Article Text

Abstract
Background Mesenchymal stem/stromal cells (MSC) accumulate and reside in tumor sites.
Methods Taking advantage of this feature in anticancer therapy, immortalized murine MSC (iMSC) were genetically altered to produce chemokine (C-C motif) ligand 19 (iMSC/CCL19), which attracts dendritic cells (DC) and T lymphocytes. Thereafter, iMSC/CCL19 were examined for their therapeutic efficacy using a syngeneic CT26 colon carcinoma cell line.
Results Co-injection of iMSC/CCL19 into mice significantly suppressed the in vivo growth of CT26 cells compared with that of CCL19-expressing immortalized fibroblasts (iFib/CCL19). This anticancer effect was not observed when injected in CT26-bearing nude mice. Co-injected iMSC/CCL19 survived longer than iFib/CCL19 in the tumor sites. In a therapeutic model, local injection of iMSC/CCL19 suppressed the tumor growth, and increased IFN (interferon)-γ+ CD8+ T cells and CCR7+ DC infiltration in tumor site was observed when treated with iMSC/CCL19, but not with iMSC. This antitumor effect was completely negated by depletion of CD4+ cells and partially negated by depletion of CD8+ cells. Furthermore, the antitumor effects induced by local injection of iMSC/CCL19 were augmented by additional therapy with anti-programmed death (PD)-ligand 1 (PD-L1) antibody, but not with anti-PD-1 antibody. This combination therapy cured most of the tumors in CT26-bearing mice.
Conclusion These results suggest that local therapy with iMSC/CCL19 can suppress tumor growth via effective recruitment of CCR7+ DC into tumor sites and increase IFN-γ+ CD8+ T cells, and that combination with anti-PD-L1 antibody therapy can be a powerful anticancer therapy.
- cell engineering
- dendritic cells
- immunotherapy
- programmed cell death 1 receptor
- tumor microenvironment
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Mesenchymal stem/stromal cells (MSC) are multipotent cells that can differentiate into osteoblasts, chondrocytes, and adipocytes.1 2 Therefore, these cells show potential as a source for cell therapy. Although the cell surface markers of MSC require further elucidation, highly purified MSC can be isolated from adult mouse bone marrow.3 Several studies have reported that MSC accumulate to injured areas and hypoxic tumor microenvironments.4 Taking advantage of these features, MSC have been employed as tumor-accumulating cells for anticancer therapy in various mouse models.5–9 Although several studies have combined human MSC and immunodeficient mice, few studies have developed models with mouse MSC and syngeneic mouse tumors. Syngeneic tumor models are critical for investigating in vivo antitumor T cell immunity after MSC therapy.
Chemokine (C-C motif) ligand 19 (CCL19) attracts T cells and dendritic cells (DC) through its receptor C-C chemokine receptor type 7 (CCR7),10 11 thereby regulating cell homing and adaptive immunity.12 13 The expression of CCL19 in human tumors correlates with intratumoral accumulation of CD8+ T cells and patient survival.14 15 In addition, CCL19-producing chimeric antigen receptor (CAR) T cells and endothelial progenitor cells can provide effective anticancer therapies.16 17
In recent years, immune checkpoint blockade (ICB) antibody therapy has received attention as a promising anticancer treatment.18 19 Several ICB antibodies targeting programmed death-1 (PD-1), PD-1 ligand (PD-L1), and cytotoxic T-lymphocyte associated protein 4 (CTLA4) can induce antitumor effects in certain cancer patients.20–22 Given that ICB therapy targeting PD-1 and PD-L1 is likely to restore exhausted antitumor T cells in tumor sites, the presence of T cells in tumor tissues is essential for ICB therapy. Indeed, T cell infiltration in tumor sites is correlated with the response to anticancer immunotherapy.23 Although promising, the therapeutic efficacy of ICB therapy is limited. Therefore, new strategies are needed to enhance the therapeutic efficacy of ICB. Given that success in anticancer ICB therapy is based on the premise of tumor-infiltrating immune cells, including T cells and DC, MSC-mediated local production of CCL19 could promote the infiltration of those cells and exert an antitumor effect.
In this study, we prepared immortalized murine MSC (iMSC) that produce CCL19 (iMSC/CCL19) and investigated their therapeutic efficacy using a CT26 colon carcinoma mouse model. Co-injection of iMSC/CCL19 into mice suppressed the in vivo growth of CT26 compared with that of CCL19-expressing immortalized fibroblasts (iFib/CCL19) in a T cell-dependent manner. In a therapeutic model, local injection of iMSC/CCL19 suppressed CT26 tumor growth; furthermore, T cell and DC infiltration increased in mice treated with iMSC/CCL19, but not with iMSC. Moreover, local injection of iMSC/CCL19 augmented the antitumor effects by combination therapy with anti-PD-L1 antibody, but not anti-PD-1 antibody, and this combination therapy cured most CT26-bearing mice.
Materials and methods
Mice
BALB/c 6-week-old female mice were purchased from CLEA Japan (Tokyo, Japan) and maintained under specific pathogen-free conditions. The experiments were carried out according to the Ethical Guidelines for Animal Experiments of the Shimane University Faculty of Medicine (IZ28-72, IZ28-55, IZ29-42, and IZ30-131).
Isolation of MSC and Fib
Murine MSC were isolated from the bone marrow of BALB/c 6-week-old female mice as described previously.3 Crushed bones from femurs and tibias were treated with 0.2% collagenase (Wako Chemicals USA,) in Dulbecco's Modified Eagle Medium (DMEM) for 1 hour at 37°C. Then, the cell suspension was filtered through a cell strainer (Falcon 2350). The cells were suspended in Hank’s balanced salt solution (HBSS) and incubated for 30 min on ice with the following monoclonal antibodies (mAbs) (purchased from eBioscience): biotinylated anti-PDGFRα, FITC-conjugated anti-Sca-1, PE-conjugated anti-CD45, and PE-conjugated anti-TER119. Biotinylated antibodies were visualized with APC-conjugated streptavidin (Invitrogen). PDGFRα+ Sca-1+ CD45−TER119− cells were sorted by a triple-laser Moflo (Dako). Murine primary fibroblasts were isolated from back skin; 10 mm diameter skin samples were collected, washed with PBS and treated with 0.2% collagenase I solution for 30 min.
Cell lines
CT26 is a murine colon carcinoma cell line of BALB/c mice origin. CT26 was maintained in RPMI-1640 medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% FBS and 20 µg/mL gentamycin (Sigma-Aldrich). iFib, iFib/CCL19, iMSC and iMSC/CCL19 were maintained in DMEM (Nacalai Tesque) supplemented with 10% FBS and 20 µg/mL gentamycin (Nacalai Tesque).
Vector construction and gene transduction
The vector containing SV40 T antigen and the EGFP gene, X3.1-I-SV40T-I-EGFP-I, was constructed by isolating SV40T via digestion with BamHI from the pLenti CMV/TO SV40 small +large T vector (clone w612-1, Addgene) and inserting it into the BglII site of V913YM vector. The SV40T gene was then inserted into the V907-pCAGY vector at the EcoRI site (pCAGY-SV40T). The CAG-SV40T gene was isolated from the pCAGY-SV40T vector with NotI and BamHI digestion and ligated with the NotI/BamHI-digested pBluescript I-I (NYSLT I-I). The CAG-SV40T and chicken HS4 insulator were then transferred to the X3.1-I-EGFP-I vector by digestion and insertion at the SalI site. The X3.1-I-EGFP-I vector was generated as described previously.24 The SV40T-EGFP gene was transfected into murine fibroblasts and MSC using Lipofectamine 3000 (Invitrogen).
Next, the retroviral vector containing the murine CCL19 gene, pWZLneo-CCL19, was constructed. The murine CCL19 cDNA (359 bp) including the EcoRI site was synthesized by Integrated DNA Technologies, and the CCL19 cDNA was then digested with EcoRI and ligated to the EcoRI-digested pWZLneo vector (Cell Biolabs). The pWZLneo-CCL19 vector was transfected into Plat-E packaging cells (Cell Biolabs). At 48-hour post-transfection, the supernatant was collected, filtered with a 0.45 µm filter (Millipore), and used to infect iFib and iMSC. At 24 hours after retroviral infection, transfectants were cultured in medium with G418 (400 µg/mL).
Treatment protocol
In a co-injection model, 5×105 CT26 cells and 1×105 iFib, iFib/CCL19, iMSC or iMSC/CCL19 cells were co-inoculated subcutaneously (s.c.) into the flank of BALB/c mice and tumor size was measured (mm2) for 20 days. In a therapeutic model, BALB/c mice were injected s.c. with 5×105 CT26 cells in the right flank, followed by intraperitoneal (i.p.) injection of cyclophosphamide (CP, 100 mg/kg) and/or intratumoral (i.t.) injection of 5×105 iFib, iFib/CCL19, iMSC or iMSC/CCL19 cells on the indicated days. Thereafter, tumor size (mm2) was measured for 29 or 30 days.
In a combination therapy model, mice received i.t. injection of 5×105 iMSC/CCL19 cells on days 13 and 15, and i.p. injection of anti-PD-L1 blockade antibodies (200 µg, clone 10F.9G2, Bio X cell) on days 15 and 17. Thereafter, tumor size (mm2) was measured for 29 or 30 days. All in vivo experiments were performed at least twice.
Flow cytometric analysis
Flow cytometry was performed using FACS Calibur (BD Biosciences). CT26 cells were cultured with or without 50 ng/mL interferon (IFN)-γ (Tonbo Biosciences) for 48 hours and stained with anti-CCR7 (clone 4B12), anti-PD-L1 (clone 10F.9G2), and isotype-matched control antibodies (BioLegend). iFib, iMSC, and iMSC/CCL19 were stained with the following antibodies as described previously (3): anti-PDGFRα (clone APA5), anti-PDGFRβ (clone APB5), anti-CD34 (clone HM34), anti-Sca-1 (clone D7), and anti-CD29 (HMβ1–1) (all antibodies from BioLegend). Anti-CD44 (clone IM7), anti-CD45 (clone 30-F11), and anti-CD117 (clone ACK2) antibodies were purchased from Tonbo Biosciences. Cell suspensions from tumor or spleen were stained with the following antibodies: PE-conjugated anti-CD3, PerCP-Cy5.5-conjugated anti-CD4, FITC-conjugated anti-CD8α, PerCP-Cy5.5-conjugated anti-CD11c, APC-conjugated anti-CCR7, FITC-conjugated anti-PD-1, and FITC-conjugated anti-F4/80 (antibodies purchased from BioLegend).
ELISA iFib, iFib/CCL19, iMSC, and iMSC/CCL19 cells were cultured for 48 hours. Then, the supernatants were collected, and the levels of CCL19 in the supernatants were measured using the mouse MIP3 beta ELISA Kit (#ab100729, Abcam) and Multiskan FC Basic plate-reader at 450 nm (Thermo Fisher).
Reverse transcription PCR
Total RNA was extracted from iFib, iFib/CCL19, iMSC, and iMSC/CCL19 using the PureLink RNA Mini Kit (Thermo Fisher). The cDNA was then synthesized using the Superscript III First-Strand Synthesis System (Invitrogen). Template cDNA was amplified for 30 cycles of PCR using KAPATaq Extra HS ReadyMix PCR Kit (NIPPON Genetics) with the following primers: egfp, 5′-ACGTAAACGGCCACAAGTTC-3′ (sense) and 5′-AAGTCGTGCTGCTTCATGTG-3′ (anti-sense); β-actin, 5′-TGGAATCCTGTGGCATCCATG-AAAC-3′ and 5′-TAAAACGCAGCTCAGTAACAGTCCG-3′. The PCR products were verified using electrophoresis on 1.5% agarose gels followed by staining with ethidium bromide.
Immunohistochemical staining
Tumors (1 day after i.t. injection of PBS, iMSC, or iMSC/CCL19) were snap frozen in Optimal Cutting Temperature (OCT)compound (Sakura Finetek Japan Co.) by using liquid nitrogen. Sections were cut with a cryostat and fixed in cold 4% paraformaldehyde (Wako). Sections were stained with Rat isotype control mAbs (IgG2a: clone eBR2a, IgG2b: clone eB149/10H5, eBioscience), anti-CD3 (clone 17A2, TONBO), anti-CD4 (clone RM4-5, eBioscience), or anti-CD8α (clone 53-6.7, TONBO)+anti-CD8b (clone H35-17.2, eBioscience), and then with ImmPRESS Goat anti-Rat IgG peroxidase polymer detection kit (Vector Laboratories). mAbs were visualized with ImmPACT DAB peroxidase substrate (Vector Laboratories) and sections were counterstained with Hematoxyline QS (Vector Laboratories). For CD11c staining, endogenous biotin blocking with an avidin/biotin blocking kit (Vector Laboratories) was added after fixation. Sections were stained with biotinylated anti-CD11c (clone N418, AbD Serotec) or a Hamster isotype control IgG (clone eBio299Arm, eBioscience), biotinylated goat anti-Hamster IgG (Vector Laboratories), and then VECTASTAIN Elite ABC HRP Kit (Vector Laboratories), and visualized with ImmPACT DAB substrate.
Results
Generation of CCL19-expressing MSC and fibroblasts
Murine PDGFR+ Sca-1+ MSC were isolated from the bone marrow of BALB/c mice, as described previously3; skin fibroblasts (Fib) were isolated as control. The cells were immortalized using the SV40T and EGFP gene. Freshly prepared primary MSC and Fib were designated as pMSC and pFib, respectively, and immortalized MSC and Fib were designated as iMSC and iFib, respectively. iMSC and iFib were positive for GFP, as they were transfected with SV40T-EGFP (figure 1A). While iFib showed decreased expression of H-2Kd, no change was observed between pMSC and iMSC (online supplementary figure S1). In addition, based on a previous study,3 we examined iFib, iMSC, and iMSC/CCL19 for their cell surface expression of PDGFRα, PDGFRβ, CD34, Sca-1, CD44, CD29, CD45, and CD117 (online supplementary figure S2). iMSC showed an expression pattern similar to that of MSC,3 and no expression differences were observed between iMSC and iMSC/CCL19.
Supplemental material
Supplemental material
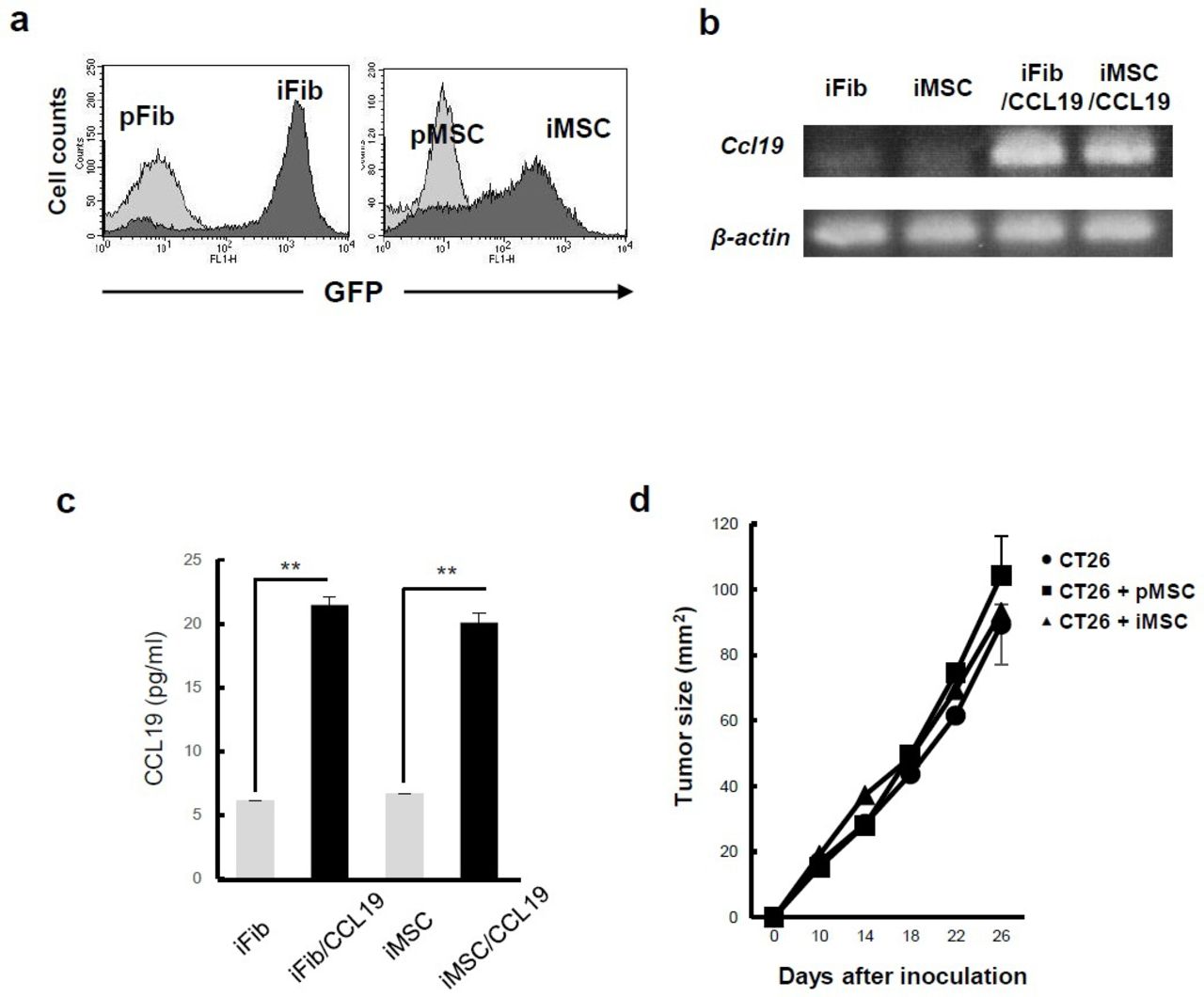
Generation of CCL19-expressing Fib and MSC. (A) Murine Fib and MSC were immortalized by transfection with SV40T-GFP vector. GFP expression in primary (p)Fib, immortalized (i)Fib, pMSC, and iMSC was examined by flow cytometry. (B) mRNA expression of iFib, iMSC, CCL19-expressing iFib (iFib/CCL19), and CCL19-expressing iMSC (iMSC/CCL19) was examined by RT-PCR. β-Actin was used as a control. (C) The levels of CCL19 in the supernatants were determined by ELISA. Data are presented as the mean±SEM. **P<0.01 by Student’s t-test. (D) CT26 colon carcinoma cells (5×105) were inoculated subcutaneously into BALB/c mice with or without pMSC or iMSC. Tumor size was measured every 4 days. Data are presented as the mean±SEM. CCL19, chemokine (C-C motif) ligand 19; iFib, immortalized fibroblasts; iMSC, immortalized MSC; MSC, mesenchymal stem/stromal cells; pFib, primary fibroblasts; pMSC, primary MSC; RT, reverse transcription.
CCL19-expressing MSC (iMSC/CCL19) and Fib (iFib/CCL19) were generated by retroviral transduction of the murine CCL19 gene. Transcription and secretary production of CCL19 were detected by RT-PCR (figure 1B) and ELISA (figure 1C), respectively. To determine whether MSC affect tumor growth in vivo, pMSC or iMSC were co-injected with CT26 cells into BALB/c mice. The results showed that MSC did not affect CT26 tumor growth (figure 1D).
Co-injection of iMSC/CCL19 suppresses CT26 tumor growth
To determine whether iFib/CCL19 or iMSC/CCL19 can suppress in vivo tumor growth, CT26 cells were co-injected into BALB/c mice with either of iFib, iFib/CCL19, iMSC, or iMSC/CCL19. The results showed that significant tumor suppression was observed in iMSC/CCL19 co-inoculated mice compared with CT26 alone mice and iMSC co-inoculated mice, whereas such significant difference was not observed in mice co-injected with iFib, iFib/CCL19, or iMSC compared with those that received CT26 alone (figure 2A,B). To examine whether CCL19 affect on cancer cell survival, CT26 cells were cultured with recombinant CCL19 (rCCL19). We observed no effect of CT26 cell viability in various doses of rCCL19 (online supplementary figure S3a), and CCR7 was not expressed on CT26 cells with or without IFN-γ stimulation (online supplementary figure S3b). Therefore, it is suggested that the observed cancer growth suppression is not due to humoral effect of CCL19/CCR7 signaling between iMSC and CT26 but may be caused by the intervention of another lineage of cell. Although iFib/CCL19 and iMSC/CCL19 produce CCL19 at similar levels in vitro (figure 1C), CT26 co-injection with iFib/CCL19 failed to suppress tumor growth in vivo. To investigate the survival duration of iFib/CCL19 and iMSC/CCL19 at the tumor sites, DNA from the tumor tissues was isolated on days 8, 12, 16, and 20 after co-inoculation, and genomic PCR was performed to confirm the in vivo survival of iFib/CCL19 and iMSC/CCL19. The results showed that iMSC/CCL19 survived at the tumor sites for at least 20 days post-inoculation, whereas iFib/CCL19 decreased by day 12 and nearly disappeared by day 16 (figure 2C). These results suggest that continuous production of CCL19 in the tumor sites is essential for the tumor growth suppression in vivo.
Supplemental material

Locally injected iMSC/CCL19 survive in tumor sites longer than iFib/CCL19, and lead to tumor suppression. (A) BALB/c mice were inoculated s.c. into the right flank with CT26 (5×105 cells) with or without iFib, iFib/CCL19, iMSC, or iMSC/CCL19 (1×105 cells). Tumor size was measured every 4 days. (B) Left, mean tumor size. Right, mean tumor size on day 20. **P<0.01 by two-tailed t-test. (C) BALB/c mice were injected s.c. with CT26 with or without iFib/CCL19 or iMSC/CCL19. On the indicated days, mice were sacrificed, and the DNA of the tumor tissues was collected. Genomic PCR with GFP gene primer sets was used to detect iFib/CCL19 and iMSC/CCL19. β-Actin was used as a control. CCL19, chemokine (C-C motif) ligand 19; iFib, immortalized fibroblasts; iMSC, immortalized MSC; MSC, mesenchymal stem/stromal cells; s.c., subcutaneously.
Local injection of iMSC/CCL19 suppresses tumor growth
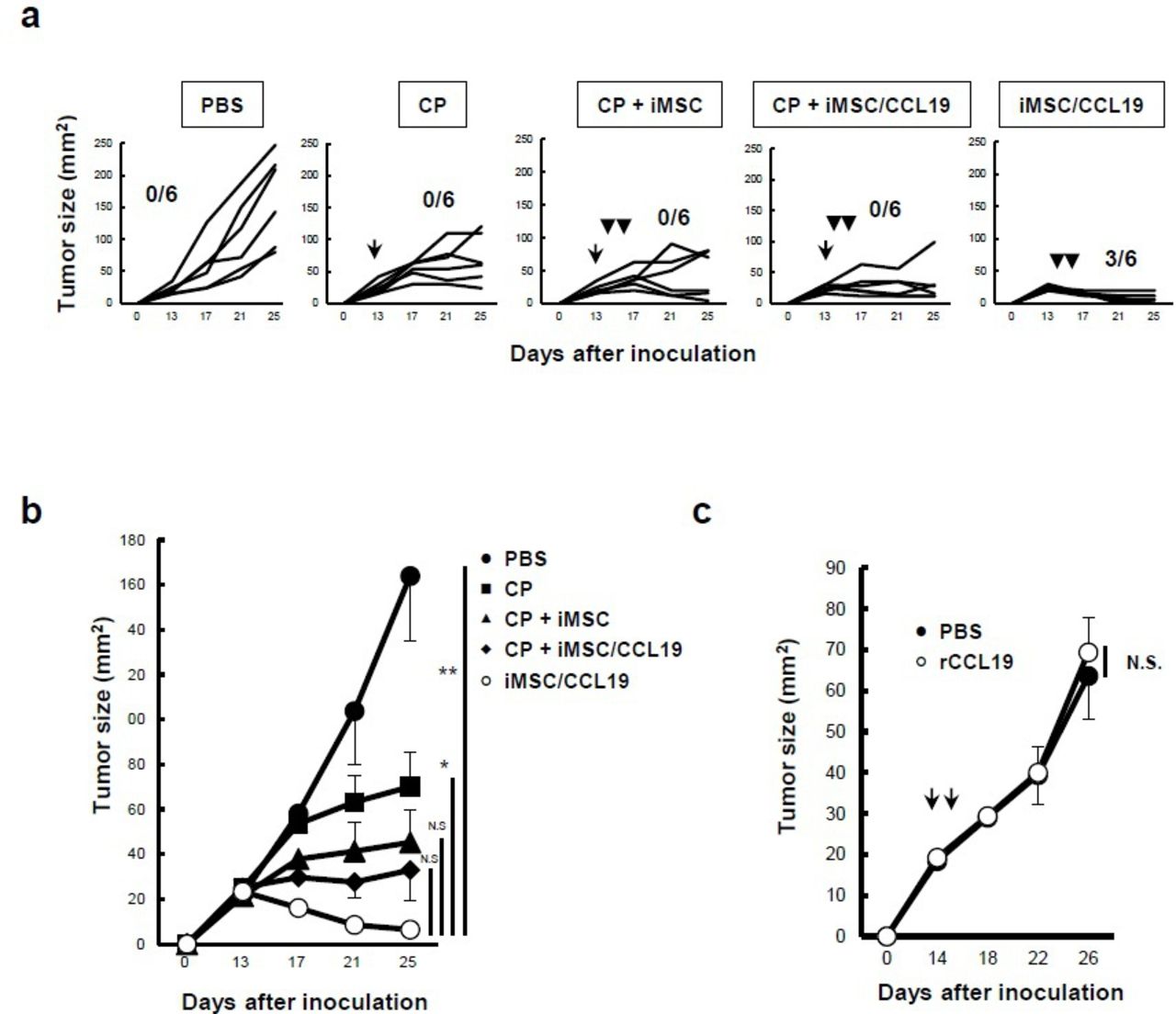
To assess the utility of iMSC/CCL19 in anticancer therapy, iMSC/CCL19 were injected into established tumor sites. Since cyclophosphamide (CP), an alkylating agent that binds to DNA strands, has immunomodulating abilities, including the induction of immunogenic cancer cell death and mitigation of immunosuppression by regulatory T cells (Treg),25 we administered CP with or without a local injection of iMSC/CCL19. Although treatment with CP, CP +iMSC, or CP +iMSC/CCL19 suppressed tumor growth, iMSC/CCL19 injection alone significantly suppressed the tumor growth and cured three out of six mice (figure 3A,B). When the cured mice were re-challenged with CT26 cells 2 months after the first inoculation, all cured mice rejected the re-challenged CT26 cells (online supplementary figure S4). In contrast, local injection of recombinant murine CCL19 into the tumor sites showed no effect on tumor growth (figure 3C) probably due to short half-life of CCL19.26 These results suggest that cell-mediated continuous CCL19 production by MSC-recruited immune cells is important for tumor suppression.
Supplemental material

Local injection of iMSC/CCL19 alone can regress tumor growth. (A) BALB/c mice were inoculated s.c. into the right flank with CT26 (5×105 cells). Thereafter, 100 mg/kg CP was administered by i.p. injection on day 13, and iMSC or iMSC/CCL19 (5×105 cells) were used for i.t. injection on days 14 and 16. Tumor size was measured every 4 days. Numbers of cured mice are shown. The mean tumor size is indicated in (B). Mean±SEM, *P<0.05. **P<0.01 by two-tailed t-test. (C) On days 14 and 16 after CT26 inoculation, i.t. injection with PBS or 0.2 µg recombinant murine CCL19 at a volume of 100 µL. The tumor size was measured every 4 days and the mean±SEM was indicated. N.S., not significant. n=5. CCL19, chemokine (C-C motif) ligand 19; CP, cyclophosphamide; iMSC, immortalized MSC; i.p., intraperitoneal; i.t., intratumoral; MSC, mesenchymal stem/stromal cells; s.c., subcutaneously.
Antitumor effect triggered by local injection of iMSC/CCL19 is T cell-dependent
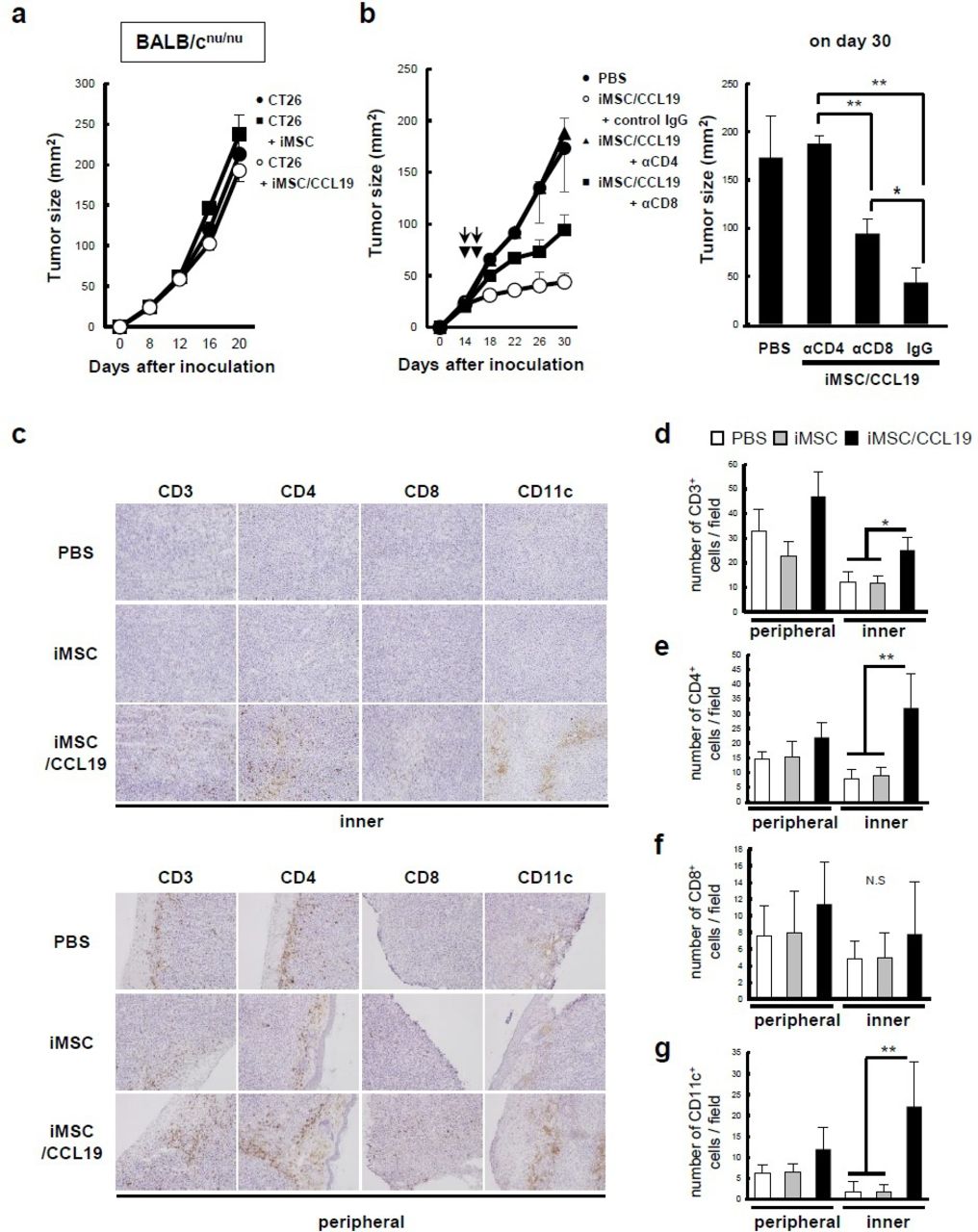
To test whether the antitumor effect is dependent on T cells, CT26 cells were co-inoculated with iMSC or iMSC/CCL19 into BALB/c nude mice. No antitumor effect of iMSC/CCL19 was observed in BALB/c nude mice (figure 4A). In addition, the antitumor effect of iMSC/CCL19 local therapy was completely or partially negated by the in vivo depletion of CD4+ or CD8+ cells, respectively (figure 4B). On the other hand, depletion of CD4+ or CD8+ cells in control-treated and iMSC-injected mice promoted the tumor growth slightly, but not significantly, in both groups (online supplementary figure S5). These results suggest that CD4+ and CD8+ cells both contribute to the antitumor effect induced by local injection with iMSC/CCL19. Next, to identify the types of infiltrating immune cells in the tumor site after local therapy with iMSC/CCL19, tumor cryosections were subjected to immunohistochemical analysis on day 1 after local therapy. The results were that infiltration of CD3+, CD4+, and CD11c+ cells, but not of CD8+ cells, into the inner regions of tumor was significantly increased by local injection of iMSC/CCL19 (figure 4C–G). On the other hand, no significant difference was observed at the peripheral regions of tumor. In addition, there was no apparent difference in infiltration of CD11b+, F4/80+, Gr-1+, and CD19+ cells among three groups (online supplementary figure S6).
Supplemental material
Supplemental material

Antitumor effect induced by iMSC/CCL19 local therapy is T cell-dependent. (A) BALB/c nu/nu mice were inoculated s.c. with CT26 (5×105 cells) with or without iMSC or iMSC/CCL19. Thereafter, tumor size was measured every 4 days. n=5, respectively. (B) CT26 tumor-bearing mice received i.t. injection with iMSC/CCL19 (arrowheads) and i.p. injection with 100 µg of control IgG, anti-CD4 (depletion) or anti-CD8 (depletion) antibody (arrows). Tumor size on subsequent days after CT26 inoculation (left), the mean tumor size on day 30 (right). **P<0.01. *P<0.05 by Tukey-Kramer test (ANOVA). n=5, respectively. (C) iMSC or iMSC/CCL19 were injected i.t. on days 14 and 16 after tumor inoculation. Cryosections of the tumor were prepared 17 days after tumor inoculation and stained with anti-CD3, anti-CD4, anti-CD8, and anti-CD11c antibodies. Representative images of inner (upper) and peripheral (lower) regions of tumor were shown. (D–G) Number of CD3, CD4, CD8, and CD11c-positive cells were shown. **P<0.01. *P<0.05 by Tukey-Kramer test (ANOVA). N.S., not significant. CCL19, chemokine (C-C motif) ligand 19; iMSC, immortalized MSC; i.p., intraperitoneal; i.t., intratumoral; MSC, mesenchymal stem/stromal cells; s.c., subcutaneously.
iMSC/CCL19 local therapy attracts F4/80− CD11c+ CCR7+ DC and increases IFN-γ+ CD8+ T cells
We used flow cytometry to examine the infiltrating immune cells and their expression of CCR7, a receptor for CCL19, after iMSC/CCL19 therapy. On day 1 after local injection with iMSC/CCL19, CCR7+ cells were increased among CD45+ cells (online supplementary figure S7a). After gating CD45+ cells from tumor tissues, we found that local injection of iMSC/CCL19 increased the percentage of CCR7+ cells in F4/80− CD11c+ DC compared with injections with PBS or iMSC (figure 5A). Interestingly, local injection of iMSC/CCL19 increased the percentage of CD45+ F4/80CD4+ CD11c+ cells at tumor sites (figure 5B), but depletion of CD4+ cells in the iMSC/CCL19 therapy group significantly decreased the percentage of CD45+ F4/80− CD11c+ cells compared with control IgG (figure 5C). Although infiltration of CD8+ cells was not increased (figure 4F), local injection of iMSC/CCL19 increased the proportion of IFN-γ+ CD8+ T cells at the tumor sites compared with PBS control, but this increase was reversed by depletion of CD4+ cells (figure 5D and online supplementary figure S7d). In contrast, no difference was observed in the percentages of IFN-γ+ CD4+ T cells and TNFα+ CD4+ T cells (online supplementary figure S8). In addition, no difference was observed in the percentages of CD3+ CD4+ CCR7+, CD3+ CD8+ CCR7+, and CD3+ CD4+ FoxP3+ cells (online supplementary figure S7b, c and f).
Supplemental material
Supplemental material
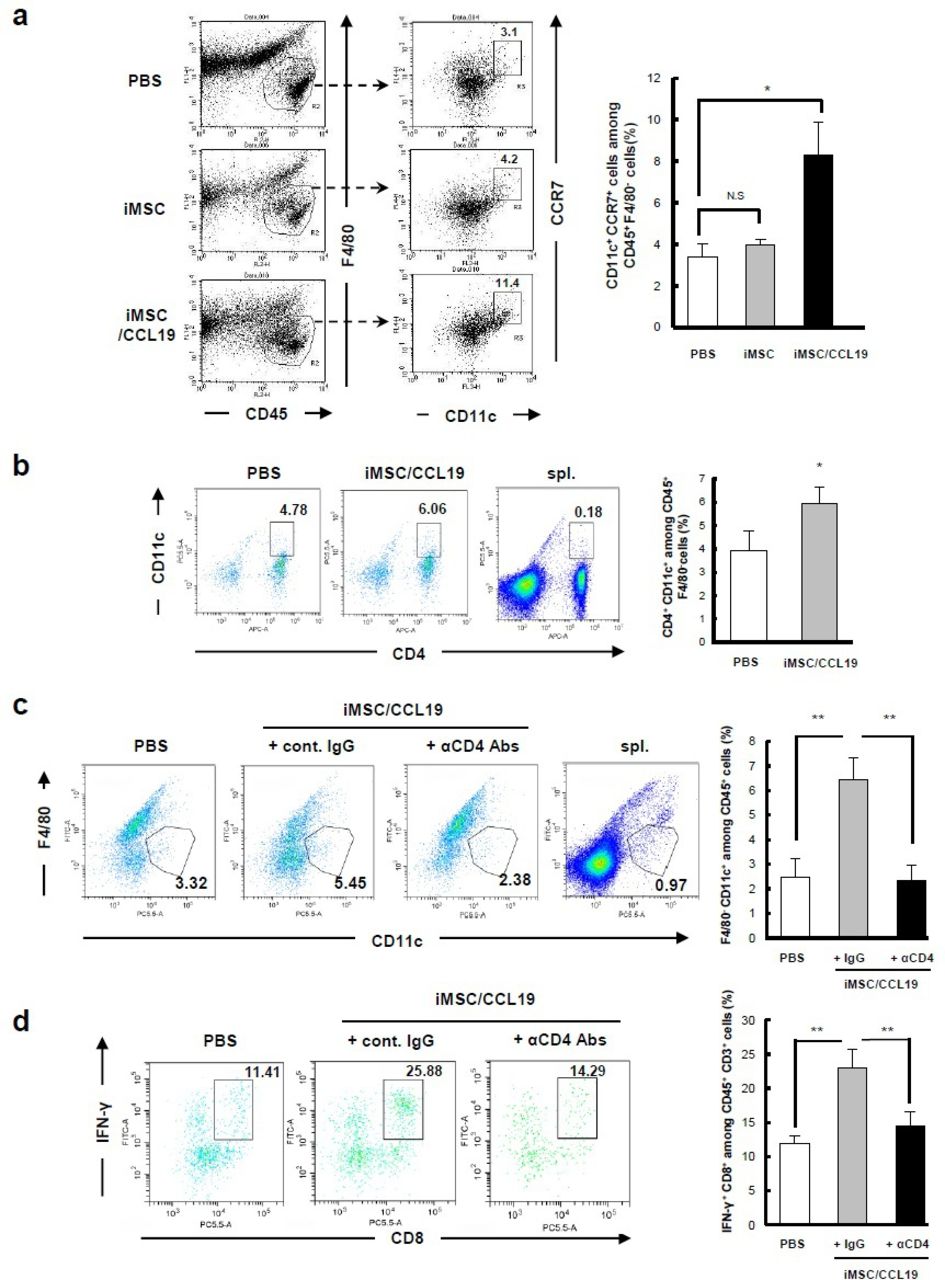
iMSC/CCL19 local therapy attracts F4/80− CD11c+ CCR7+ DC and increases IFN-γ+ CD8+ T cells iMSC or iMSC/CCL19 were injected i.t. on days 14 and 16 after tumor inoculation. Tumor cells were collected 17 days after tumor inoculation, and tumor-infiltrating immune cells were analyzed by flow cytometry. (A) Representative plots (left) and the percentages of indicated CD11c+ CCR7+ among CD45+ F4/80cells are shown (right). *P<0.05 by Tukey-Kramer test (ANOVA). N.S., not significant. (B) Representative plots (left) and the percentages of CD4+ CD11c+ cells among CD45+ F4/80cells are shown (right). (C,D) CT26 tumor-bearing mice were received i.t. injection with iMSC/CCL19 (arrowheads) and i.p. injection with 100 µg of control IgG or anti-CD4 (depletion) antibody as described in figure 4b. (C) Represent plots (left) and the percentages of CD11c+ F4/80 cells gated on CD45+ cells were shown. (D) The percentages of IFN-γ+ CD8+ T cells among CD3+ T cells are shown (represent plots: left, bar graph; right). **P<0.01 by Tukey-Kramer test (ANOVA). n=3. Splenocytes collected from tumor-bearing mice were analyzed as a control. CCL19, chemokine (C-C motif) ligand 19; IFN, interferon; IgG, immunoglobulin G; iMSC, immortalized MSC; i.p., intraperitoneal; i.t., intratumoral; MSC, mesenchymal stem/stromal cells.
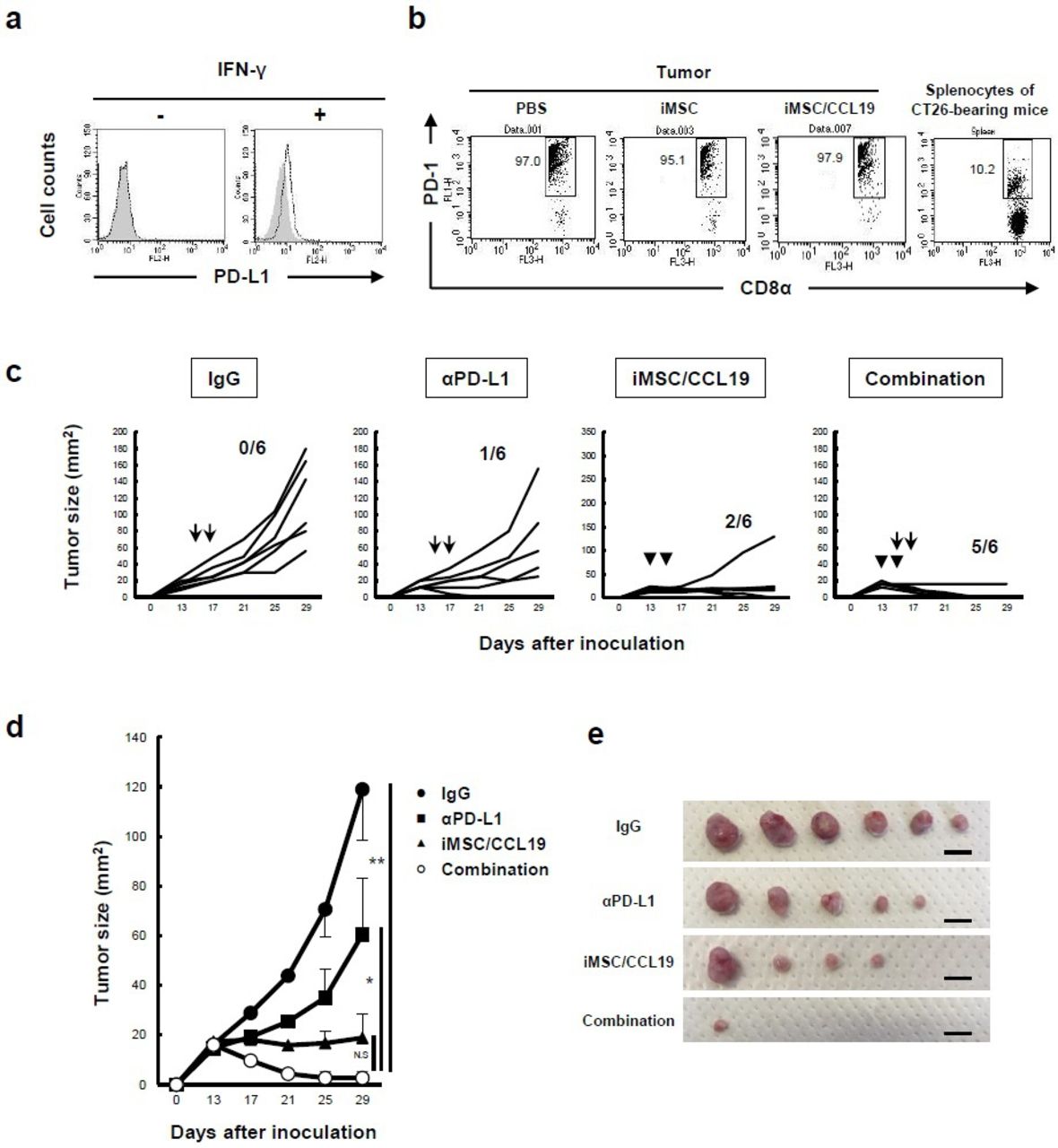
Local injection of iMSC/CCL19 augments the antitumor effect by anti-PD-L1 blockade therapy
CT26 cells expressed PD-L1 when treated with IFN-γ in vitro (figure 6A) and >90% of CD8α+ T cells in tumor tissues expressed PD-1 (figure 6B). The positive percentage of PD-1 on CD4+ T cells was about 4% with or without iMSC/CCL19 treatment (online supplementary figure S7e). Therefore, we next examined the antitumor effects of iMSC/CCL19 local therapy combined with anti-PD-L1 or anti-PD-1 blockade antibody therapy. Although anti-PD-L1 therapy or local therapy with iMSC/CCL19 alone suppressed tumor growth, only one and two out of six mice were cured, respectively (figure 6C). However, the combination of iMSC/CCL19 and anti-PD-L1 blockade therapy suppressed tumor growth more effectively, curing five out of six mice (figure 6C–E). In contrast, the combination of iMSC/CCL19 and anti-PD-1 blockade failed to enhance the antitumor effect induced by iMSC/CCL19 local therapy (online supplementary figure S9).
Supplemental material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Local injection of iMSC/CCL19 augments the antitumor effect by anti-PD-L1 blockade therapy. (A) PD-L1 expression on CT26 cells that were cultured in the presence or absence of recombinant 50 ng/mL IFN-γ for 48 hours. Gray: isotype control, black dotted: PE anti-PD-L1 antibody. (B) CT26-bearing mice were i.t. injected with iMSC or iMSC/CCL19 on days 14 and 16; on day 17 after tumor inoculation, TILs were collected and stained with anti-CD3, anti-CD8α, and anti-PD-1 antibodies. Splenic cells from CT26-bearing mice were analyzed as control cells. The numbers represent percentages of PD-1+ CD8+ cells among CD3+ CD8+ T cells. (C) CT26-bearing mice were i.t. injected with iMSC/CCL19 on days 13 and 15 after tumor inoculation (arrowhead). Control IgG (200 µg) or anti-PD-L1 (200 µg) antibody were i.p. injected on days 15 and 17 (arrow). The tumor growth in individual mice is shown. (D) The mean (±SEM) tumor size. *P<0.05. **P<0.01 by two-tailed t-test. N.S., not significant. (E) Photograph of tumor tissues is shown. Scale bar=10 mm. CCL19, chemokine (C-C motif) ligand 19; IFN, interferon; iMSC, immortalized MSC; i.t., intratumoral; MSC, mesenchymal stem/stromal cells; TILs, tumor-infiltrating lymphocytes.
Discussion
Human MSC promote the development/progression of cancer cells via several mechanisms: immunosuppression by indoleamine 2,3-dioxygenase, interleukin (IL)-10, and nitric oxide,27 promotion of metastasis,28 and inhibition of apoptosis of cancer cells by production of tumor growth factor-β29 in the tumor microenvironment. Alternatively, MSC suppress tumor growth by promoting recruitment of inflammatory cells,30 inhibiting neovascularization,31 and inducing apoptosis of cancer cells.32 The varying results reported with human MSC could be due to the purification methods employed and the tissue source. However, few studies have investigated the effects of murine MSC on tumor growth.33
In this study, we found that freshly purified and immortalized MSC showed no effects on the in vivo growth of syngeneic tumor cells (figure 1D). As it is difficult to examine how human MSC affect in vivo growth of autologous cancer cells in vivo, our investigation involving murine MSC and a syngeneic therapy model may be informative.
Antitumor effects induced by local therapy with iMSC/CCL19 were not observed in nude mice (figure 4A) and canceled by in vivo depletion of CD4+ cells or CD8+ cells (figure 4B). Notably, in vivo depletion of CD4+ cells completely abrogated the antitumor effects of the iMSC/CCL19 local therapy. Since CT26 cells were negative for MHC class II molecules (data not shown), CD4+ T cells could not kill CT26 cells directly. Rather, we suppose the following mechanisms. The administration of anti-CD4 antibody depleted CD4+ T cells as well as CD4+ CD11c+ cells. Given that CD4+ T cells are essential for maturation of DC, the depletion of CD4+ T cells resulted in impaired maturation of DC, leading to insufficient cross-priming of tumor-specific CD8+ T cells. Indeed, the numbers of tumor-infiltrating IFN-γ+ CD8+ T cells were increased in the tumors of mice that received the iMSC/CCL19 local therapy (figure 5C), whereas such IFN-γ+ CD8+ T cells were decreased when CD4+ cells were depleted (figure 5D). In contrast, this increase was not observed in tumor-infiltrating IFN-γ+ CD4+ T cells and TNF-α+ CD4+ T cells (online supplementary figure S8). On the other hand, the administration of anti-CD4 antibody decreased the percentages of CD45+ F4/80- CD11c+ cells at tumor sites (figure 5C). On the basis of these findings, we assume another possibility that CD45+ F4/80CD4+ CD11c+ cells might participate directly or indirectly in the antitumor effects of the iMSC/CCL19 local therapy. Interestingly, Kim and colleagues have previously reported that a part of CD3− CD4+ accessory cells express OX40L and CD30L and suggested that those cells activate T cells.34 35 CD4+ cells may also play a role in antitumor effect of the iMSC/CCL19 local therapy.
iMSC/CCL19 local therapy increased the percentages of CCR7+ DC in tumor tissues (figure 5A). Mature DC express CCR7, and this receptor plays a role in recruiting DC to secondary lymphoid tissues.36 Immune responses are suppressed in the tumor microenvironment, for example, downregulation of CD80/86 by Treg and decreased expression of MHC class II by IL-10.37 As we observed no increase in IFN-γ+ or TNF-α+ CD4+ T cells in the tumor sites, DC (F4/80- CD11c+ CCR7+) may activate CD8+ T cells in draining lymph nodes, resulting in the increased IFN-γ+ CD8+ T cells in the tumor sites.
We examined the antitumor effects induced by the combination of iMSC/CCL19 local therapy with either anti-PD1 or anti-PD-L1 antibody therapy. Our result showed that anti-PD-L1 antibody therapy, but not anti-PD-1, augmented the antitumor effects induced by iMSC/CCL19 local therapy (figure 6 and online supplementary figure S9). As for the reason why the combination of PD-L1 mAb and iMSC/CCL19 was superior to that of PD-1 mAb and iMSC/CCL19, we could propose the following possibilities. First, given that cis-PD-L1 interacts with CD80 on antigen-presenting cells and inhibits them from providing a co-stimulatory signal to T cells,38 anti-PD-L1 antibody might inhibit this interaction and restore the potential of co-stimulatory molecules. Second, given that IgG subclass was different between the two antibodies and that T cells express PD-1 on their cell surface, anti-PD-1 antibody (rat IgG2a) could show higher complement-dependent cytotoxicity than anti-PD-L1 antibody (rat IgG2b). Further studies are still needed to elucidate the precise mechanism.
CP has several immunomodulatory capacities, such as the relief of Treg cells and induction of immunogenic cancer cell death.25 39 40 We previously reported that CP augments antitumor effects in mice that were treated with anti-CTLA-4 antibody.41 However, the antitumor effects were more apparent in the iMSC/CCL19 local therapy alone than with the combination of CP and iMSC/CCL19 local therapy (figure 3). In this study, no synergistic effect was observed when combined with CP, possibly because administration of CP might have damaged the iMSC/CCL19.
This study investigated the antitumor effects induced by iMSC/CCL19 local therapy. This therapy can effectively recruit T cells and DC into tumor sites via continuous production of CCL19 at the tumor microenvironment. iMSC/CCL19 local therapy augmented the antitumor effects of anti-PD-L1 antibody therapy. In this study, we demonstrated the therapeutic efficacy of iMSC/CCL19 by local injection, whereas local injection has limitation in terms of clinical application. However, other routes of administration, for example, arterial injection, might be useful for therapeutic purposes, as reported by Uchibori and colleagues.6 Given that antitumor effects by anti-PD or anti-PD-L1 antibody therapy essentially require the presence of antitumor T cells in the tumor sites, local therapy with iMSC/CCL19 is therapeutically meaningful. However, the practicality of this strategy is limited, as these cells must be administered locally. Nevertheless, our findings indicate that local manipulation is an efficient strategy to elicit antitumor immunity in tumor-bearing hosts.
Acknowledgments
We thank Ms Hiromi Miyauchi for technical assistance, and Dr Tamio Okimoto and Dr Ryosuke Tanino for fruitful discussion.
References
Footnotes
Twitter @pentadou
Contributors Conceived and designed the experiments: YI and MH. Performed the experiments: YI, RY, and AM. Analyzed the data: YI and MH. Contributed reagents/materials/analysis tools: YI, RY, HK, YK, MO, and YM. Wrote the paper: YI and MH.
Funding This study was supported in part by JSPS KAKENHI Grant (no. JP19K16713 to Y.Iida), Shimane University “WAKATE” supporting project and “SUIGAN” project.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All experimental procedures were approved by the Animal Center of Shimane University.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.