Article Text

Abstract
Background Checkpoint inhibitors can induce profound anticancer responses, but programmed cell death protein-1 (PD-1) inhibition monotherapy has shown minimal activity in prostate cancer. A published report showed that men with prostate cancer who were resistant to the second-generation androgen receptor inhibitor enzalutamide had increased programmed death-ligand 1 (PD-L1) expression on circulating antigen-presenting cells. We hypothesized that the addition of PD-1 inhibition in these patients could induce a meaningful cancer response.
Methods We evaluated enzalutamide plus the PD-1 inhibitor pembrolizumab in a single-arm phase II study of 28 men with metastatic castration-resistant prostate cancer (mprogressing on enzalutamide alone. Pembrolizumab 200 mg intravenous was given every 3 weeks for four doses with enzalutamide. The primary endpoint was prostate-specific antigen (PSA) decline of ≥50%. Secondary endpoints were objective response, PSA progression-free survival (PFS), time to subsequent treatment, and time to death. Baseline tumor biopsies were obtained when feasible, and samples were sequenced and evaluated for the expression of PD-L1, microsatellite instability (MSI), mutational and neoepitope burdens.
Results Five (18%) of 28 patients had a PSA decline of ≥50%. Three (25%) of 12 patients with measurable disease at baseline achieved an objective response. Of the five responders, two continue with PSA and radiographic response after 39.3 and 37.8 months. For the entire cohort, median follow-up was 37 months, and median PSA PFS time was 3.8 months (95% CI: 2.8 to 9.9 months). Time to subsequent treatment was 7.21 months (95% CI: 5.1 to 11.1 months). Median overall survival for all patients was 21.9 months (95% CI: 14.7 to 28 .4 months), versus 41.7 months (95% CI: 22.16 to not reached (NR)) in the responders. Of the three responders with baseline biopsies, one had MSI high disease with mutations consistent with DNA-repair defects. None had detectable PD-L1 expression.
Conclusions Pembrolizumab has activity in mCRPC when added to enzalutamide. Responses were deep and durable and did not require tumor PD-L1 expression or DNA-repair defects.
Trial registration number clinicaltrials.gov (NCT02312557).
- biomarkers
- tumor
- drug therapy
- combination
- immunotherapy
- lymphocytes
- tumor-infiltrating
- prostatic neoplasms
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Despite advancements in the management of metastatic castration-resistant prostate cancer (mCRPC), prostate cancer remains the second most common cause of cancer death in US men.1 Current therapies that extend survival for mCRPC include second-generation androgen receptor antagonists (enzalutamide), inhibitors of androgen synthesis (abiraterone), chemotherapy (docetaxel, cabazitaxel), a cellular vaccine (sipuleucel-T) and a radiopharmaceutical (radium-223).2–9 There are few data to guide sequencing of these agents and no data supporting routine use of combination therapies. Management of mCRPC after enzalutamide and/or abiraterone is a particularly challenging clinical problem.
To date, immune checkpoint inhibitors used as monotherapy in mCRPC have shown mixed activity. Two phase III studies of the anti-Cytotoxic T Lymphocyte-Associated Protein-4 (CTLA-4) antibody ipilimumab failed to meet their primary endpoint of improved overall survival, and early studies of programmed cell death protein-1 (PD-1) inhibitors showed no objective radiographic responses in men with mCRPC.10–12 Two recent studies examined single-agent pembrolizumab for programmed death ligand 1 (PD-L1) positive and PD-L1 negative mCRPC. One enrolled only PD-L1 positive mCRPC (n=23), most of which had received prior docetaxel (91%), and a 17.4% durable response rate was observed.13 In the second study, all patients had received docetaxel, but there was no clear difference in response rate between those with PD-L1 positive (n=131) staining versus negative staining (n=67).14 Combinations of PD-1 inhibition with CTLA-4 and the poly-ADP ribose polymerase (PARP) inhibitor olaparib have also demonstrated activity.15
Studies showed that PD-L1, a ligand for PD-1, was upregulated on dendritic cells in men with mCRPC either progressing on or refractory to enzalutamide.16 Primary prostate cancer tumors are considered ‘cold tumors’ poorly infiltrated with T cells, and there is a general lack of PD-L1 expression on prostate cancer cells.17 However, a recent analysis revealed that one-third of mCRPC biopsies exhibit PD-1 staining18 suggesting that strategies to enhance CD8+ T cell function and decrease inhibitory signals in the tumor may underlie the success of immunotherapies for prostate cancer. Since enzalutamide is associated with increased expression of PD-L116 and enzalutamide augments castration, we hypothesized that enzalutamide would be a strong partner to PD-1 inhibitors. Additionally, we previously reported a profound response to immunotherapy (anti-CTLA-4) in a man with mCRPC on enzalutamide with biochemical progression.19
Here we present the results of a phase II study examining the antitumor effect of pembrolizumab added to enzalutamide in men with mCRPC whose cancer is progressing on enzalutamide alone.
Methods
Study design and conduct
All human investigations were carried out after approval by a local Human Investigations Committee and in accord with an assurance filed with and approved by the Department of Health and Human Services. The data have been anonymized to protect the privacy of the participants. Investigators obtained informed consent from each participant.
The detailed clinical trial design has been described elsewhere.20 In brief, men with mCRPC progressing on enzalutamide by prostate-specific antigen (PSA) and/or imaging studies continued enzalutamide and received pembrolizumab 200 mg intravenously every 3 weeks for four doses. Subjects could have previously received sipuleucel-T and could have received abiraterone or chemotherapy for castration-sensitive disease, but previous treatment with PD-1, PD-L1 or CTLA-4 antibodies for any disease state was prohibited. Furthermore, subjects with a history of autoimmune disease or pneumonitis were excluded.
Assessments
During treatment, subjects were followed every cycle for efficacy and toxicity. After the active treatment phase, subjects were followed every 6 weeks and were imaged with a Technicium99 bone scintigraphy and computed tomography8 scan of the chest, abdomen, and pelvis every 12 weeks. PSA was measured at each cycle during the treatment phase and then every 6 weeks. Any PSA progression or radiographic progression during the first 12 weeks required confirmation after week 12 on study. All adverse events were recorded from the time the consent form was signed through 30 days following cessation of treatment. This follow-up schedule continued until disease progression. Following progression, patients were followed for survival by telephone contact every 3 months.
Endpoints
The primary endpoint of the study was a confirmed serum PSA decrease ≥50% at any time on treatment. Secondary endpoints were PSA progression-free survival (PFS), objective response, radiographic PFS per RECIST V.1.1, time to next treatment and overall survival. Exploratory endpoints were the fraction of PD-L1 expressing tumor cells and the number of genetic mutations per biopsy, including testing for microsatellite instability (MSI) and mutations associated with DNA-repair defects.
All baseline tumor biopsies underwent analysis by whole exome sequencing (WES) for samples containing ≥10% tumor or by genotyping using the Knight Diagnostic Laboratory’s GeneTrails Solid Tumor Panel of 124 genes frequently altered in cancer, which assessed single nucleotide variations, multinucleotide variations, indels, and copy number variations if the sample contained <10% tumor.21 Two patients were tested using the Personal Genome Diagnostics panel (Baltimore, Maryland, USA), which tested 203 genes for the same features as the GeneTrails test.22 All responders underwent WES regardless of tumor content. MSI was assessed by PCR of BAT-25, BAT-26, NR-21, NR-24, MONO-27 for all patients, with orthogonal MSI analysis of almost 3000 different markers using microsatellite instability detection by next generation sequencing (mSINGS).23 DNA-repair genes examined included well-described markers (BRCA2, ATM, FANCA, CHEK2, BRCA1, PALB2, HDAC2, RAD51, MLH3, ERCC3, MRE11, NBN) as well as markers that could potentially have clinical relevance (BARD1, BRIP1, CDK12, RAD51B, RAD51C, RAD51D, RAD54L).
WES reads were aligned against the GrCH37d5 genome using the Sanger cppmap workflow (https://github.com/cancerit/dockstore-cgpmap) with realignment around indels and base recalibration performed using the Open Genomics GATK cocleaning workflow (https://github.com/OpenGenomics/gatk-cocleaning-tool). The Mbp of genome covered by WES was determined using bedtools genomecov (V.2.26.0), where any base pair covered by at least six aligned reads was considered covered,24 human leukocyte antigen (HLA) typing was performed from WES reads using Optitype (V.1.3.1).25 Somatic variants were called using a collection of callers via the mc3 workflow26 (https://github.com/opengenomics/mc3), retaining all variants produced by Pindel and all variants reported by two or more tools that were not overlapped by a Pindel variant. Germline variants were called using GATK HaplotypeCaller (V.3.7-0-gcfedb6),27 using VariantFiltration with cluster size 3 and cluster window size 15 to flag and subsequently eliminate variants with total coverage of <10.0 reads, quality by read depth of <2.0, and Phred quality of <100.0. HapCUT2 was used for patient-specific haplotype phasing28 by first combining germline and consensus somatic variants into a single variable call format (VCF) using neoepiscope’s merge functionality, then running HapCUT2’s extractHAIRS, allowing for extraction of reads spanning indels, to produce the fragment file used with HapCUT2 to predict haplotypes. Neoepitopes for each patient were predicted using neoepiscope29 (V.0.3.5), enumerating peptides of 8–11 in length from phased variants on protein coding, nonsense-mediated decay, polymorphic pseudogene, T cell receptor variable, and immunoglobulin variable transcripts. Binding predictions for neoepitopes were performed using netMHCpan (V.4.0). Only peptides which bound at least one of a patient’s major histocompatibility complex (MHC) Class I alleles predicted by Optitype were retained.
Formalin-fixed, paraffin-embedded tumor samples were sent to a commercial vendor (Qualtek) for PD-L1 quantification by immunohistochemistry (clone 22C3). Additional methods details are described elsewhere.30
Levels of serum cytokines and chemokines pretreatment and post-treatment were measured in triplicate using a Cytokine/Chemokine/Growth Factor 45-Plex Human ProcartaPlex Panel 1 kit (ThermoFisher Scientific, Waltham, Massachusetts, USA) and Luminex 200 (Austin, Texas, USA). Among the list of 45 unique analytes, the following 11 analytes were detectable: MIP-1 alpha/CCL3, LIF, IP-10/CXCL10, IL-7; Eotaxin/CCL11, PIGF-1, IL-1RA, RANTES/CCL5, HGF, BDNF, and PDGF-BB.
Whole blood immune profiling assays were conducted as previously described.31 Briefly, heparinized whole blood was stained with a cocktail of antibodies to identify T cell subsets as well as monocytic myeloid-derived suppressor cells (MDSCs). Stained cells were then incubated with BD fluorescence-activated cell sorting (FACS) lysing solution (BD Biosciences, Franklin Lakes, New Jersey, USA) followed by washes. For intracellular antigens (eg, Ki-67, granzyme B, and perforin), cells were permeabilized with the Permeabilizing Solution 2 (BD Biosciences) followed by incubation with staining antibodies. Samples were then acquired with a BD LSRFortessa (BD Biosciences). Data were analyzed with FlowJo V.9 (FlowJo, LLC, Ashland, Oregon, USA). The levels of activation (CD38, HLA-DR), proliferation (Ki-67), and functional (granzyme B, perforin) markers were assessed on effector memory (CD45RA−CCR7−) CD4 and CD8 T cell subsets. Monocytic MDSCs were defined as lineage (CD3, CD7, CD19, and CD20) negative, CD15−CD11b+CD33+CD14+HLA-DRlow and the frequency of MDSCs within the lymphocyte plus monocyte gate was used to assess their relationship to treatment effect.
Power analysis and statistical methods
Patients with PSA progression on enzalutamide are unlikely to have a spontaneous PSA response without a change in therapy. We deemed that a 25% response rate (defined as a PSA decrease ≥50%) to pembrolizumab observed here would be worthy of further study. Using a null hypothesis of 5% and alternate hypothesis of 25%, 25 evaluable patients were needed with 90% power and a one-sided alpha of 0.05. To account for potential drop-out, we enrolled 28 subjects. Descriptive statistical analyses were conducted for all variables of interest. Kaplan-Meier method was used to estimate the distribution of overall survival and PFS. Exact binomial test was used to determine whether the response rate was significantly >5%. Statistical analysis for clinical endpoints were conducted in R V.3.4.3.
The following statistical analyses were done using Prism V.7 (GraphPad Software; La Jolla, California, USA). The effect of the treatment on serum CXCL10 levels and CD38, HLA-DR, and Ki-67 expression on CD4 and CD8 effector memory T cells were first determined by a repeated measures one-way analysis of variance test. Once statistical differences in means were confirmed, the difference between two timepoints was assessed by the paired Student’s t-test. A p value of <0.05 was considered significant. The difference between non-responders and responders in baseline perforin or granzyme B expression in CD8 effector memory T cells as well as a baseline frequency of MDSCs were evaluated using an unpaired Student’s t-test. Survival curves are depicted using a Kaplan-Meier method and the relationship between the baseline perforin, granzyme B, or MDSCs and PFS was determined by log-rank test.
Results
Patients
Twenty-eight men were enrolled at the Knight Cancer Institute, Oregon Health & Science University, from April 2015 to August 2016. The median age was 72 years (range 52–90, table 1). Seventeen patients underwent a biopsy of a metastatic tumor prior to treatment. The median number of treatments for mCRPC prior to study entry was 2. Median time on enzalutamide prior to study entry was 52 weeks (range 23–230). Twenty-three (82%) patients had a PSA response on enzalutamide prior to progression. Five patients received sipuleucel-T as standard therapy prior to study entry.
Baseline characteristics
Treatment
The median number of pembrolizumab doses was 4 (range 2–12), and patients with best response of stable disease and no evidence of radiographic or clinical progression were treated with a second course of pembrolizumab for progressive disease. None of the non-responders converted to responders with a second course of pembrolizumab.
Anti-tumor activity
Five (18%, 95% CI: 0.06 to 0.37) of 28 men met the primary endpoint of PSA decrease of 50% or greater from start of treatment. The response rate is significantly greater than 5%, our planned null hypothesis (p=0.01171, exact binomial test). Of the five PSA responders, three had measurable soft tissue disease. All three of these patients had a partial radiographic response (figure 1); the other two responders were not assessable via radiographic metrics such as RECIST because they had bone-only disease. One of the responders subsequently died of pancreatic cancer with an undetectable PSA, 22.5 months following enrollment. Another responder had PSA progression 36 months after cycle 1, but died of a cerebrovascular event 42.4 months. One patient developed PSA progression after 10 months, and subsequently received a second and third course of pembrolizumab without a PSA response. However, that patient did not require a new antineoplastic treatment for 26.8 months after enrollment. The remaining two responders continue to have a sustained PSA response after 39.3, and 37.7 months without any additional therapy. Of note, one of them has not continued enzalutamide therapy, but the other one has.
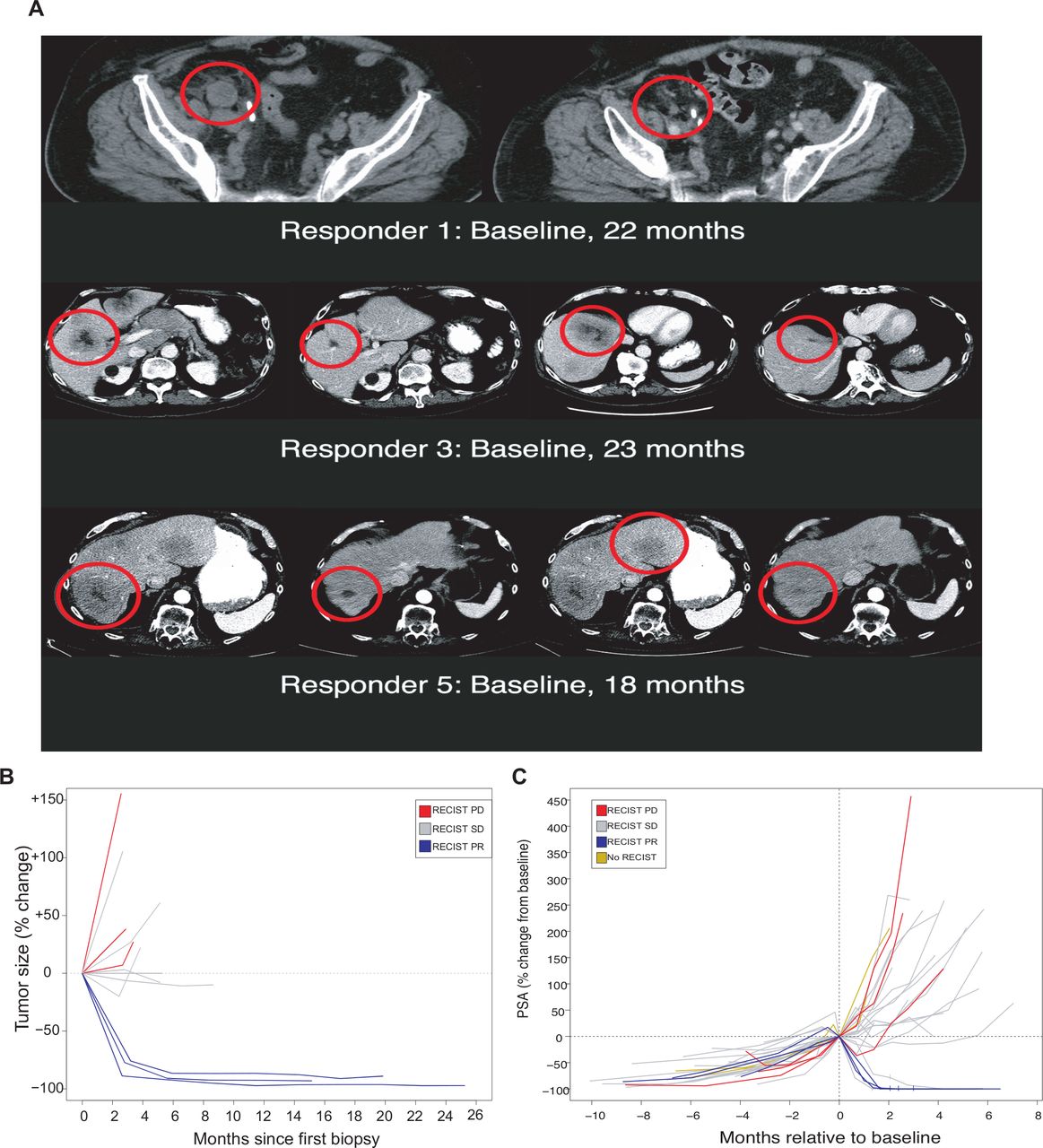
Radiographic and PSA changes. (A) Radiographs of the responders who had measurable disease. (B) Spider plot for changes in radiographic response for participants with measurable disease. (C) Changes in PSA for all participants. PSA, prostate-specific antigen; RECIST PD, RECIST progressive disease; RECIST SD, RECIST stable disease.
Twelve (43%) of the patients had RECIST measurable disease. Three patients achieved a partial response (25%), four had disease progression and five had stable disease. One of the three partial responders has not relapsed after 39.3 months. Another partial responder progressed by imaging after 36.4 months, while the third partial responder did not show radiographic progression prior to starting a new cancer treatment. However, RECIST is an incomplete metric of response in this cohort, as there were many patients with stable disease by RECIST criteria that exhibited clear PSA progression or durable PSA response (figure 1). Among patients with radiographically measurable disease, we note additional small discrepancies between overall RECIST scoring and biopsied lesion-specific measurements (figure 1). For instance, at least two patients with stable disease by RECIST criteria exhibited clear growth of biopsied lesions. These same patients also exhibited PSA progression, with 16%–20% increase over baseline by their last cycle of pembrolizumab therapy.
For the entire cohort, the median follow-up was 37 months, and the median PSA PFS time was 3.8 months (95% CI: 2.8 to 9.9 months). Median time to subsequent treatment was 7.2 months (95% CI: 5.1 to 11.1 months). Median overall survival was 22.2 months (95% CI: 14.7 to 28.4 months). For the five responders, median follow-up was 38 months and the duration of response to date is 21.9 to NR months. Two are still in partial response after 39.3 and 37.8 months of follow-up. Median PSA PFS time was 36.1 months (95% 9.9 to NR), and time to subsequent treatment was not achieved. The median overall survival for the responders was 41.7 months (95% CI: 22.2 to NR). For the 23 non-responders, median follow-up time was 36.5 months; median PSA PFS time was 3.1 months (95% CI: 2.8 to 4.4 months); median time to subsequent treatment was 5.2 months (95% CI: 3.8 to 8.5 months); and overall survival was 18.6 months (95% CI: 13.4 to 26.4 months). Twenty-two of 28 men have died, and all but three of those deaths were attributable to prostate cancer. PSA histories and response trajectories are distinct between responders and non-responders (figure 1). None of the five patients who had received sipuleucel-T responded to pembrolizumab
Exploratory endpoints
Seventeen (61%) of 28 participants had a baseline biopsy of a metastatic lesion. Three of the five responders had a baseline biopsy, and there were 14 biopsies among the non-responders, 13 of which had identifiable tumor. One of the responders exhibited substantial MSI (assessed by both PCR and mSINGS) with over 4000 mutations in his tumor (table 2). He also had mutations in several DNA-repair genes, with at least one known heterozygous deleterious (cancer-predisposing) variant in Ataxia-Telangiesctasia Mutated (ATM). The other two responders had no markers of MSI and 352 and 210 total mutations in their tumors, respectively. One non-responder had a single marker of MSI by PCR but was not deemed MSI high by mSINGS,23 and three non-responders had heterozygous mutations in DNA-repair pathways (two of which are thought to be cancer-associated). All 17 baseline biopsies were sent for PD-L1 staining, and none were found to have PD-L1 present on cancer cells. Gene expression profiling of total tumor confirmed PD-L1 expression was not different between responders and non-responders (data not shown). In one responder and two non-responders, PD-L1 was detected on tumor-infiltrating lymphocytes (TILs, table 2).
Characteristics among subjects with baseline biopsies (n=16)
To compare mutational burden and possible neoepitopes among patients with WES data, normalized values (ie, the number of somatic variants/genomic coverage) were compared between responders (n=3) and non-responders (n=6), with no statistically significant difference observed between groups (median 6.5 vs 4.9, respectively, p=0.42, online supplementary figure 1). Comparison of predicted neoepitope burden between groups, predicted from WES data, demonstrated a trend toward higher numbers of predicted neoepitopes among responders (679 vs 405.5, p=0.42, figure 2).
Supplemental material
Supplemental material

Distribution of mutational burden in responder’s versus non-responders. Boxplot showing the distributions of mutational burden (left) and neoepitope count (right) for responders versus non-responders. The center line in each box represents the median, with bottom and top boundaries of the box representing the first and third quartiles, respectively. Lines extending down and up from the box represent the minimum and maximum values, respectively. Non-responders are shown in light blue, and responders are shown in dark blue. There was no statistically significant difference between responders and non-responders in terms of mutational burden (median 6.5 vs 4.9 variants per megabase pair covered, p=0.42) or predicted neoepitope burden (679 vs 405.5 neoepitopes, p=0.42).
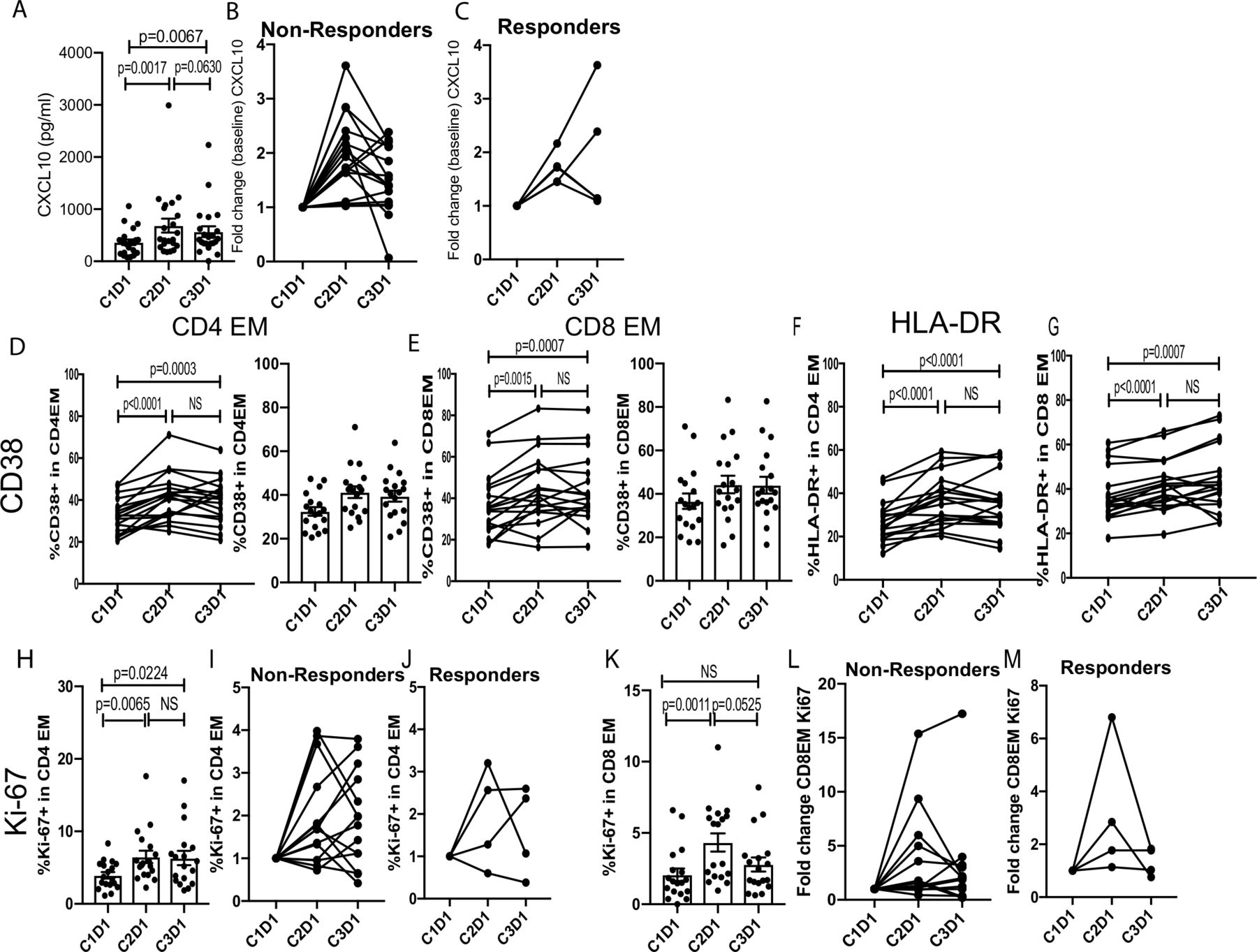
Markers of pharmacological activity and immunological activity to pembrolizumab/enzalutamide were measured by examining changes in serum cytokines and chemokines as well as circulating leukocytes pretreatment and post-treatment. We detected elevated CXCL10 levels following treatment (figure 3A), suggesting that this approach effectively induced a proinflammatory (type-1; Th1) immune response.32 There was no significant difference in CXCL10 levels between non-responders and responders at baseline or fold change over the course of treatment suggesting that CXCL10 is a pharmacodynamic marker rather than an immune correlate of clinical responsiveness. Next, we assessed the activation and proliferation of effector memory (CCR7−CD45RA−) CD4 and CD8 T cells by flow cytometry. We observed increased activation of effector memory CD4 and CD8 T cells as assessed by changes in CD38 and HLA-DR at the end of cycle 1 (C2D1; Cycle 2 Day 1). Moreover, these T cell subsets maintained a similar level of activation at the C3D1 timepoint (figure 3B, top two rows). We also detected increased proliferation of effector memory CD4 and CD8 T cells on treatment (figure 3B, bottom row). Interestingly, proliferation of effector memory CD4 T cells was sustained even at the C3D1 time point, whereas effector memory CD8 T cell proliferation returned to the pretreatment levels by this time. The extent of effector memory T cell activation and proliferation was similar between the non-responder and responder cohorts (data not shown). We also examined MIP-1alpha, RANTES, and Eotaxin expression, but did not find any significant changes in these cytokines/chemokines following treatment (online supplementary figure 1). The percent of B cells and natural killer cells also did not change (online supplementary figure 2).
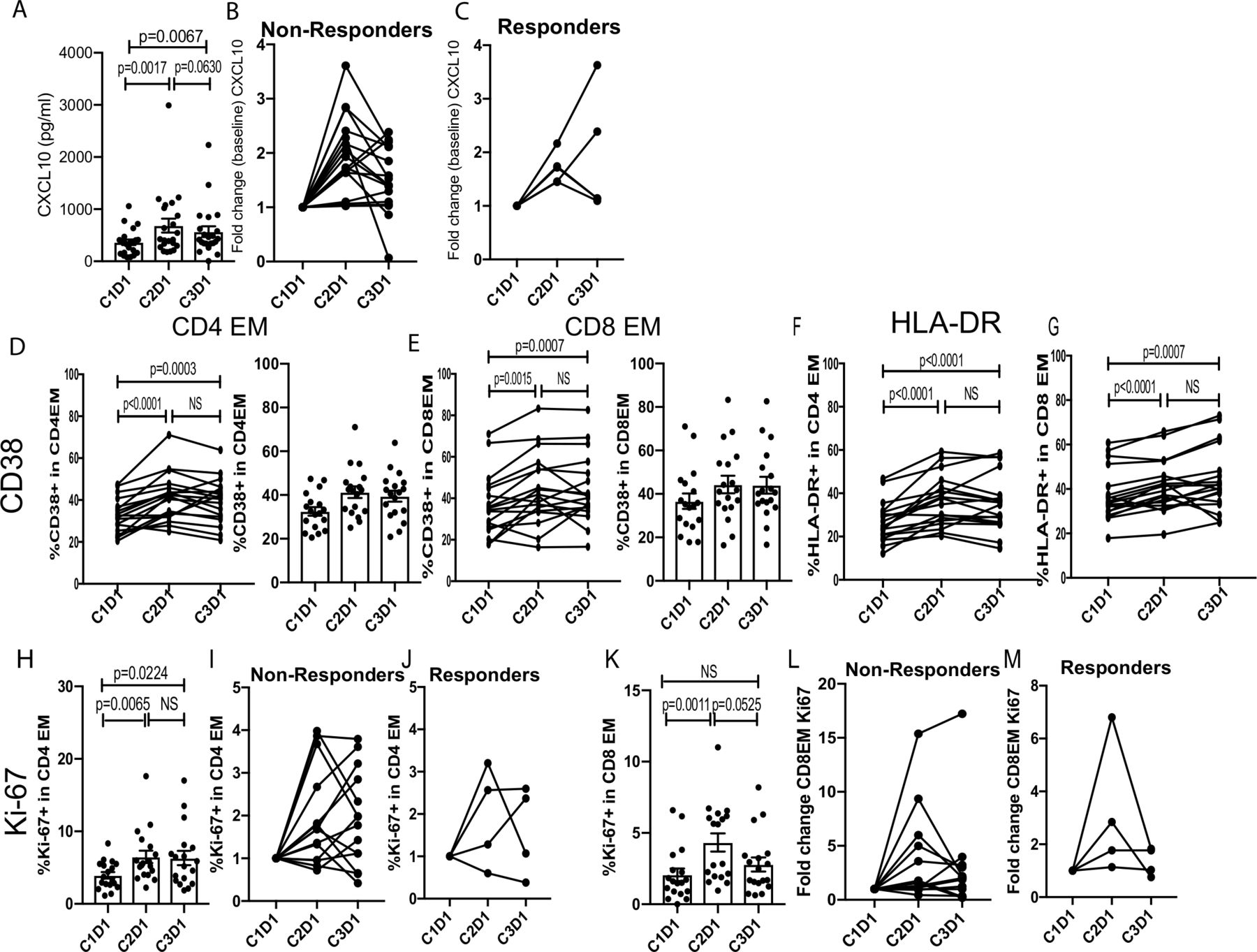
Pembrolizumab plus enzalutamide treatment induced systemic immune responses. (A) Serum CXCL10 levels pretreatment (C1D1; Cycle 1 Day 1) and post-treatment (C2D1, Cycle 2 Day1; C3D1, Cycle 3 Day 1) are shown (n=22). (B) The percent expression of CD38, HLA-DR, or Ki-67 in EM CD4 or CD8 T cells is shown (n=18). EM, effector memory; NS, not significant.
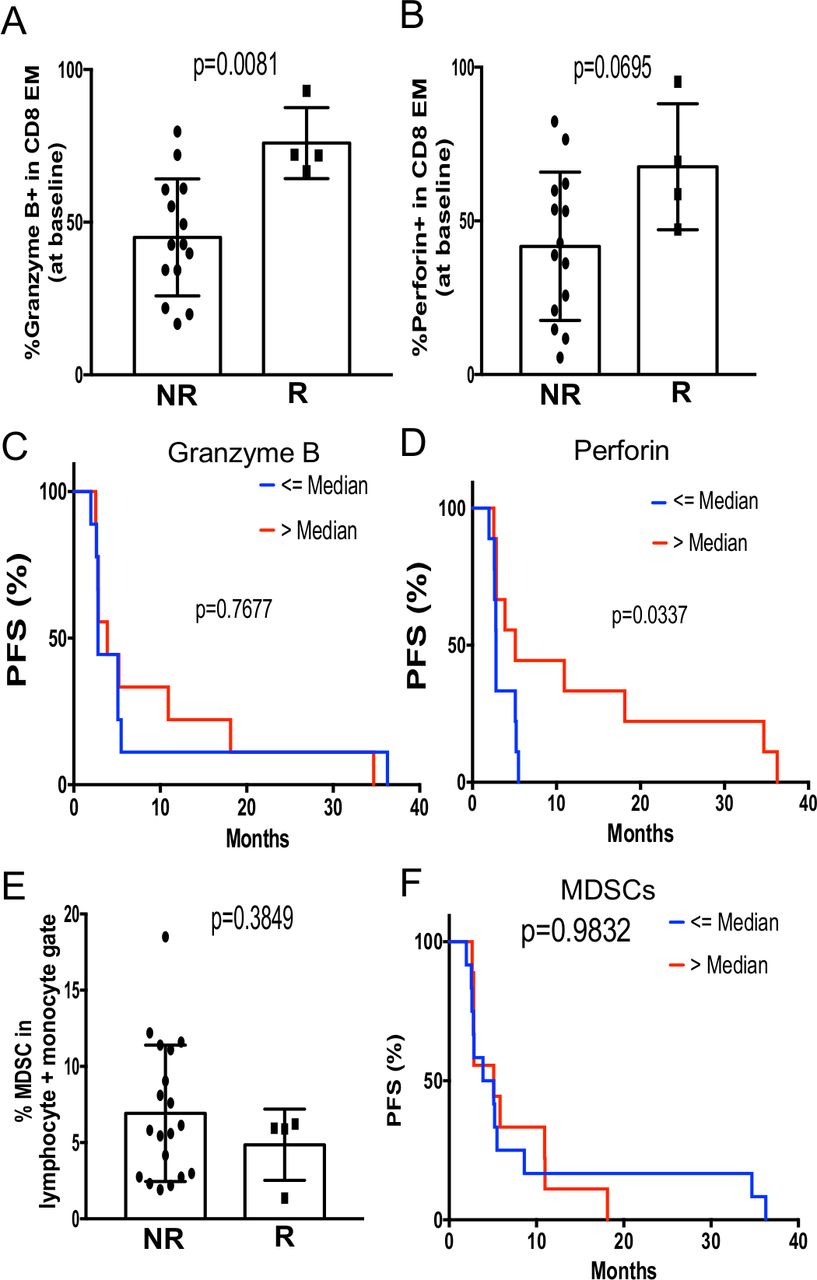
We asked whether there were any correlations between pre-existing immune subsets and clinical responses. Interestingly, we found that the frequency of granzyme B+effector memory CD8 T cells was significantly higher in responders than non-responders (figure 4A, left). Similarly, we observed a trend of increased perforin+effector memory CD8 T cells in responders (figure 4A, right), suggesting that responding patients may harbor a pre-existing pool of cytotoxic tumor-reactive T cells. Further analysis revealed that when patients were grouped based on the frequency of granzyme B or perforin-expressing effector memory CD8 T cells, patients with increased perforin expression had improved PFS (figure 4B). In contrast, there was no correlation between the frequency of MDSCs with clinical response or outcome (figure 4C). Taken together, these data suggest that the clinical benefit of pembrolizumab plus enzalutamide may require the presence of pre-existing tumor-reactive CD8 T cells.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The functional status of EM CD8 T cells as a potential immune correlate with clinical response and outcome. (A) The percent expression of GzmB or perforin in EM CD8 T cells is compared between non-responders (NR; n=14) and responders (R; n=4). (B) Kaplan-Meier curves demonstrating that patients with higher median percentage of perforin, but not GzmB, EM CD8 T cells at baseline had prolonged PFS. (C) The frequency of MDSCs at baseline did not correlate with clinical response or outcome. EM, effector memory; GzmB, granzyme B; MDSCs, myeloid-derived suppressor cells; PFS, progression-free survival.
Safety
There were seven patients with immune-related adverse events (irAE, see table 3). Two patients had colitis, three had hypothyroidism, one had hyperthyroidism, and two had myositis. The colitis resolved for both patients after aggressive treatment with high-dose steroids plus mesalamine or infliximab. Patients with hypothyroidism continue on thyroid replacement, and the patient with hyperthyroid received methimazole with resolution. The patients with myositis received high-dose steroids with a taper lasting 3 months. Four patients discontinued treatment due to irAEs after 2–4 doses of pembrolizumab. No patient developed an irAE on the second course of pembrolizumab. Most adverse events were as expected with enzalutamide and prostate cancer, except that two of the responders were diagnosed with a second malignancy while on therapy. One patient developed metastatic pancreatic cancer. A second patient, who was a non-smoker and had received radiation to the prostate bed 12 years prior to pembrolizumab treatment, developed superficial bladder cancer. Neither event was felt to be related to pembrolizumab. One responder had immune-related colitis and myositis.
Adverse events
Discussion
This study is the first to demonstrate that PD-1 checkpoint inhibition in mCRPC can produce profound and durable responses as assessed by both PSA and radiographic criteria in a subset of patients. This study also introduces a novel paradigm for assessing biopsied lesion-specific responses when interpreting tumor-sequencing data.
Recently, PD-1 inhibitors were approved for any tumor type that exhibits high MSI.20 In prostate cancer, estimates of MSI vary, but may be as high as 12%.33 One responder in this study was found to have MSI, but it is clear that this is not the principal driver of response as two other responders with baseline tissue did not exhibit MSI. Only one metastatic lesion per patient was biopsied for this study, and these lesions responded to pembrolizumab despite the absence of MSI. While there has been some discussion about the best markers to detect MSI in prostate cancer tumors,34 current MSI measurements do not appear to be a universal predictor of PD-1 response in the enzalutamide-refractory prostate cancer setting. Prostate cancers with CDK12 loss may have more susceptibility to checkpoint inhibition,35 but we did not find this mutation in the three responders who had baseline biopsies.
In contrast to recent publications,13 18 this study showed that mCRPC does not routinely express PD-L1, even after the development of resistance to enzalutamide. Notably, our initial publication described PD-L1 expression on tumor cells, but on further study, only TILs showed expression. This study therefore demonstrates that PD-L1 expression in tumor biopsies is not necessary for antitumor activity of pembrolizumab, consistent with several prior studies in other tumor types.36
Another study examined the effect of AR-V7 positivity on the effectiveness of checkpoint inhibition using ipilumumab and nivolumab.37 The investigators hypothesized that AR-V7 positivity correlated with DNA-repair defects and might make them more sensitive to checkpoint inhibitors. They treated 15 men with mCRPC with both ipilimumab and pembrolizumab and found that of the six patients who had DNA-repair defects, PSA PFS was longer than for the nine who did not. In our group of patients, 4 of the 16 evaluable patients had a putative DNA-repair defect,38 and only one responded to treatment, likely because he also had MSI. An additional study explored nivolumab and ipilimumab in non-molecularly selected patients. They found a 21% PSA response rate and 26% radiographic response rate in men who had not received chemotherapy for mCRPC.38
This study has several limitations. Many clinically approved regimens of PD-1 inhibitors allow for indefinite use, but only four cycles of pembrolizumab were used in this study design. While the vast majority of subjects had already demonstrated a clear disease trajectory after four cycles of therapy, there were several patients with stable disease whose PSA began rising after cessation of therapy. It is possible that we could have seen more prolonged stable disease with continued use of PD-1 inhibitors in some patients, but it is also notable that our responders enjoyed continued remissions long after completion of four doses of pembrolizumab. Retreatment of non-responders with initial stable disease did not yield responses. Our study is small, and there is a company sponsored trial (KEYNOTE-199, cohorts 4 and 5) exploring response rates to pembrolizumab following progression on enzalutamide in 120 additional patients (NCT02787005).
Since our initial study design, multiple additional trials for mCRPC have been launched to explore combination therapy of checkpoint inhibitors with enzalutamide and other partners such as PARP inhibitors, radium-223, chemotherapy and other immunotherapies.15 39 40 Preliminary results with PARP inhibitors in combination with a PD-L1 inhibitor show that a subset of patients respond, even if their tumors do not harbor DNA-repair defects.40 These results provide the rationale for further testing of that combination and correlative studies to reveal determinants of response. The impact of single agent enzalutamide on the immune system is being examined in an additional study (NCT02484404).
Conclusion
PD-1 inhibition has activity in mCRPC—including in patients whose tumors appear to lack MSI, DNA-repair defects, or PD-L1 expression. Further studies are warranted to more accurately reveal the activity of PD-1 inhibition in men with mCRPC and to clarify markers of response. Observed responses were deep, durable and achieved after only four doses of pembrolizumab.
Acknowledgments
Kristi Eilers, Jennifer Podolak, Claire Turina, Shawna Bailey, Shaadi Tabatabaei, Matthew McCallum, Emily Martinez, Mandy Burns, Vicky Cheng, Yolanda Prado, Carrie Symmes ran the operations of this study. We would like to thank Brooke Beckett, Alice Fung, Eric Foss and Bryan Foster for their expertise in doing tumor biopsies for research. Neel Patel prepared the pictures of the radiographs. Jeremy Cetnar, Jacqueline Vuky, Fred Ey, Allean Johnson, Justina Lynch contributed patients to this study. Histopathology Shared Resource at OHSU for their pathology support. Erik Liederbauer performed part of the analysis.
References
Footnotes
Twitter @WWredmond4
Contributors JG designed the trial, enrolled patients, interpreted results and drafted the manuscript. RES, JJA, RCB enrolled patients and drafted the manuscript. GVT reviewed specimens for adequacy of prostate adenocarcinoma tissue, interpreted genetic results and aided in drafting the manuscript. RFT and MAW conducted genetic analysis, reviewed data and aided in drafting the manuscript. WLR and YK performed immunoassays, interpreted results and aided in drafting the manuscript. YC and EL performed statistical analysis and aided in drafting the manuscript. CD contributed to study design, interpretation of results and aided in drafting the manuscript. TMB contributed to study design, enrolled patients, interpreted results and aided in drafting the manuscript. AEM reviewed genetic and immunoassay results and drafted the manuscript. All authors read and approved the final manuscript.
Funding This research was funded by Merck Sharp & Dohme Corporation and OHSU Foundation. Funds from the Bloomberg Kimmel Institute (BKI) supported a portion of the laboratory work. Additional laboratory funding was funded by Merck.
Competing interests JG has received research funding from Astellas/Medivation, Merck, Sanofi, Janssen Biotech, and travel support from Sanofi. CD has received research funding from Bristol Myers Squibb (BMS). He has received consulting fees from BMS, Merck, AstraZeneca (AZ) and Medimmune. He has patents licensed to AZ, BMS and Medimmune. WLR has received research grants, consulting fees, and/or royalties from Bristol-Myers Squibb, Merck, Galectin Therapeutics, and Nektar Therapeutics. TMB has research funding from Astellas/Medivation, Alliance Foundation Trials, Boehringer Ingelheim, Corcept Therapeutics, Endocyte Inc, Janssen Research & Development, OncoGenex, Sotio, and Theraclone Sciences/OncoResponse. TMB receives consulting fees from AbbVie, AstraZeneca, Astellas Pharma, Bayer, Boehringer Ingelheim, Clovis Oncology, GlaxoSmithKline, Janssen Biotech, Janssen Japan, Merck, and Pfizer and has stock ownership in Salarius Pharmaceuticals, and Arvinas Inc. JJA, GVT, and RS have no conflicts. RCB is a co-owner of Third Coast Therapeutics Inc, which has an option to license patents of which he is an owner. AEM has a sponsored research agreement with AstraZeneca and has received reagents from Genetech. JJA has received consulting fees from Merck.
Patient consent for publication Not required.
Ethics approval All participants signed a valid consent form for OHSU IRB approved trial #11 025. No study-related activities were performed prior to consent.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.