Article Text

Abstract
Background Despite some successes with checkpoint inhibitors for treating cancer, most patients remain refractory to treatment, possibly due to the inhibitory nature of the tumor stroma that impedes the function and entry of effector cells. We devised a new technique of combining immunotherapy with radiotherapy (XRT), more specifically low-dose XRT, to overcome the stroma and maximize systemic outcomes.
Methods We bilaterally established 344SQ lung adenocarcinoma tumors in 129Sv/Ev mice. Primary and secondary tumors were irradiated with either high-dose or low-dose of XRT with systemic anti-programmed cell death protein 1 and anti-cytotoxic T-lymphocyte associated protein 4 administration. Survival and tumor growth were monitored for the various groups, and secondary tumors were phenotyped by flow cytometry for immune populations. Tumor growth factor-beta (TGF-β) cytokine levels were assessed locally after low-dose XRT, and specific immune-cell depletion experiments were conducted to identify the major contributors to the observed systemic antitumor effect.
Results Through our preclinical and clinical studies, we observed that when tumor burden was high, there was a necessity of combining high-dose XRT to ‘prime’ T cells at the primary tumor site, with low-dose XRT directed to secondary (metastatic) tumors to ‘modulate the stroma’. Low-dose XRT improved the antitumor outcomes of checkpoint inhibitors by favoring M1 macrophage polarization, enhancing natural killer (NK) cell infiltration, and reducing TGF-β levels. Depletion of CD4+ T cells and NK cells abrogated the observed antitumor effect.
Conclusion Our data extend the benefits of low-dose XRT to reprogram the tumor environment and improve the infiltration and function of effector immune cells into secondary tumors.
- radioimmunotherapy
- radiotherapy
- immunotherapy
- lung neoplasms
- tumor microenvironment
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Although technologic advances over the past decade have considerably improved the detection, diagnosis, and treatment of early-stage cancers, metastatic disease remains lethal in many cases, and new treatment paradigms are actively being sought to control disease that has already spread beyond the primary tumor. As an example, immunotherapy has produced some impressive clinical results, but to date its activity, particularly as monotherapy, is limited to a minority of cases and types of cancer. As another example, radiation therapy (XRT) has long been a cornerstone of cancer treatment, but until recently it has been seen largely as a means of controlling local tumor growth by its ability to directly damage tumor-cell DNA. However, radiation is also known to have a wide variety of immunomodulatory effects, and as such its potential for overcoming treatment resistance or improving the effectiveness of systemic therapies—particularly immunotherapy—is increasingly being recognized.1 2 Indeed, high-dose stereotactic XRT (H-XRT) has been used in attempts to transform tumors into in situ vaccines by eliciting the release of neoantigens from the tumor that in turn prime T cells and contribute to T cell repertoire diversity.3 Others have found that H-XRT in combination with anti-cytotoxic T-lymphocyte associated protein 4 (CTLA-4) can have a significant survival advantage in a murine 4T1 metastatic breast cancer model.4 Moreover, H-XRT directed toward oligometastatic disease sites, can provide local control and extend survival for patients with cancer of various types.5 In our own studies of mechanisms of resistance to immune checkpoint inhibitors, we discovered that H-XRT can induce interferon-beta cytokine production, thereby upregulating MHC-I molecules, initiating and stimulating T cell responses, and enhancing the sensitivity of resistant cells to anti-programmed cell death protein 1 (PD1) therapy.6 H-XRT has also been shown to prompt the release of danger signals such as Adenosine triphosphate (ATP) and high-mobility-group box 1 (HMGB1) which can activate macrophages via toll-like receptor 4,7 in addition to STING pathway activation due to cytoplasmic DNA release.8 In other mechanistic studies, we demonstrated in a murine model carrying bilateral tumors that treating primary tumors with H-XRT led to the upregulation of OX40 on CD4+ T cells and subsequent intratumoral injection of an OX40 agonist promoted abscopal effects in secondary untreated tumors.9
In contrast, we also observed unwanted side effects of H-XRT such as lymphodepletion at primary sites, increased local and circulating myeloid-derived suppressor cells (MDSCs), and upregulated T-regulatory cells (Tregs). Collectively, these effects can enhance tumor-mediated immunosuppression, which has been implicated in resistance to anti-PD1 immunotherapy. Indeed, we found in another series of studies that adding an inhibitor of indoleamine 2,3-dioxygenase 1 led to suppression of MDSCs, increased CD8+ T cell infiltration, and retarded tumor growth and metastasis in a model of anti-PD1-resistant lung cancer.10 Moreover, combining H-XRT with anti-glucocorticoid-induced TNFR-related protein (GITR), led to the depletion of Tregs at the primary tumor site, increased the numbers of CD4+ and CD8+ cells and effector memory on tumor rechallenge, and led to pronounced abscopal effects in half of the mice so treated.11 Collectively, these results suggest that combinatorial strategies targeting multiple points of tumor immune evasion may lead to more robust and lasting antitumor responses.
In efforts to enhance the effectiveness of systemic immunotherapies, we have been investigating mechanisms underlying the low response rates of secondary tumors in murine models. Our hypothesis is that these tumors shield themselves with inhibitory physical and cellular components of the surrounding stroma. Rich in cancer-associated fibroblasts, MDSCs, protumor growth M2 macrophages, and Tregs,12 these components of the stroma function collectively to mediate treatment resistance and facilitate tumor progression.13 We further hypothesize that low-dose radiation (L-XRT) directed to secondary tumors, delivered in combination with immunotherapy and H-XRT to primary tumors, can modify the stroma so as to enhance systemic antitumor benefits. We hereby showed that L-XRT treatment resulted in increased proportions of effector immune cells in the transplanted lung cancer murine tumors. Others have shown that low-dose total body irradiation can reduce Tregs, decrease tumor burden, and prolong survival in an F10 melanoma model.14 In another study, very-low-dose (four fractions of 75 mGy each) total body irradiation given 1 day before four fractions of 1 Gy each was shown to activate T cells and natural killer (NK) cells and to retard tumor growth in a Lewis lung carcinoma (LLC).10 Finally, in a pancreatic carcinoma model, L-XRT (2 Gy) led to polarization of M2 macrophages into the M1 phenotype via production of induced nitric oxide synthase. This in turn augmented the infiltration of adoptively transferred TCRCD8+ T cells.15 Ultimately, we propose that our radiation strategy with H-XRT and L-XRT (which we call ‘RadScopal’ technique) in combination with checkpoint inhibitors, modulates the tumor microenvironment (TME) of both primary and secondary tumors to maximize systemic antitumor effects in solid tumors.
Materials and methods
Patients
All procedures described were reviewed and approved by The University of Texas MD Anderson Cancer Center institutional review board, and all subjects provided written informed consent to participate.
Mice, cell lines, and antibodies
All animal experiments were conducted in accordance with The University of Texas MD Anderson Cancer Center’s Institutional Animal Care and Use Committee policies. Mouse models involved the use of 8 to 12 week old 129Sv/Ev mice, wild-type C57BL/6 mice, and nu/nu mice with deficient T cell function. The 344SQ parental lung adenocarcinoma cell line (344SQ-P) was a generous gift from Dr. Jonathan Kurie at MD Anderson Cancer Center. LLC cells were obtained from the American Type Culture Collection. The MC38-gp100 cells were a generous gift from Dr. Patrick Hwu at MD Anderson Cancer Center. 344SQ-P cell line was validated by DDC Medical (http://ddcmedical.com; Fairfield, OH) by short-tandem-repeat DNA fingerprinting. 344SQ-P cells tested mycoplasma negative. The anti-PD1 and anti-CTLA-4 IgG2b antibodies were provided by Bristol-Myers Squibb. For the immune-cell depletion studies, anti-CD4 (500 µg/injection, clone GK1.5, Bio X Cell), anti-CD8 (500 µg/injection, clone 53–6.7, Bio X Cell), and anti-NK (30 µL/injection, Asialo) antibodies were given intraperitoneally on days 2, 7, 13, and 20 to maintain the depletion pressure during the experiment.
Tumor establishment and treatment
Mice were implanted with tumor cells either in one leg (the one-tumor model) or both legs (the two-tumor model) to test the experimental conditions described. In the two-tumor model, 129Sv/Ev mice were injected with 344SQ-P tumor cell lines in both hind legs, 0.5×106 cells in the right leg (the ‘primary’ tumor) and 0.1×106 cells in the left leg (the ‘secondary’ tumor), 4 days apart, and then treated with various combinations of XRT and immunotherapy per specified. The immunotherapy agents were delivered intraperitoneally at titrated doses of 50 µg/injection for anti-CTLA-4 and 200 µg/injection for anti-PD1. Primary and secondary tumors were measured twice a week with digital calipers. When the average diameter of the primary tumors reached 7 mm, those tumors were irradiated locally with H-XRT (three fractions of 12 Gy each) from a Cesium source. Secondary tumors were irradiated with L-XRT (two fractions of 1 Gy each) 3 days later. Mice were euthanized when either the primary or secondary tumors reached 14 mm in diameter. At defined experimental endpoints, mice were killed, their lungs collected, and metastatic nodules counted after staining with Bouin’s fixative solution. Tumor-infiltrating leukocytes (TILs) from secondary tumors, draining lymph nodes (dLNs), and spleens were harvested and processed. Anesthesia, analgesia, and euthanasia were accomplished with isoflurane, buprenorphine, and CO2, respectively.
Flow cytometry
Samples were obtained from blood, spleen, or tumors as indicated. Tumor samples were weighed after harvest, digested/dissociated, and total cells were counted. TILs were then stained for lymphoid and myeloid panels using cell surface as well as intracellular markers. Briefly, cells were incubated with corresponding surface markers for 30 min at 4°C, then washed and resuspended in FACS buffer. For intracellular staining, cells were fixed with BD Cytofix for 15 min, washed, and then incubated with 1× permeabilization buffer for 30 min at room temperature. Intracellular antibodies were added for an additional 30 min, after which cells were washed and evaluated on an LSR-II flow cytometer, and data analyzed with FlowJo software. Fluorochrome-conjugated antibodies were obtained from BioLegend. The lymphoid panel included anti-CD4 (100449), anti-CD8 (100734), anti-CD49b (108907), anti-CD44 (103012), anti-CD62L (104418), anti-NKG2D (130208), anti-Foxp3 (126406), and anti-Granzyme B (515407). The myeloid panel comprised anti-CD45 (103126), anti-Gr-1 (108412), anti-CD11b (101262), anti-F4/80 (123108), anti-CD11c (117308), anti-CD103 (121423), anti-CD38 (102718), and anti-CD206 (141716).
NanoString analysis
Tumors subjected to L-XRT were harvested, TILs were isolated, and RNA was extracted and subjected to NanoString analysis with the nCounter Immunology Panel of 561 mouse genes including 15 internal reference genes. This panel includes major cytokines and chemokines with their receptors, interferons with their receptors, and TNF receptor superfamily genes. Data were analyzed and heatmaps generated with nSolver V4.0 software.
GP-100 protein ELISA
Serum samples from C57BL/6 mice implanted with MC38-gp100 tumor cells irradiated with three 12 Gy fractions, three 5 Gy fractions, two 1 Gy fractions, or three 12 Gy fractions with α-PD1 were collected at 24 hours after XRT and subjected to sandwich ELISA against gp-100 protein (LSBio, Cat#LS-F19711). Briefly, 96-well plates coated with capture antibody were incubated with serum for 2 hours at 37°C, followed by incubation for 1 hour with biotinylated detection antibody. Plates were then washed, incubated with streptavidin–HRP complex for 1 hour, washed again several times, and incubated with TMB substrate for 20 min at 37°C, after which the reaction was stopped and the plates read with a microplate reader at 450 nm.
Reverse transcription polymerase chain reaction (RT-PCR)
TILs from the secondary tumors of the various treatment groups were harvested on day 14, 1 day after the second fraction of L-XRT, and the RNA extracted for RT-PCR quantification. Primers used were murine TGFB1_Fw CCACCTGCAAGACCATCGAC and TGFB1_Rv CTGGCGAGCCTTAGTTTGGAC; PPIA_Fw GAGCTGTTTGCAGACAAAGTTC and PPIA_Rv CCCTGGCACATGAATCCTGG.
TGF-β ELISA
Sera were collected from defined experimental groups 3 days after the last fraction of low-dose radiation and tumor growth factor-beta (TGF-β) levels were quantified using Bio-Plex Pro TGF-b 3-plex Panel (BioRad, Cat#171W4001M) according to manufacturer’s instructions.
Statistics
GraphPad Prism V8.0 software was used for statistical analyses. Student’s t-tests were used to assess significance between individual groups. Survival percentages were analyzed by using the Kaplan-Meier method and compared with log-rank tests. Two-way analysis of variance (ANOVA) was used where applicable to compare tumor growth curves for experimental and control groups. Where indicated, one-way ANOVA followed by Tukey’s multiple comparison tests were conducted. Statistical significance was defined as p≤0.05.
Results
Low-dose XRT boosts antitumor immunity through T cell activation, NK cell infiltration, M1 macrophage polarization, and reduction of TGF-β cytokine
In an effort to study the efficacy of L-XRT and identify the underlying mechanisms, we established 344SQ-P tumors in the right hind legs of 129Sv/Ev mice (one-tumor model) and irradiated the tumors to various doses when they reached 7–8 mm. We found that L-XRT (1 Gy × 2 fractions) alone was effective in extending survival by controlling tumor growth (figure 1A and online supplemental figure S1). Additionally, combination of L-XRT with immunotherapy agents such as anti-PD1 and anti-CTLA-4 significantly enhanced the antitumor efficacy of these systemic checkpoint inhibitors. L-XRT monotherapy extended survival from day 24 to day 35; L-XRT + anti-PD1 extended survival to day 38; L-XRT + anti-CTLA-4 to day 41; while L-XRT + anti-PD1 + anti-CTLA-4 scored 60% tumor-free survival over 62-day observation period (figure 1A). In a separate experiment, tumor-dLNs were harvested 3 days after L-XRT and flow cytometric analysis showed significant activation of CD4 (figure 1B) and CD8 T cells (figure 1C) after two 1 Gy fractions to the tumor. Other titrated doses such as 0.1 Gy × 2, 0.5 Gy × 2, 0.5 Gy × 4, and 1 Gy × four did not result in significant differences (data not shown). L-XRT was also found to enhance infiltration of NK cells within the TME (ie, among TILs) but not in the spleen at 48 hours after treatment (figure 1D). We further found that L-XRT led to significant increases in M1 (Gr1int. CD11b+ F4/80+ CD38hi) tumor-associated macrophages (TAMs, figure 1E). Molecular NanoString analysis for TILs 24 hours after L-XRT in the one-tumor model showed significant downregulation of TGF-β1 gene expression as compared with non-irradiated control tumors (p=0.0007, figure 1F and online supplemental figure S2). Other significantly modulated targets included complement system proteins C1s, C2, C4a, C6, C7, C8b; proinflammatory cytokines and their receptors such as IL-1; and Vcam1 anchoring molecule needed to recruit trafficking T cells and help their infiltration into tumors (p=0.04). STAT6, usually associated with Th2 Cytokines IL-4/IL-13 and M2 macrophages, was also downregulated with L-XRT.
Supplemental material

L-XRT hampers tumor growth, activates T cells, increases NK cells and M1 macrophages, and downregulates the inhibitory cytokine TGF-β. (A), The one-tumor mouse model was established by injecting 344SQ-P cells (0.5 × 106) into the right hind legs of 129Sv/Ev mice (n=5/group), after which L-XRT (1 Gy × 2 fractions) was delivered when tumors reached around 7–8 mm in diameter. Anti-PD1 (200 µg/inj.) and anti-CTLA-4 (50 µg/inj.) were administered intraperitoneally on days 5, 8, 12, and 16 post 344SQ-P injection. Mice were euthanized when tumor reached 14 mm, and survival was plotted by the Kaplan-Meier method. Experiment was repeated twice, and similar patterns were detected. (B and C) dLNs from the L-XRT dose groups were harvested at 3 days after radiation and analyzed with flow cytometry. Both CD4 (No XRT vs 1 Gy × 2, p=0.0318) and CD8 cells (No XRT vs 1 Gy × 2, p=0.0001) were activated after two 1 Gy fractions, as depicted by the activation marker CD44. (D and E), spleens (spl), dLNs, and TILs were harvested at 48 hours after two 1 Gy fractions for phenotyping by flow cytometry. (D) Cells were gated on the CD45+ population and then on CD49b+ to identify NK cells (TILs no XRT vs TILs 1 Gy × 2, p=0.0328). (E) Cells were gated on CD45+ and then on Gr1intermediate and CD11b+ to identify macrophages, and further gated on F4/80+ CD38hi to identify M1 macrophages (No XRT vs 1 Gy × 2, p=0.0463). (F) NanoString molecular counts (nCounter immunology panel) showed significant reduction in local TGF-β expression 24 hours after two 1 Gy fractions of L-XRT (p=0.0007). Data for each group was represented as mean±SD and Student’s t-tests were used to compare groups. *p≤0.05 was considered to indicate statistical significance. dLNs, draining lymph nodes; L-XRT, low-dose radiation; PD1, programmed cell death protein 1; TGF-β, tumor growth factor beta; TILs, tumor-infiltrating leukocytes.
Combining high-dose XRT with low-dose XRT (RadScopal technique) and immunotherapy is key for generating systemic antitumor responses and downregulating TGF-β
Evidence from our preclinical and clinical observations suggested that when the tumor burden is large (ie, >1 lesion or disease site), primary lesions must be controlled with H-XRT (to augment the release of tumor-associated antigens; online supplemental figure S3), and secondary lesions should receive L-XRT (to maximize systemic immune-mediated outcomes). To further confirm this hypothesis, we established the two-tumor murine model, in which we treated primary and secondary tumors with either H-XRT or L-XRT or a combination of both, with simultaneous systemic α-PD1. H-XRT to primary only plus α-PD1 effectively controlled primary tumors, but L-XRT plus α-PD1 did not (online supplemental figure S4). Moreover, either H-XRT or L-XRT to both primary and secondary tumors, with α-PD1, did not control secondary tumors (online supplemental figure S4). However, delivering H-XRT to primary tumors with L-XRT to secondary tumors and systemic α-PD1 led to significant retardation of secondary tumor growth (online supplemental figure S4).
To further enhance the effects of L-XRT and remove stromal barriers, we replicated the two-tumor model in 129Sv/Ev mice, only this time we used both α-CTLA-4 and α-PD1 as immunotherapy backbone in order to block Tregs and attenuate T cell exhaustion (figure 2A). Our goal was to optimally modulate the stroma and allow effector immune cells to infiltrate/expand, and elicit their killing functions. As expected, we found that using either α-PD1 or α-CTLA-4 with the RadScopal technique noticeably enhanced systemic abscopal responses. Adding both immunotherapies along with H-XRT and L-XRT successfully controlled primary and secondary tumors and achieved 90% survival over the 50-day observation period (figure 2B,C). To explore the effects of these treatment conditions on TGF-β expression, we harvested TILs from secondary tumors at 24 hours after the final L-XRT dose (figure 2D) and subjected the isolated RNA to RT-PCR. Although H-XRT alone significantly upregulated TGF-β1 relative to the control group (p=0.05), the addition of L-XRT in RadScopal treated group significantly downregulated TGF-β1 when compared with H-XRT only group (p=0.0275). Addition of α-CTLA-4 to RadScopal + α-PD1 further decreased TGF-β1 expression relative to the H-XRT-only condition (p=0.0102, figure 2D). Moreover, we collected sera on day 16 from various groups of the two-tumor model to assess for systemic TGF-β1 levels (figure 2E). The difference between H-XRT and L-XRT groups was significant (p=0.0173); H-XRT versus RadScopal, p=0.1196; and H-XRT versus RadScopal + α-PD1 + α-CTLA-4, p=0.0188.
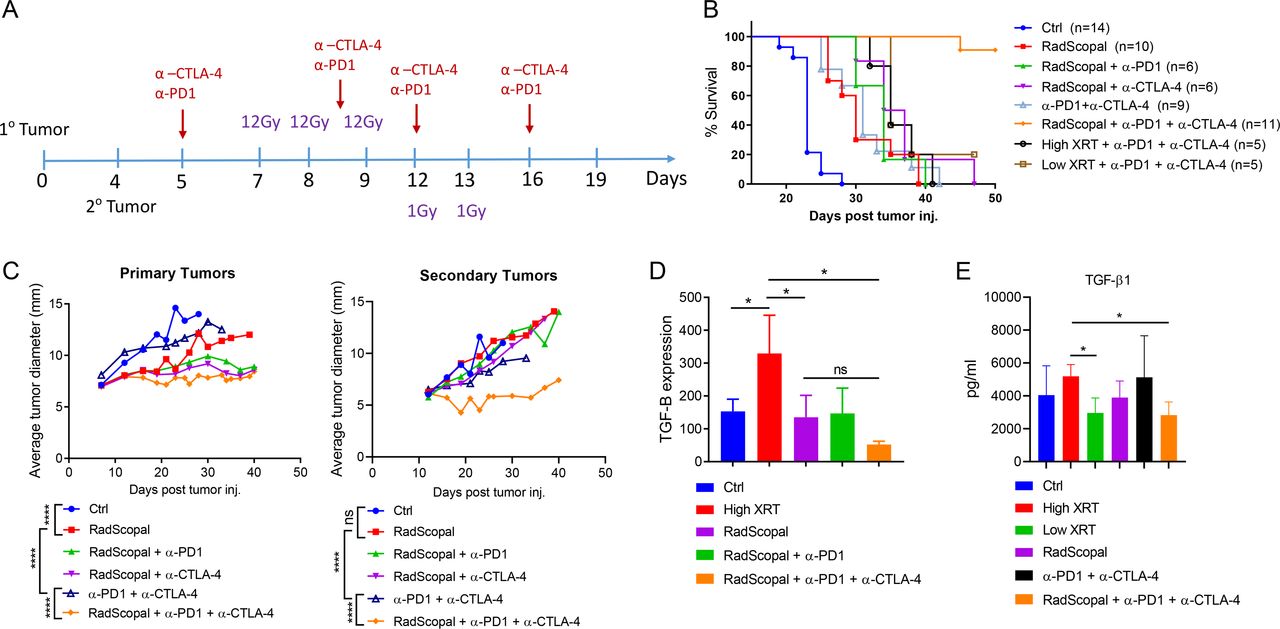
H-XRT to the primary tumor plus L-XRT to the secondary tumor (RadScopal technique) plus double-agent immunotherapy is required for optimal systemic antitumor outcomes and reduction of TGF-β at secondary sites. (A) Treatment timeline. Primary tumors were injected subcutaneously on day 0 and secondary tumors injected on day 4. H-XRT (three 12 Gy fractions) was delivered to primary tumors on days 7, 8, and 9; L-XRT (two 1 Gy fractions) was delivered to secondary tumors on days 12 and 13. Double-agent immunotherapy (anti-PD1 + anti-CTLA-4) was given intraperitoneally on days 5, 8, 12, and 16. (B) Survival curves for the indicated radioimmunotherapy treatment groups. ‘RadScopal’ refers to H-XRT to the primary and L-XRT to the secondary tumors. Experiment was repeated twice and data were pooled. Median survivals following the order of group labels were (23, 30, 34, 35.5, 31, N/A, 35, and 35 days post tumor inoculation, respectively). (C) Tumor growth curves for primary (1°) and secondary (2°) tumors after the indicated treatments. Mice were euthanized when either the primary or secondary tumors reached 14 mm in diameter. (D) Secondary tumors were harvested on day 14, 1 day after delivery of the second L-XRT fraction. TILs were enriched by Histopaque separation and RNA was isolated for RT-PCR with TGF-β primers and peptidylprolyl isomerase A (PPIA) controls. Delta–delta CT values were calculated and TGF-β expression was reported using 2−ΔΔCT values. (E) Systemic TGF-β levels were assessed by serum BioPlex assay on day 16. *p≤0.05 was considered statistically significant. H-XRT, high-dose radiation; L-XRT, low-dose radiation; PD1, programmed cell death protein 1; PPIA, peptidylprolyl isomerase A ; TGF-β, tumor growth factor beta; TILs, tumor-infiltrating leukocytes.
High-dose XRT with low-dose XRT treatment is effective across mouse models, is immune-mediated, and relies on CD4 T cells and NK cells
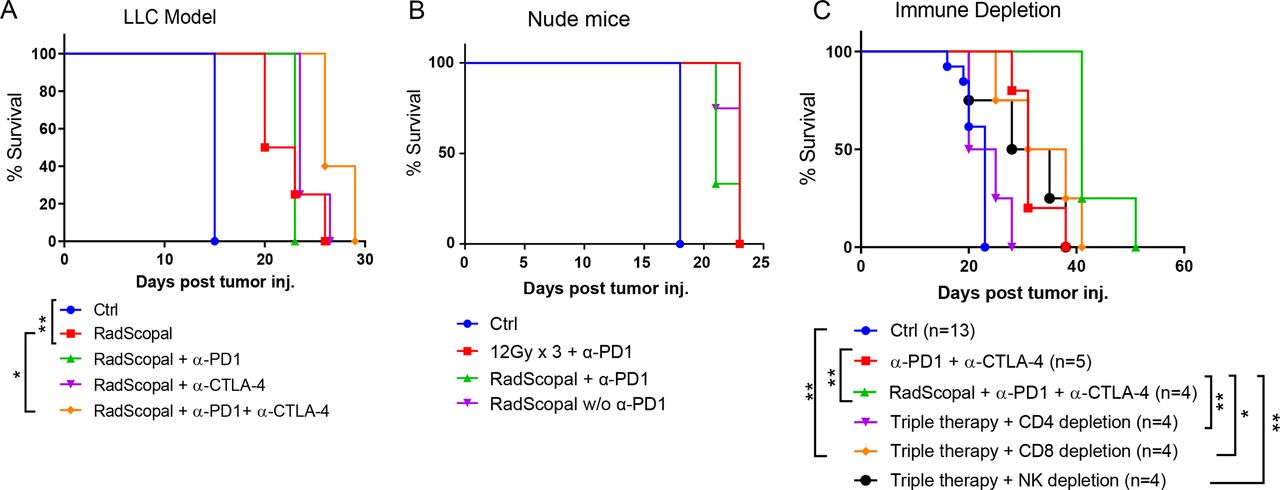
In the next set of experiments, we sought to confirm that the detected antitumor protection with RadScopal + immunotherapy is not limited to 344SQ and may be extended to other solid tumors. Therefore, we established the bilateral two-tumor model in C57BL/6 mice and challenged them with LLC tumors (figure 3A). As was true for the 344SQ model, combining H-XRT with L-XRT led to longer survival than in the control mice (p=0.0047), and the addition of anti-PD1 + anti-CTLA-4 extended survival still further (p=0.0198, figure 3A). However, similar experiments with nude mice (which lack normal T cell function) negated the antitumor response (figure 3B), indicating that adaptive immunity is essential for protection against tumors. To further dissect the immune cell population(s) involved and their role in the observed antitumor protectivity, we created 129Sv/Ev mouse models in which CD4 cells, CD8 cells, or NK cells were depleted before treatment and the depletion pressure was maintained once a week until the experimental endpoint. Depletion of CD4 cells completely negated the triple combination response of RadScopal treatment + anti-PD1 + anti-CTLA-4 (p=0.0062), and all mice expired within a period similar to that of tumor-bearing controls (figure 3C). Depletion of NK cells also significantly reduced the antitumor response (RadScopal treatment + anti-PD1 + anti-CTLA-4 vs NK depletion, p=0.0067), but the effect was not as pronounced as depleting the CD4 T cells. Surprisingly, depletion of CD8 cells interfered the least with the antitumor effect (p=0.04), but we observed higher numbers of lung metastases with the ablation of this population (online supplemental figure S5).

H-XRT to the primary tumor plus L-XRT to the secondary tumor is effective in other solid tumor models, is immune-mediated, and depends on CD4 T cells and NK cells. (A) The two-tumor LLC model was established in C57BL/6 mice (n=5/group). Survival curves are shown for the indicated groups, with log-rank tests used to compare groups (RadScopal vs control, p=0.0047; RadScopal vs RadScopal + anti-PD1 + anti-CTLA-4, p=0.0198). (B) In a two-tumor nu/nu mouse model lacking functional T cells (n=4/group), neither H-XRT + anti-PD1 nor RadScopal + anti-PD1 showed any efficacy. (C) A two-tumor 344SQ-P model was established in 129Sv/Ev mice, which were then given anti-CD4, anti-CD8, or anti-NK antibodies to deplete the corresponding immune cells. Survival was plotted by the Kaplan-Meier method and log-rank tests used to compare groups. *p≤0.05 was considered statistically significant. H-XRT, high-dose radiation; LLC, Lewis lung carcinoma; L-XRT, low-dose radiation; NK, natural killer; PD1, programmed cell death protein 1.
Combining high-dose XRT with low-dose XRT and double-agent checkpoint blockers increases CD4+ effector T cells, enhances NK cell activation, and increases M1 macrophages in secondary tumors
To further explore the effects of H-XRT, L-XRT, and immunotherapy on immune cell subpopulations, we harvested secondary tumors, spleens, and blood on day 19 (figure 2A) and used flow cytometry to phenotype the lymphoid and myeloid populations. CD4+ T cells in particular were upregulated with the triple combination of RadScopal treatment + anti-PD1 + anti-CTLA-4 (one-way ANOVA, p=0.0008, figure 4A), but not Tregs (one-way ANOVA, p=0.8810, figure 4B) in secondary tumors. The anti-CTLA-4 IgG2b isotype used is non-depleting (online supplemental figure S6) and functions by blocking Tregs. The triple combination (RadScopal treatment + anti-PD1 + anti-CTLA-4) also significantly enhanced NK cell activation and Granzyme B production both in terms of percentages (figure 4C, one-way ANOVA, p=0.0019) and in numbers of cells (figure 4D, one-way ANOVA p=0.0099, followed by Tukey’s multiple comparison test, anti-PD1 + anti-CTLA-4 vs RadScopal + anti-PD1 + anti-CTLA-4, p=0.0173). The percentages of conventional dendritic cells (CD11c+ CD11b+) were unaltered when comparing RadScopal + anti-PD1 + anti-CTLA-4 to RadScopal only or anti-PD1 + anti-CTLA-4 only (figure 4E). However, their absolute cell counts were upregulated on adding RadScopal radiation to anti-PD1 and anti-CTLA-4 treatment (one-way ANOVA p=0.0019, with post hoc Tukey’s multiple comparison analysis, anti-PD1 + anti-CTLA-4 vs RadScopal + anti-PD1 + anti-CTLA-4, p=0.0358, figure 4F). Similarly, the triple therapy also increased M1 macrophage percentages (figure 4G, one-way ANOVA p=0.0064, followed by Tukey’s multiple comparison analysis, RadScopal vs RadScopal + anti-PD1 + anti-CTLA-4, p=0.05) and M1 macrophage numbers (figure 4H, one-way ANOVA p=0.0010, followed by Tukey’s test, anti-PD1 + anti-CTLA-4 vs RadScopal + anti-PD1 + anti-CTLA-4, p=0.0054). We also observed decreased M2 percentages when adding immunotherapy to radiation (figure 4I, one-way ANOVA p=0.0006, and post hoc Tukey’s multiple comparison test for RadScopal vs triple combination, p=0.0042). The immunotherapy alone seemed to have advantage over the triple combination to decrease M2 numbers (figure 4J), but the counts did not reach significance. Collectively, findings from this phenotyping data is in agreement with our previous CD4 and NK depletion data and emphasizes innate and adaptive immune collaborative effort. Next, our examination of the excised spleens (online supplemental figure S7) also revealed that H-XRT + L-XRT + anti-PD1 + anti-CTLA-4 led to upregulation of CD4+ T cells (RadScopal vs RadScopal + anti-PD1 + anti-CTLA4, p=0.0129). However, we only saw activation of NK cells (CD49b+ NKG2D+) with RadScopal + anti-CTLA-4 vs RadScopal group alone (p=0.0111). Interestingly, migratory dendritic cells (CD11c+ CD103+) that can cross prime antigens to T cells were significantly higher in the triple combination group versus anti-PD1 + anti-CTLA-4 group (p=0.0065). In addition, both CD38+ M1 population (p=0.0352) and CD206+ M2 population (p=0.0449) were higher in the spleen with the triple combination therapy versus anti-PD1 + anti-CTLA-4 immunotherapy, which could be attributed to the dual effect of H-XRT on systemic cell populations. At last, phenotyping of lymphoid population in the blood revealed significant upregulation of CD4 central memory with the triple combination therapy (anti-PD1 + anti-CTLA-4 vs RadScopal + anti-PD1 + anti-CTLA-4, p=0.0433; RadScopal vs RadScopal + anti-PD1 + anti-CTLA-4, p=0.0094), while CD4 effector memory was upregulated by either immunotherapy alone or with triple therapy (anti-PD1 + anti-CTLA-4 vs RadScopal, p=0.0006; RadScopal vs RadScopal + anti-PD1 + anti-CTLA-4, p=0.0224, online supplemental figure S8). On the other hand, CD8 central memory was not significantly impacted by the triple therapy, but circulating CD8 effector memory was equally upregulated by both immunotherapy alone and triple combination treatments (anti-PD1 + anti-CTLA-4 vs RadScopal, p=0.0034; RadScopal vs RadScopal + anti-PD1 + anti-CTLA-4, p=0.0047). Interestingly, Tregs were also upregulated in the blood on adding anti-PD1 + anti-CTLA-4 to H-XRT + L-XRT (anti-PD1 + anti-CTLA-4 vs RadScopal, p=0.0139; RadScopal vs RadScopal + anti-PD1 + anti-CTLA-4, p=0.0025, online supplemental figure S8). However, that trend did not apply to secondary tumors treated locally with L-XRT as demonstrated earlier in figure 4B.
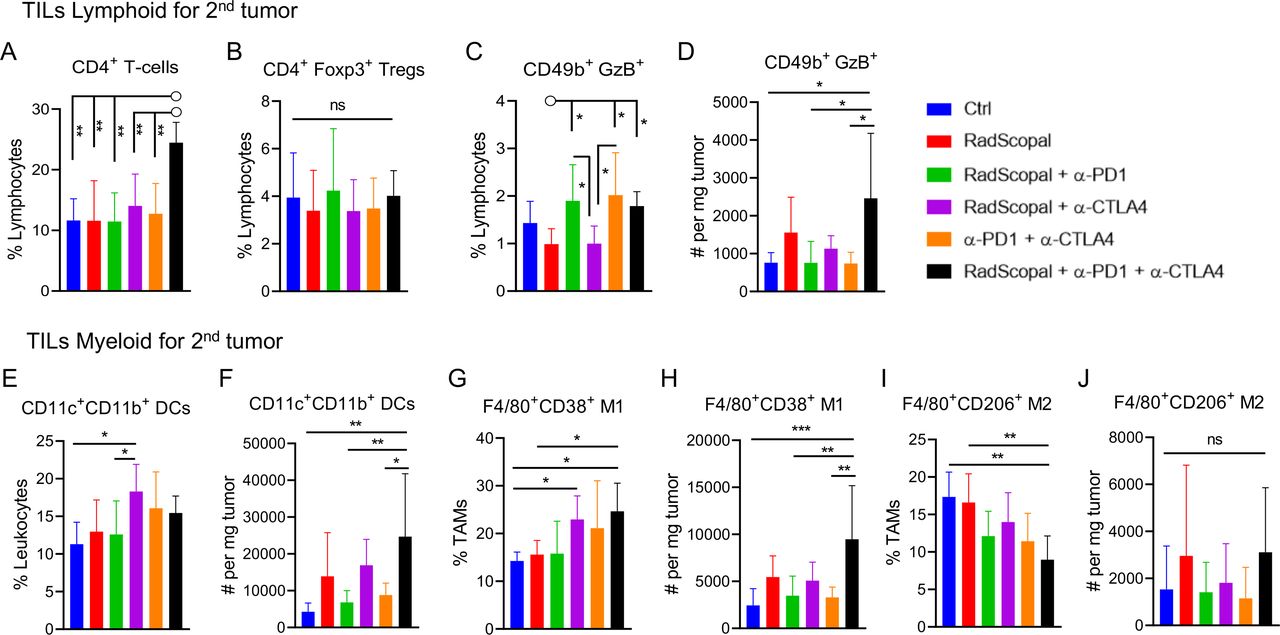
H-XRT to the primary tumor plus L-XRT to the secondary tumor plus anti-CTLA-4 and anti-PD1 checkpoint inhibitors increase CD4+ T-effector cells, enhance NK cell function, and increase M1 macrophage polarization in secondary tumors. On day 19, we isolated TILs from the secondary tumors of the two-tumor 129Sv/Ev mouse model to phenotype lymphoid and myeloid subpopulations by flow cytometry (n=5–6/group). (A) Percentages of CD4+ T cell lymphocytes. (B) Percentages of Treg lymphocytes. (C) Percentages of CD49+ NK cells producing granzyme B in the lymphocyte population. (D) Total numbers of CD49+ granzyme B+ NK cells per mg of tumor weight. (E) Percentages of DCs from leukocytes. (F) Total numbers of DCs per mg of tumor weight. (G) Percentages of F4/80+ CD38hi M1 TAMs in the Gr1intermediate CD11b+ population that was gated on the CD45+ parent population. (H) Total numbers of M1 TAMs per mg of tumor weight. (I) Percentages of F4/80+ CD206+ M2 TAMs in the Gr1intermediate CD11b+ population that was gated on the CD45+ parent population. (J) Total numbers of M2 TAMs per mg of tumor weight. Data for each group was represented as mean±SD. One-way ANOVA statistical analysis followed by Tukey’s multiple comparison tests were conducted to compare the multiple treatment groups. p≤0.05 was considered to indicate statistical significance. *p≤0.05, **p≤0.01, ***p≤0.001. ANOVA, analysis of variance; DCs, dendritic cells; H-XRT, high-dose radiation; L-XRT, low-dose radiation; NK, natural killer; PD1, programmed cell death protein 1; TAMs, tumor-associated macrophages; TILs, tumor-infiltrating leukocytes; Tregs, T regulatory cells.
Clinical observations: enhanced response of L-XRT lesions in patients who received H-XRT and immunotherapy
The benefits of L-XRT are not limited to in vivo murine experiments. To date, we have treated more than 25 patients on various trial arms with H-XRT, L-XRT, and immunotherapy to promote secondary tumor response.16 Two representative cases from our recent prospective trial (NCT02710253) are demonstrated here (figure 5A and online supplemental figure S9), followed by the tumor growth curves of nine patient cases that received RadScopal treatment from the same trial (figure 5B and table 1). Patient 1 in figure 5A was a 54-year-old man with metastatic human papillomavirus-positive oropharyngeal squamous cell carcinoma who initially reported partial response to anti-PD1 therapy. However, after 8 months, multiple lesions in the lung and a large abdominal mass had increased in size. A single lesion in the left upper lobe of the lung was treated with H-XRT (50 Gy in four 12.5 Gy fractions) and the abdominal lesion was concurrently treated with L-XRT (6 Gy in four 1.5 Gy fractions) and ongoing anti-PD1 therapy (pembrolizumab) with no other treatments in the interim. Two months later, the abdominal lesion (prospectively treated with L-XRT) had not yet responded and showed elevated avidity on positron emission tomography (PET) scanning (standardized uptake value (SUV) 11.9). However, a PET scan obtained 6 months after L-XRT showed an interval decrease in lesion volume (from 5.2 cm in diameter to 2.5 cm, >50% from that on the baseline CT scan) and near-complete resolution of avidity on PET (minimal residual SUV), indicating a robust response.

Prospective treatment of secondary tumors with L-XRT shows significant therapeutic benefit in patients. (A) Scans from a 54-year-old male with metastatic human papillomavirus (HPV)-associated oropharyngeal squamous cell carcinoma refractory to previous treatment with anti-PD1 therapy. A single lesion in the upper left lobe of the lung was treated with H-XRT (four 12.5 Gy fractions) and a large abdominal lesion was concurrently treated with L-XRT (four 1.5 Gy fractions) and ongoing pembrolizumab (anti-PD1). A PET scan obtained 6 months after L-XRT showed a decrease of more than 50% in the volume of the abdominal lesion with near-complete resolution of avidity. (B) Spider plot depicting tumor response curves of secondary lesions that received L-XRT in nine patients who received concurrent H-XRT to primary tumors and systemic anti-PD1 or anti-PD-L1 immunotherapy. H-XRT, high-dose radiation; L-XRT, low-dose radiation; PD, progressed disease; PD1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; PET, positron emission tomography; PR, partial response.
Patient 2 was a 73-year-old male with metastatic melanoma who received anti-PD1 therapy for 8 months, where his best response was stable disease, until he progressed and presented with lesions in the lung and spleen (online supplemental figure S9). A single lesion in the left lobe of the lung was treated with H-XRT (45 Gy in 15 3 Gy fractions), and part of a metastasis in the anterior spleen was concurrently treated with L-XRT (5 Gy in five 1 Gy fractions), and ongoing pembrolizumab. Partial radiation to the spleen was provided due to potential for poor organ function if total lesion was irradiated at that time. Scans obtained 4 months after treatment showed decreased density of the splenic lesion treated with L-XRT (from 78 to 30 Hounsfield units) and decreased size (from 5.2 cm to 3.1 cm in the largest diameter), suggesting a partial response. Only the portion of the splenic metastasis treated with L-XRT showed signs of response (online supplemental figure S9).
To emphasize the translational benefits of the RadScopal treatment, we have graphed the tumor response curves of secondary lesions that received L-XRT treatment in nine patients who received concurrent H-XRT and mainly anti-PD1 or anti-programmed death-ligand 1 (PD-L1) immunotherapy (figure 5B and table 1). Eight out of the nine patients had their secondary lesions partially respond over the 40 weeks observation period. It is interesting to note the one patient who did not show any response to L-XRT had progressed on OX40 and 4-1BB. All patients had their primary tumor treated with stereotactic H-XRT and either one or two secondary lesions treated with L-XRT including but not limited to inguinal lymph nodes, liver lesions, bone metastasis, peritoneal implant, and upper/lower lung lesions. The growth of secondary lesions was monitored over time along with non-irradiated lesions if any. Patients who received L-XRT to all lesions were excluded from analysis as they continued to progress which is in agreement with our preclinical observations. An additional patient with single lesion who showed complete response to L-XRT was also excluded from analysis due to the absence of secondary lesions.
Discussion
Immunotherapy, with checkpoint inhibitors in particular, has had profound effects on the treatment of cancer; nevertheless, most patients do not experience full benefit from such therapy, reasoning to the inability of lymphocytes to penetrate solid tumors and/or get activated efficiently. We developed a unique form of combination therapy in which H-XRT is delivered to primary tumors (to release antigens and prime T cells) and L-XRT is given to secondary tumors (to modulate the stroma and allow the infiltration and expansion of effector T cells and NK cells). The proposed effects of this double form of radiation therapy, which we call ‘RadScopal’ treatment, are illustrated in figure 6. We recently completed a post-hoc clinical analysis of 26 patients who received L-XRT either in the form of radiation scatter from nearby H-XRT lesion or intentional L-XRT was delivered to the tumor lesion (lung or liver mainly) with immunotherapy backbone. Lesions that received L-XRT showed an enhanced response rate of 36% while control lesions that received no L-XRT had only 4% response rate.16 To understand the underlying mechanisms, we first established a lung adenocarcinoma one-tumor model transplanted in 129Sv/Ev mice and evaluated the optimal L-XRT dose per fraction that may correspond to this murine model. We found that giving two 1 Gy fractions on subsequent days was optimal for controlling tumor growth, while higher or lower doses of radiation did not prove efficacious, as higher doses especially could be deleterious to the immune cells.17–19 A closer look at the TME revealed significant infiltration of NK cells after 1 Gy × 2 L-XRT and polarization of TAMs to the M1 phenotype. This antitumor modulation of the TME and stroma was shown to be coupled with reduction in TGF-β in TILs of subsequent experiments on L-XRT treatment.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Illustration of the proposed effects from ‘RadScopal’ therapy. At the primary tumor site, high-dose stereotactic radiation (H-XRT) releases tumor-associated antigens and upregulates MHC-I molecules to initiate T cell priming. APCs then present those neoantigens to CD4+ and CD8+ T cells in the dLNs for further activation. At the secondary (abscopal) tumor site, L-XRT modulates the stroma to overcome its inhibitory barriers. Pronounced mechanisms involve M1 macrophage polarization, downregulation of TGF-β, activation and infiltration of CD4+ T cells, and increases in proportion and activity of NK cells. The addition of double-agent checkpoint-inhibitor immunotherapy (anti-PD1 plus anti-CTLA-4) further augments the systemic effects of the radiation by blocking Tregs and attenuating T cell exhaustion, thereby prolonging and enhancing the antitumor response. APCs, antigen-presenting cells; dLNs, draining lymph nodes; L-XRT, low-dose radiation; PD1, programmed cell death protein 1; TGF-β, tumor growth factor beta; Tregs, T-regulatory cells.
In the next set of experiments, we investigated the effects of L-XRT in a two-tumor murine model, representing a larger tumor burden. The rationale was that the presence of an additional tumor may hinder the effectiveness of treatment directed to the L-XRT site as well as the existence of cross-communication between the two tumors.20 Indeed, treating both the primary and secondary tumors with L-XRT was not effective regardless of whether anti-PD1 was used or not. Similarly, delivering H-XRT to both tumors seemed to accelerate tumor growth, which could be attributed to H-XRT’s ability to induce higher levels of Tregs.11 Optimal antitumor control was achieved by treating the primary tumor with H-XRT while subjecting the secondary tumor to L-XRT, especially in combination with anti-PD1, hence giving birth to the ‘RadScopal’ concept and opening the way for bringing radiation therapy to a new realm of systemic disease control.
Despite the positive immune-regulatory aspects of H-XRT, it is shown by others to upregulate TGF-β levels and elicit negative side effects.21 22 We thus combined RadScopal therapy (H-XRT plus L-XRT) with anti-PD1 and anti-CTLA-4 immunotherapy, with the goals of quenching Tregs, further downregulating TGF-β, and preventing long-term T cell exhaustion. This triple combination led to 90% survival over the 50-day observation period, significant control of both primary and secondary tumors, and reduction of TGF-β in the TILs of secondary tumors treated with L-XRT. We deliberately chose a Treg-blocking antibody (rather than a Treg-depleting antibody) for anti-CTLA-4, and confirmed the lack of depletion with blood phenotyping—results that directly correlate with current human studies with ipilimumab and tremelimumab.23
To demonstrate that the triple combination therapy (RadScopal treatment + anti-PD1 + anti-CTLA-4) is histology agnostic, we tested this regimen in a bilateral LLC model where RadScopal treatment alone had significant antitumor efficacy over control, which was further enhanced by adding anti-PD1 and anti-CTLA-4 immunotherapy. Conversely, the antitumor effects were negated in nude mice lacking functional T cells, which emphasizes the importance of systemic immune effectors over local tumor killing by radiation. Indeed, selective depletion experiments in 129Sv/Ev wild-type mice showed that CD4+ T cells and NK cells were crucial for the antitumor effect while CD8+ T cells were subsidiary.
Next, our flow cytometry analyses of TILs from secondary tumors exposed to L-XRT in the two-tumor model confirmed the importance of the CD4+ T and NK cell lymphoid populations. The triple combination therapy significantly upregulated the percentage of CD4+ T cells but did not alter Treg percentages, indicating that the upregulated CD4 cells were T-effectors, and confirming that the anti-CTLA-4 used did not deplete Tregs. The triple therapy also increased NK cell percentages and absolute cell numbers, and a closer look at cell counts revealed that RadScopal only or immunotherapy only (α-PD1 + α-CTLA-4) did not significantly increase activated NK-counts. This also applied to the dendritic cells in the myeloid population of the analyzed TILs, where absolute cell counts increased only with the triple combination therapy but not with RadScopal or immunotherapy treatments alone. Concomitant with the ability of L-XRT to reduce TGF-β cytokine, the TILs phenotyping data also demonstrated reprogramming of the TME to generate more M1 macrophages and reduce M2 macrophages in the two-tumor model. Absolute cell counts marked a synergistic increase in M1 population with the triple therapy at secondary tumors while individual therapies with radiation or checkpoint inhibitors used did not score similar results. On the other hand, M2 macrophage percentages were high with RadScopal treatment alone since the H-XRT component is known to upregulate this population as well as MDSCs that may migrate systemically and impact secondary tumor sites.24 25 Therefore, the addition of double-agent immunotherapy (α-PD1 + α-CTLA-4) was necessary for reducing this M2 population. Collectively, our findings suggest that triple therapy (RadScopal treatment plus double-agent immunotherapy) can overcome the inhibitory effects of the tumor stroma and its associated barriers.
With the encouraging data obtained on the preclinical level, we have treated patients prospectively with RadScopal XRT. The two patients represented here were on anti-PD1 therapy, but were refractory to treatment prior to L-XRT. In patient 1 we treated the lung lesion with H-XRT and treated the abdominal lesion with L-XRT. In patient 2 we treated the lung lesion with H-XRT and treated a portion of the spleen tumor with L-XRT. Both patients significantly responded to L-XRT and patient 2 in particular proved L-XRT non-ablative to the splenic immune cells. The portion of the spleen that received L-XRT responded while the other untreated portion served as a negative control and continued growing. Notably, this absence of deleterious effects from L-XRT on immune cells illustrates the transformation of radiotherapy from a means of local control to an immune modulator. The L-XRT dose-per-fraction given to all of the patients who we report was between 1 Gy and 2 Gy which is in relevance to the preclinical dose used. In our recently published post hoc analysis of successful L-XRT-mediated abscopal responses, the dose-per-fraction intentionally prescribed ranged between 1 Gy and 2 Gy.16 Therefore, a similar L-XRT dose and fractionation schedule was used in this study at the discretion of the treating radiation oncologist. While our collective clinical cases suggest a potential dosage strategy, evidence from further prospective trials is warranted before establishing the most optimal dosage and fractionation to elicit L-XRT-mediated abscopal responses.
Numerous clinical trials are currently underway to test combinations of various targeted immune treatments with radiotherapy (‘iRT’).26 27 Recently completed phase I/II iRT studies with anti-CTLA-4 have demonstrated abscopal responses in 20%–30% of patients28–31 per RECIST or irRC criteria.32 33 Another recent prospective trial testing concurrent radiotherapy and granulocyte-macrophage colony-stimulating factor (GM-CSF) for patients with metastatic solid tumors had a promising abscopal response rate of 27%.34 Other iRT trials have shown toxicity profiles to be within acceptable limits, as might be suggested by the continued use of iRT in clinical trials.35 36 Other current trials with radiation plus immunotherapies include anti-CTLA-4 (eg, NCT02097732, NCT01970527), anti-PD1/PD-L1 (eg, NCT02402920, NCT02635360), GM-CSF (eg, NCT02677155) and TGF-β blockade (eg, NCT02538471). Abscopal effects thus far remain limited, but some studies illustrate that abscopal effects can be significant if the stroma is addressed. For example, in one study of non-small cell lung cancer, patients whose tumors were ‘stroma-poor’ (the ratio of tumor to stroma being measured on H&E stained tissue specimens) had better survival and lower disease recurrence than patients with ‘stroma-rich’ tumors.37 We believe that the RadScopal technique will also lead to control of microscopic and macroscopic disease in tumors with considerable stroma.
While to our knowledge the clinical cases reported here are the first to respond to H-XRT and L-XRT with immunotherapy, clinical trials (NCT02710253, NCT03085719) testing this concept are ongoing at this time to identify additional benefits or limitations. The post hoc analysis of our recently completed phase II trial of ipilimumab (anti-CTLA-4), in which stereotactic radiation therapy was given to lung or liver metastases, showed that tumors near the targets that received low-dose scatter radiation had significantly higher response rates (31%) compared with distant tumors that did not receive radiation (5%).38 While both prior retrospective studies16 38 and the present study provide exciting evidence for the therapeutic potential of L-XRT, a prospective randomized trial is warranted to confirm the role of L-XRT. In some of the patient cases we report here, there was regrowth of the tumors, suggesting delayed immune evasion. Therefore, further clinical data are needed to understand the efficacy and durability of this approach. Indeed, L-XRT treatments have been used by others in the context of whole-lung irradiation for Ewing’s sarcoma, suggesting that L-XRT can be easily translated in clinics worldwide.39 40 In our view, L-XRT is safe and easy to implement, and if a tumor does not respond with low-dose-mediated immunologic mechanisms, then H-XRT can be used to target the same tumor without violating dose constraints for retreatment. Finally, current cell-based therapies are proven efficacious for liquid tumors such as leukemia, but they fail to penetrate solid tumors due to the surrounding stroma. Since L-XRT can reprogram the TME to enhance the penetration of effector immune cells, it could be used in combination with adoptive cell therapies (eg, chimeric antigen receptor-T (CAR-T), CAR-NK, TCR-engineered cells) to extend the benefits of immunotherapy to greater numbers of patients.
Acknowledgments
The authors thank BMS for providing the anti-CTLA-4 and anti-PD1 antibodies. The authors also thank Dr Evan Cohen of the Hematopathology Research Department at MD Anderson for running multiplex plates, and Chen KueiHua Liu at University of Houston Downtown for assisting with data analysis. The authors are also grateful for the editorial suggestions from Christine F Wogan, MS, ELS, of MD Anderson’s Division of Radiation Oncology, in the development of this manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @mauscaetano, @hmenonz, @GlitzaMDPhD
Contributors HBB composed the original manuscript. Generating and interpreting preclinical data were done by HBB, AIY, JES, MSC, DC, FM, AL, AC, and TPR. Generating and interpreting clinical data were done by JWW, DSH, RF, AD, RR, NIC, HM, TRC, ICG, SGC, and QNN. All authors reviewed and proof read the manuscript. Supervision and oversight were done by JWW and MAC.
Funding This study received financial support from Bristol-Myers Squibb (BMS), Varian, and Cancer Center Support (Core) Grant P30 CA016672 from the National Cancer Institute, NIH, to the University of Texas MD Anderson Cancer Center.
Competing interests JW serves on the science advisory boards for Alpine Immune Sciences, RefleXion Medical, Mavu Pharma, MolecularMatch and Checkmate Pharmaceuticals, and he is the founder of Healios Oncology, MolecularMatch, and OncoResponse companies. MD Anderson Cancer Center has a trademark for RadScopal (TM). JW has research support collaborations with Bristol Myers Squibb, Checkmate Pharmaceuticals, Mavu pharma, Merck, and Nanobiotix. AD is an advisory board member for Nektar Therapeutics. IG is a consultant for Bristol Myers Squibb and Array Biopharma. DSH has consulting/advisory role in Alpha Insights, Axiom, Adaptimmune, Baxter, Bayer, Genentech, GLG, Group H, Guidepoint Global, Infinity, Janssen, Merrimack, Medscape, Numab, Pfizer, Seattle Genetics, Takeda, Trieza Therapeutics. DSH has research/grant funding from AbbVie, Adaptimmune, Amgen, AstraZeneca, Bayer, Bristol Myers Squibb, Daiichi-Sankyo, Eisai, Fate Therapeutics, Genentech, Genmab, Ignyta, Infinity, Kite, Kyowa, Lilly, LOXO, Merck, MedImmune, Mirati, MiRNA, Molecular Templates, Mologen, NCI-CTEP, Novartis, Pfizer, Seattle Genetics, Takeda. DSH's ownership interests include MolecularMatch (Advisor), OncoResponse (founder), Presagia Inc (Advisor).
Patient consent for publication Not required.
Ethics approval MD Anderson Cancer Center's Institutional Animal Care and Use Committee (IACUC).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.