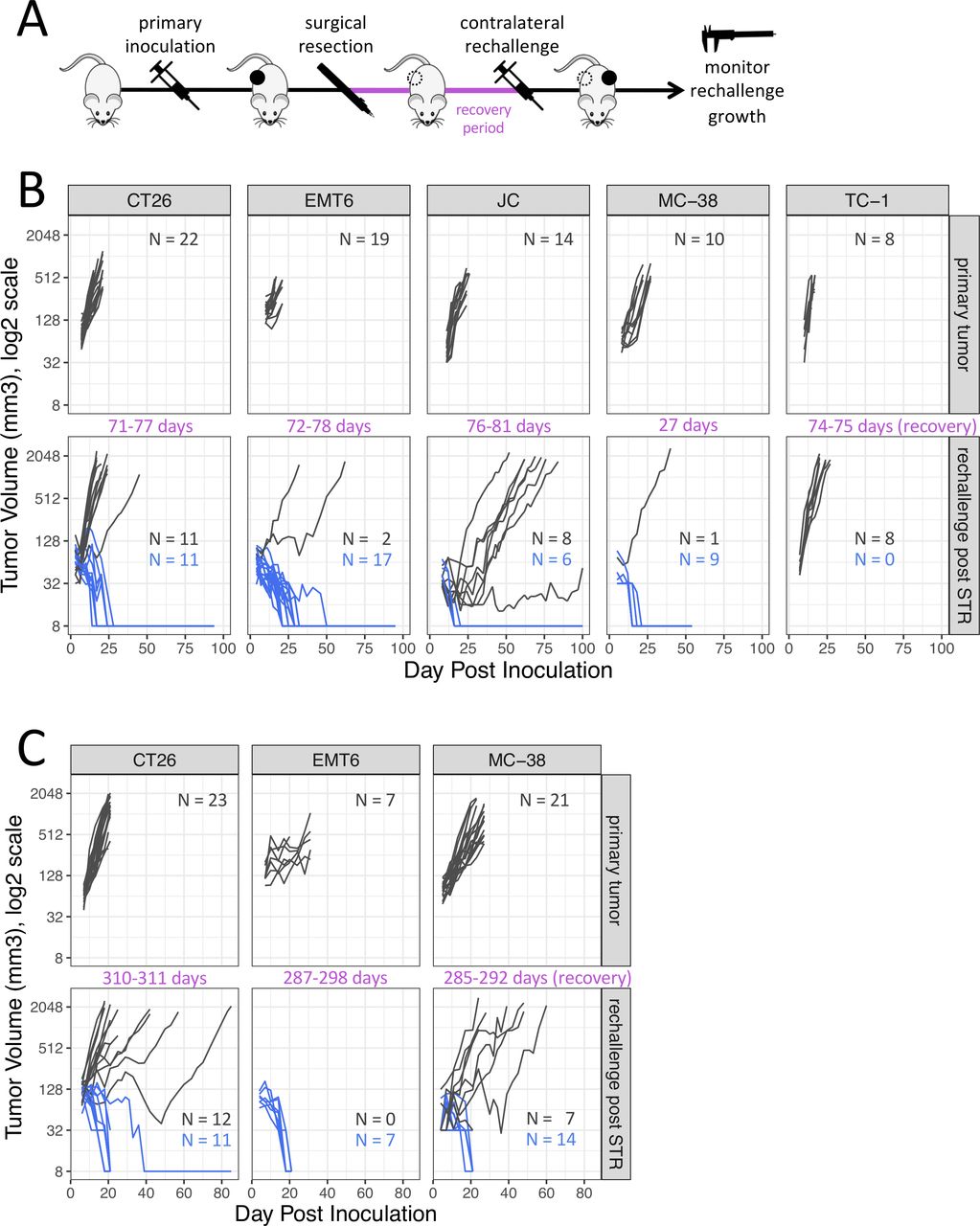
Subcutaneous tumor growth in the same syngeneic hosts following a primary tumor inoculation and following a repeat tumor challenge after surgical resection of the primary tumor (STR). (A) Diagram of experimental design. (B) Rechallenge administered 27–81 days after tumor resection. (C) Rechallenge administered 285–311 days after tumor resection. Dark gray traces represent animals exhibiting tumor growth, blue traces represent animals exhibiting tumor rejection. The complete absence of a tumor is displayed as 8 mm3 in the log2 scale. STR, surgical tumor resection.
Materials and methods
In vivo inoculation of tumor cell lines
All in vivo studies were conducted in compliance with Genentech’s Institutional Animal Care and Use Committee in South San Francisco, California, USA. The murine carcinoma cell lines of the colon (CT26)4, breast (JC)5 and lung (TC-1)6 were obtained from American Type Culture Collection (Manassas, Virginia, USA). The murine breast carcinoma EMT6-Luc7 cell line is a variant of the wild-type obtained from ATCC that was engineered in-house to express luciferase. The murine colon carcinoma cell line MC-384 was obtained from Leiden University Medical Center, The Netherlands. All cells were cultured in vitro to confluence and harvested into HBSS:Matrigel (BD Biosciences; Franklin Lakes, New Jersey, USA) (1:1, v:v) for subcutaneous inoculation into syngeneic, immunocompetent female mice (Charles River Laboratories, Hollister, California, USA) aged 8–26 weeks. MC-38 and TC-1 cells were inoculated into C57BL/6 mice whereas CT26, EMT6-Luc and JC cells were inoculated into BALB/c mice. Tumor cell inoculations were made at 0.1 million cells in a volume of 0.1 mL. Naive, age-matched animals were employed as controls for rechallenge inoculations. Tumor growth was monitored with calipers and tumor volumes were calculated from perpendicular length and width measurements using the formula: Tumor Volume (mm3)=0.5 × length × width2.
Surgical tumor resection
STR was performed under isoflurane anesthesia after tumor volume exceeded 200 mm3 and a minimum of 15 days had elapsed since tumor cell inoculation. Tumors were exposed for excision via opposing elliptical skin incisions made over the tumor and then carefully disconnected from their hosts with a disposable cautery to prevent bleeding. Surgical incisions were closed with wound clips which were removed 7–10 days later. Analgesia was induced immediately prior to STR and maintained thereafter as needed with carprofen (5 mg/kg, s.c.). Animals that experienced primary tumor regrowth due to incomplete STR were not used for subsequent studies.
Adoptive transfer
Draining axillary, brachial and inguinal lymph nodes, spleen and bone marrow were collected from animals determined to be rejecting a 21-day-old secondary tumor which was inoculated >9 months after STR of the primary tumor (CT26, n=10; EMT6-Luc, n=7; MC-38, n=13). Lymph nodes and spleens were gently crushed and disaggregated into a single cell suspension by passing through a 70 µm filter and pooled with bone marrow cells. Pooled material was resuspended for intravenous injection into 10 syngeneic recipients subjected to lethal whole-body irradiation 4–6 hours prior to cell injection. Adoptive transfer was similarly performed from 10 naïve (non-tumor-bearing) donor mice into an equal number of control recipients. Bone marrow ablation in BALB/c and C57BL/6 mice was achieved via one or two exposures to 525 rads staged 4 hours apart using a Cesium-137 irradiator, respectively. Recipients of adoptive transfer were each given 4–10 × 107 cells, corresponding to roughly half the number of cells isolated from each donor mouse. Mice were maintained on medicated drinking water containing glucose (2 g/L), neomycin (1.1 g/L) and polymyxin B (110 mg/L) for 14 days after irradiation. Twenty-one days after bone marrow reconstitution, adoptive transfer recipients were challenged with the standard subcutaneous tumor inoculum.
Identification of tumor antigen-specific CD8+ T cells in mice bearing MC-38 tumors
Draining axillary, brachial and inguinal lymph nodes and spleen were collected from 10 naïve animals as well as from mice bearing subcutaneous MC-38 tumors at 3, 10, 14 and 21 days post a primary tumor cell inoculation (n=10/time point). At each time point tissues were collected, gently crushed and disaggregated into a single cell suspension by passing through a 70 µm filter. Cells were plated at 1×106 per well in complete RPMI-1640 media supplemented with 10% Fetal Bovine Serum, 25 mM HEPES, 4 mM L-Glutamine, 55 µM β-mercaptoethanol (GIBCO) and 10 U/mL of human interleukin-2 (SIGMA). Cells were stimulated by the addition of either dimethyl sulfoxide or a mixture of peptides including the tumor-specific neoantigens M86 and ADPGK as well as p15E (KSPWFTTL, MBL Life Science).8–11 Cells were cultured for 6 hours in the presence of brefeldin A (GolgiPlug, 1 µL/mL, BD Biosciences) to prevent secretion of cytokines. Responding CD8+T cells were identified by flow cytometry using antibodies specific for CD3 (17A2), CD4 (GK1.5) and CD8 (53–6.7). Cells were fixed and permeabilized (CytoFix/Perm, BD Biosciences) prior to staining with anti-interferon-γ (XMG1.2). Samples were analyzed using a BD LSRFortessa X-20 analyzer (BD Biosciences) and FlowJo software (Tree Star).
Results
Tumor rejection rates rise following surgical resection of a primary tumor
To test whether growth of a primary tumor is sufficient to lead to the rejection of a tumor rechallenge in the absence of therapy, tumor rejection rates for five commonly used syngeneic tumor models (CT26, EMT6-Luc, JC, MC-38 and TC-1) were assessed 27–81 days following complete surgical resection of an established primary tumor (figure 1B). Rejection of tumor rechallenge after STR was defined as the absence of a palpable mass exhibiting progressive growth for as long as animals were observed after the repeat inoculation, a period lasting more than 7 weeks at a minimum to ensure a delayed onset of tumor growth was not missed as per historical experience with such models. With the exception of animals inoculated with TC-1 tumors, animals that previously harbored CT26, EMT6-Luc, MC-38 or JC rejected a secondary subcutaneous tumor inoculation when the same tumor cell line was reinoculated. The highest incidence of rejection was observed for MC-38 (9/10, 90%) and EMT6-Luc (17/19, 89%), followed by CT26 (11/22, 50%) and JC (6/14, 43%). Rejection rates for primary tumor inoculations was generally less than 10% (CT26, 0%; EMT6-Luc, 4%±4%; JC, 0%; MC-38, 8±5%; TC-1, 2±2%; mean±SEM).
Immunity induced by a primary tumor is long-lived and transferable
To examine whether tumor-immunity was durable beyond 3 months we assessed the growth on secondary tumor rechallenge more than 9 months following resection of primary tumors (≥310 days CT26; ≥287 days EMT6-Luc; ≥285 days MC-38) (figure 1C). Rejection rates were similar to the rates observed in mice 1–3 months after STR in all models tested with EMT6-Luc showing 100% rejection (7/7), followed by MC-38 (14/21, 67%) and CT26 (11/23, 48%).
We next set out to determine if the anti-tumor immunity seen in these mice could be transferred via transfer of lymphocytes (figure 2). Protection against a primary tumor challenge was afforded to tumor-naïve mice 3 weeks after lethal bone marrow ablation and reconstitution by adoptive transfer of immune cells obtained from the bone marrow, spleen and lymph nodes of mice experiencing tumor rejection 21 days following their repeat inoculation given more than 9 months after excision of their primary tumor. Lymphocytes were transferred from mice following rejection of a secondary tumor to confirm that these mice indeed possessed functional antitumor immunity. Tumor rejection in such adoptive transfer recipients was greater than 70% for CT26 (9/10, 90%), EMT6-Luc (9/10, 90%) and MC-38 (7/10, 70%) far greater than the maximum of 8% spontaneous rejection expected from these cell lines in naïve animals. No protection was afforded to mice that were reconstituted with lymphocytes from non-tumor-bearing controls.

Experimental design diagram and growth of subcutaneous tumors in adoptive transfer recipients of pooled immune cells from either naïve control mice or from mice rejecting a 21-day-old tumor rechallenge given >9 months after primary tumor resection (STR). Dark gray traces represent animals exhibiting tumor growth, blue traces represent animals exhibiting tumor rejection. The complete absence of a tumor is displayed as 8 mm3 in the log2 scale. BM, bone marrow; LN, lymph node; SP, spleen; STR, surgical tumor resection.
Tumor-specific CD8+ T cells are detected in the absence of therapy
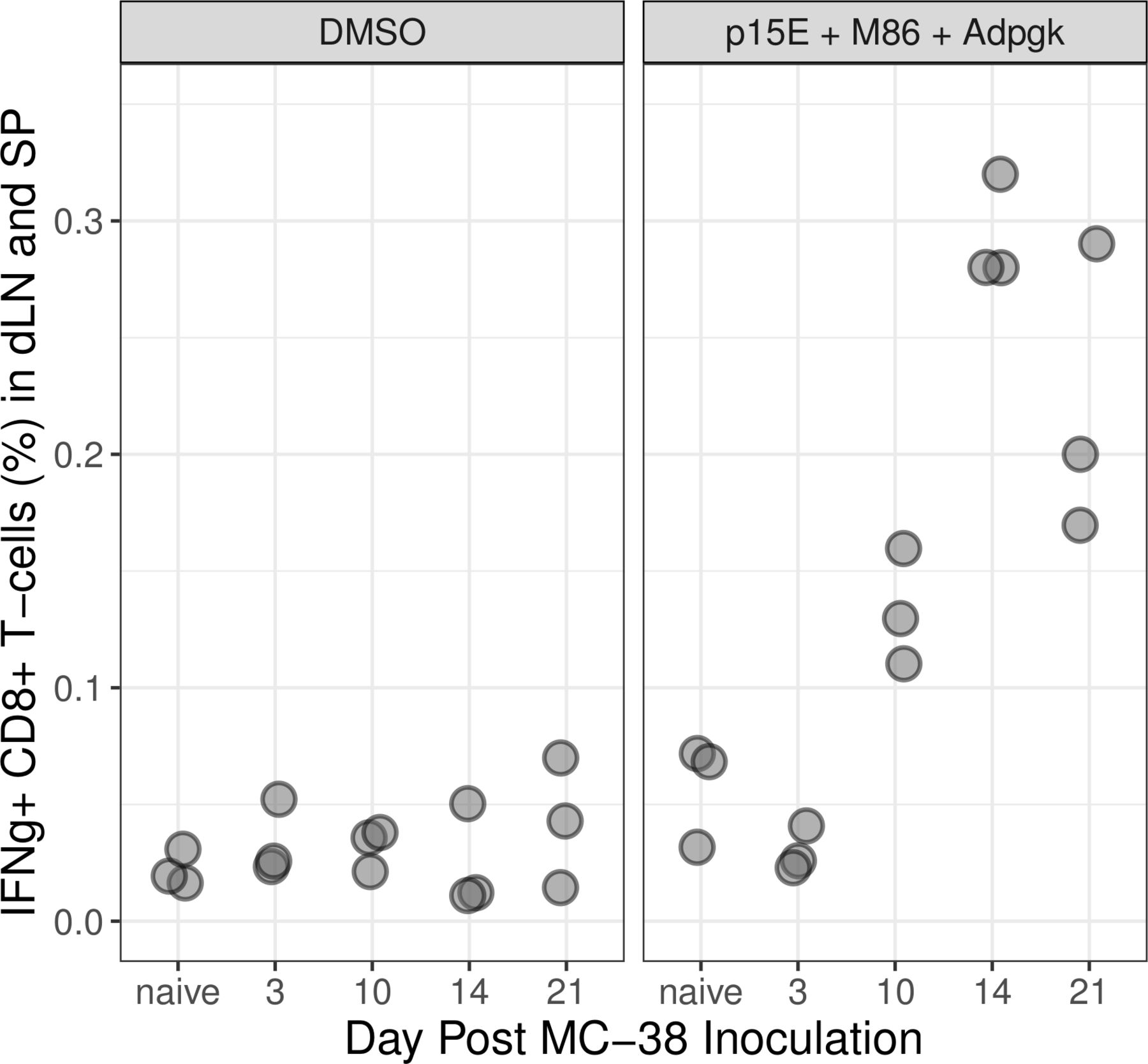
We rationalized if primary tumor inoculation is sufficient to immunize mice to a rechallenge then we should be able to detect the presence of tumor-reactive CD8+T cells in peripheral lymphatic tissues following the first subcutaneous inoculation. To test this, we analyzed CD8+T cells harvested from lymph node and spleen of mice inoculated with MC-38 tumors for their ability to recognize tumor neoantigens (figure 3). CD8+T cells were judged to be tumor reactive if they were capable of producing interferon-gamma on recognition of the well-defined tumor neoantigens present in the MC-38 model: p15E, M86 and ADPGK ex vivo.4–7 Tumor-specific CD8+T cells were detected in pooled spleens and lymph node preps from mice bearing MC-38 tumors 10, 14 and 21 days following inoculation, whereas such T cells were not detected in mice bearing tumors for 3 days nor in naïve control mice.

{kind=link}
{kind=link}
{kind=link}
CD8+ T cells producing IFNγ in draining lymph nodes (dLN) and spleens (SP) of mice bearing primary MC-38 tumors. Lymph nodes and spleens from 10 naïve animals and 10 tumor-bearing animals were collected at each time point post inoculation and disaggregated into a single-cell suspension. All cells were pooled and triplicate aliquots (gray circles) were stimulated with either DMSO or the tumor neoantigens M86, Adpgk and p15E. Responding CD8+ T cells were identified by flow cytometry for intracellular IFNγ. DMSO, dimethyl sulfoxide; IFNγ, interferon-γ.
Discussion
We provide evidence that a very common experimental design employed to demonstrate the formation of antitumor immunological memory following experimental therapy is flawed and may inadvertently report antitumor immunity established by primary tumor inoculation alone. In such tumor rechallenge studies, naive animals constitute inadequate controls as they have never been exposed to the tumor being assessed. We tested secondary tumor growth following STR of primary tumors in five syngeneic models (CT26, EMT6-Luc, JC, MC-38 and TC-1) in the absence of therapy and found elevated rates of tumor rejection versus those seen in naïve animals in four of these models. TC-1 was the only model not observed to have elevated tumor rejection rates following STR. The CT26, EMT6-Luc and MC-38 models were examined further in the absence of therapy and anti-tumor immunity was observed to be long-lived, lasting more than 9 months following STR in each model. We also demonstrated that the confirmed long-term immunity post-STR in these three models could be adoptively transferred with lymphocytes affording protection against the same tumor type in recipient mice verifying adaptive immunity.
While STR is a therapeutic intervention that itself may lead to tumor cell death, liberation of tumor antigens and some level of antitumor immunity, it is unlikely to contribute significantly. The fact that we observe CD8+T cells that are tumor antigen-specific 10 days following primary tumor inoculation in the MC-38 model suggests that STR is not required to release antigens. Additionally, the use of resection-rechallenge methodology was once a mainstay of investigators studying how the immune system responds to a tumor. In these early studies, rejection post-STR was not observed to be uniformly widespread, but determined to be dependent on the timing of excision relative to the ‘age’ of primary tumor, the timing of rechallenge relative to STR and the size of the rechallenge inoculum.12 Importantly, the degree to which different models rejected a rechallenge following STR was considered a measure of their immunogenicity, with models derived from chemical carcinogenesis exhibiting the highest rejection levels in this experimental setting.12–17 It is now widely accepted that mutational burden and in particular neoepitope presentation drive tumor immunogenicity and are predictive for a tumor’s response to immunotherapy.18 19 The model-specific differences in rechallenge rejection rates post-STR we observed most likely reflect the inherent differences in immunogenicity of each model. CT26 and MC-38 were developed through chemical induction4; TC-1, viral transformation6; EMT6 and JC, from a spontaneous tumor.5 7 The higher rejection rates observed for MC-38 and CT26 can be understood based on their relatively high mutation burden11 due to their mode of initiation. While the parental EMT6 line contains between threefold and fivefold fewer coding variant compared with CT26 or MC-3811 consistent with being derived from a spontaneous tumor the EMT6-Luc line is rejected at the higher overall rate in our study. A luciferase variant of EMT6, EMT6-Luc was chosen for our study as a positive control for immunogenicity as lineage-tracing markers such as eGFP and firefly luciferase have been demonstrated to be immunogenic in immunocompetent murine hosts.20. Beyond mutational burden and neoepitope presentation tumor lines may exhibit immunogenicity thorough genetic drift of either the cell lines themselves over extensive passage in vitro, genetic drift of the mouse strains maintained in isolated colonies over the years, or both.16 While we have primarily focused our attention on tumor models that are inherently immunogenic, as these are often the models used to assess the activity of novel immunotherapeutic agents, it would be of interest to determine whether similar observations are made using tumors that are less immunogenic such as B16F10 or 4T1. Similarly, it would also be of great interest to determine if the rates of rejection of secondary tumors are similar between subcutaneous tumors and metastases and if the site of the primary tumor (subcutaneous vs orthotopic) impacts the outcome.
Given the strong interest in identifying novel therapeutics that extend the reach and activity of checkpoint inhibitors, it is clear that researchers will continue to perform preclinical studies in these highly immunogenic models. Based on these results and the numerous studies that predate checkpoint inhibition, investigators need to leverage the recent advances in the understanding of cancer immunology and should include proper controls to fully demonstrate that long-term antitumor activity is induced by the therapeutic being investigated and not solely the primary tumor inoculum.
References
Footnotes
Contributors BA: planning, conduct, reporting. AMB: planning, conduct. A-HC: planning. RAC: planning. SML: planning, conduct, reporting. JMS: planning, reporting. KT: planning. SEG: planning, reporting.
Funding All work described herein was funded by Genentech, a member of the Roche group.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All in vivo studies were conducted ethically in compliance with Genentech's Institutional Animal Care and Use Committee in South San Francisco, California, USA.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as online supplementary information. The data presented in this report are available from the corresponding author on request.