Article Text

Abstract
Background We report the clinical activity and safety of bintrafusp alfa, a first-in-class bifunctional fusion protein composed of the extracellular domain of the transforming growth factor β (TGF-β)RII receptor (a TGF-β ‘trap’) fused to a human IgG1 monoclonal antibody blocking programmed death-ligand 1 (PD-L1), in patients with heavily pretreated squamous cell carcinoma of the head and neck (SCCHN).
Methods In this phase I dose-expansion cohort, patients with advanced SCCHN not amenable to curative therapy that progressed/recurred after platinum therapy in the recurrent/metastatic setting, or <6 months after platinum therapy in the locally advanced setting, received bintrafusp alfa 1200 mg intravenously every 2 weeks. The primary endpoint was confirmed best overall response (BOR; Response Evaluation Criteria for Solid Tumors (RECIST) 1.1) per independent review committee (IRC); other endpoints included BOR per investigator and safety.
Results As of August 24, 2018, 32 patients had received bintrafusp alfa (median follow-up 86.4 weeks; range 2–97). Per IRC, the confirmed objective response rate (ORR) was 13% (95% CI 4% to 29%; 4 partial responses (PR)); 4 patients had stable disease (SD) (disease control rate 34%; 95% CI 19% to 53%). Per investigator, there were 5 PRs (ORR, 16%), including 2 patients who developed delayed PRs after initial disease increase (total clinical response rate 22%). Responses (ORRs) were observed in patients with PD-L1-positive (12%), PD-L1-negative (17%; 73-10 antibody for immunohistochemistry), human papillomavirus (HPV)-positive (33%) and HPV-negative tumors (5%). Grade 3 treatment-related adverse events (TRAEs) were reported in 11 patients (34%), with no grade 4 TRAEs or treatment-related deaths.
Conclusions Bintrafusp alfa showed clinical activity across subgroups of PD-L1 expression and in HPV-positive tumors and had a manageable safety profile in patients with heavily pretreated advanced SCCHN. Activity in HPV-positive tumors is favorable compared with historical data from PD-L1 inhibitors and is being further investigated in an ongoing study of HPV-associated tumors.
Trial registration number NCT02517398.
- head and neck neoplasms
- immunotherapy
- clinical trials as topic
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Squamous cell carcinoma of the head and neck (SCCHN) causes nearly 300 000 deaths per year as of 2012.1 Smoking, alcohol use and infection with human papillomavirus (HPV) are leading risk factors for SCCHN.2 3 Patients with advanced or recurrent SCCHN have a poor prognosis and a median overall survival (OS) of <1 year in the first-line setting, and an even lower median OS in the second-line or later-line settings.4–7 OS results from KEYNOTE-048 (NCT02358031) support the use of pembrolizumab plus a platinum agent and 5-fluorouracil or as monotherapy in patients with combined positive score (CPS) >1, as a frontline regimen for patients with recurrent/metastatic disease.6 8 9 Patients who experience progression on cetuximab with chemotherapy have few treatment options, with low response rates (<15%)10 11 and response durability or disease stabilization rarely exceeding 3 months.12–15 Patients with advanced SCCHN that progressed within 6 months after the last dose of platinum-based therapy in either the locally advanced or recurrent/metastatic setting have aggressive tumor biology and a poor prognosis and represent an unmet clinical need.8 9 16 Anti-programmed cell death protein 1 (anti-PD-1) immune checkpoint inhibitors, including nivolumab and pembrolizumab, have shown clinical activity in the second-line setting for the treatment of SCCHN—objective response rates (ORRs) of 13%–16%4 7 17—and are approved for the treatment of patients with SCCHN who experienced disease progression on or after platinum-containing chemotherapy.
Transforming growth factor β (TGF-β) signaling is implicated in multiple tumorigenic processes, including immune-related and non-immune-related pathways. TGF-β is overexpressed in multiple tumor types and can have suppressive effects in early stages of tumorigenesis; however, in the tumor microenvironment, TGF-β loses its antiproliferative, tumor-suppressing activity and instead promotes tumor progression via processes such as immune evasion and epithelial-to-mesenchymal transition (EMT), once a malignancy has been established.18 Although TGF-β is integral in mediating immune self-tolerance in normal tissues,18 its immunosuppressive activities assist in immune evasion of tumor cells, and TGF-β inhibition can facilitate T-cell invasion in the tumor microenvironment and increase susceptibility to anti-PD-1/programmed death-ligand 1 (PD-L1) therapy in mouse tumor models.19 20 Furthermore, TGF-β can promote drug resistance and metastasis via increased fibrosis, angiogenesis and EMT.21
Increased plasma TGF-β1 negatively correlates with survival in patients with SCCHN who received cetuximab-based therapy.22 Furthermore, there is a link between dysregulation of TGF-β signaling and the development of HPV-positive tumors,23–25 and HPV status is a prognostic biomarker in SCCHN.26 Consequently, it is possible that dysregulation of TGF-β signaling may drive tumorigenesis in HPV-positive cancers. Virus-associated cancers are more likely to be immunologically inflamed and thus susceptible to immunotherapy-based treatments.27 Therefore, sequestering TGF-β from the tumor microenvironment while simultaneously inhibiting the PD-L1 immune checkpoint may provide a novel treatment approach.
Bintrafusp alfa (M7824) is a first-in-class bifunctional fusion protein composed of the extracellular domain of the human TGF-β receptor II (TGF-βRII or TGF-β ‘trap’) fused via a flexible linker to the C-terminus of each heavy chain of an IgG1 antibody blocking PD-L1 (anti-PD-L1).28 Preclinical studies in murine models showed that bintrafusp alfa more effectively suppressed tumor growth and metastasis than either a TGF-β trap or an anti-PD-L1 antibody alone.29 Bintrafusp alfa also inhibited spontaneous metastasis in multiple preclinical models,29 30 presumably due to its suppressive effect on EMT. Data from a phase I, dose-escalation study suggested that bintrafusp alfa—administered at doses of up to 20 mg/kg—had a manageable safety profile and showed early signs of clinical efficacy in patients with heavily pretreated advanced solid tumors.28 Bintrafusp alfa also showed activity in patients with HPV-associated malignancies, including cervical cancer, anal cancer and SCCHN.31 Here, we report the efficacy and safety results of a phase I trial of bintrafusp alfa monotherapy in patients with advanced SCCHN that progressed after the last dose of platinum-based therapy.
Methods
Study design and participants
NCT02517398 is an ongoing, phase I, open-label trial of bintrafusp alfa that includes multiple solid tumor expansion cohorts. Planned enrollment for the advanced SCCHN cohort was 30 patients. With 30 subjects treated, the study has approximately 87% power to rule out a ≤15% ORR (null hypothesis) when the true ORR is 35% at the 10% type I error rate (one-sided). Meeting this endpoint would have warranted the clinical development of a phase II or III trial based on a very strong phase I signal. Eligible patients were aged ≥18 years and had histologically confirmed, advanced SCCHN not amenable to curative therapy that progressed/recurred after prior platinum therapy in the recurrent/metastatic setting, or within 6 months after the last dose of platinum received in combination with radiotherapy for the treatment of locally advanced disease. Patients may have received prior cetuximab, but prior treatment with any antibody/drug-targeting T-cell coregulatory proteins such as anti-PD-1, anti-PD-L1 or anticytotoxic T-lymphocyte-associated protein-4 antibody was not allowed. Patients had an Eastern Cooperative Oncology Group performance status of 0 or 1; life expectancy of ≥12 weeks; adequate renal, hepatic and hematological function; measurable disease by RECIST 1.1 and available archival tumor material or fresh biopsies taken within 28 days of the first administration of study drug. Patients were not selected on the basis of PD-L1 expression or other biomarkers.
Procedures
Integrated analysis of bintrafusp alfa exposure, response and progression-free survival (PFS) supported a flat dose of 1200 mg every 2 weeks,32 and this dose was used in the expansion cohorts. Patients received bintrafusp alfa via intravenous infusion over 1 hour once every 2 weeks until confirmed progressive disease (PD), unacceptable toxicity or trial withdrawal. Treatment beyond PD was permitted if adverse events (AEs) were manageable, performance status was maintained, and no further treatment was indicated.
Premedication with an antihistamine and acetaminophen approximately 30–60 min prior to each dose of bintrafusp alfa was mandatory for the first 2 infusions and was optional afterward. Dose reduction was not permitted; however, interruption or discontinuation of bintrafusp alfa was allowed as described in online supplementary table S2.
Supplemental material
Tumor response was assessed by radiographic imaging 6 weeks after initiating treatment, then every 6 weeks for the first year and every 12 weeks thereafter. Response was confirmed by repeated radiographic assessment no sooner than 4 weeks from the first documentation of response. AEs were monitored throughout treatment, for the first 28 days after the last study dose, at 10 weeks post-treatment and every 12 weeks thereafter and were assessed according to NCI-CTCAE V.4.03. Immune-related AEs were identified using a prespecified list of MedDRA preferred terms followed by medical review.
Outcomes
The primary endpoint for this expansion cohort was to assess the confirmed best overall response (BOR) according to RECIST 1.1 as adjudicated by an independent review committee (IRC). Secondary endpoints included BOR per investigator and safety and tolerability. Exploratory endpoints included PFS, duration of response (DOR) and disease control rate (DCR) according to RECIST 1.1 as adjudicated by IRC, and OS. Exploratory biomarker analyses of efficacy were also performed.
Statistical analysis
Efficacy and safety were analyzed in all patients who received ≥1 dose of bintrafusp alfa. DCR—defined as the proportion of patients with a BOR of complete response (CR), partial response (PR), non-CR/non-PD or stable disease (SD) —was tabulated with an exact 95% Clopper-Pearson CI. PFS, DOR and OS were analyzed using the Kaplan-Meier method. Safety and tolerability were analyzed using descriptive statistics.
Biomarker analyses
Tumor PD-L1 protein expression was measured by immunohistochemical staining of formalin-fixed, paraffin-embedded (FFPE) tumor tissue blocks using a proprietary assay (Dako, Carpinteria, California, USA) and an anti-PD-L1 rabbit monoclonal antibody clone 73-10 under license from Merck KGaA (Darmstadt, Germany). PD-L1 positivity in tumor cells was scored as the proportion of tumor cells showing membranous PD-L1 staining.
HPV status was determined by RNA sequencing (RNASeq) using FFPE tissue samples and standard protocols. Kallisto33 (V.0.43.1) was used to align sequencing results to a transcriptome containing the human Ensembl transcripts (V.91, GRCh38) and viral genomes from the National Center for Biotechnology Information collection. HPV content in each sample was assessed as the fraction of reads mapping to any papillomavirus genome. To determine a cut-off (for fraction of viral reads), the same process was applied to SCCHN samples from SRP063006/GSE72536 and SRP066090/GSE74927. The HPV statuses deposited with these sequences were used to choose a cut-off that provided perfect separation between the 28 positive and 51 negative training samples.
For gene expression quantification, RNASeq data were generated from FFPE tissue samples and standard protocols. Bowtie2 V.2.2.334 was used to align sequencing results to the Ensembl 75 human genome (GRCh37 February 2014). Gene expression was determined using RSEM V.1.2.31 with the Ensembl gene annotations.35 Hypothesis testing was performed by comparing RSEM-computed expected counts. Transcript-per-million (TPM) values were upper-quartile normalized and log-transformed for further analysis. Signature scores were defined as the mean log 2 (fold-change) among all genes in each gene signature (online supplementary table S3).19 36 These were calculated by adding a pseudocount of 0.5 TPM to all genes and samples, determining the log 2 (TPM), then subtracting the median log 2-TPM for each gene across all samples from the log 2-TPM for each gene.
Supplemental material
Tumor samples were used to determine immune phenotype based on available immunohistochemistry data (PD-L1 stain and PD-L1 negative control) and H&E staining. An exploratory classification system was used to categorize tumors as having an immune-inflamed (immune cells in direct physical contact with tumor cells), immune-excluded (≥1% of tumor stroma area populated by lymphocytes, immune cells possibly located in immediate vicinity of tumor cells but not efficiently infiltrating tumor cell clusters, and very infrequent physical contact between lymphocytes and tumor cells), or immune desert (<1% of tumor stroma area populated by lymphocytes, no dense immune cell infiltrates and no contact of immune cells with tumor cells) phenotype. This system was developed from methods previously described.19 A pathologist who was masked to the response data scored the scanned slides and determined the corresponding immune phenotype.
Results
Patient population and baseline characteristics
Between October 6, 2016 and January 12, 2017, 46 patients were screened. Of these, 32 patients were enrolled and treated (full analysis set) at 20 sites in the Asia-Pacific region, Europe and the USA. At the time of data cut-off on August 24, 2018, patients had been followed for a median of 86.4 weeks (range 2–97 weeks); 4 patients (13%) remained on treatment and the median duration of therapy was 12.1 weeks (range 2–96 weeks). The primary reason for treatment discontinuation was disease progression (n=20 (63%)), AE (n=5 (16%)), consent withdrawal (n=1 (3%)), death unrelated to study treatment (hemorrhage; n=1 (3%)) and investigator decision (n=1 (3%)). The death unrelated to study treatment occurred 2 weeks after the patient received the first and only dose of bintrafusp alfa. Twenty-eight patients (88%) had progressed on platinum-based therapy in the second-line or later-line, recurrent and/or metastatic setting, and 4 patients (13%) had progressed on platinum-based therapy in the locally advanced setting (table 1).
Patient demographics and baseline characteristics
Efficacy
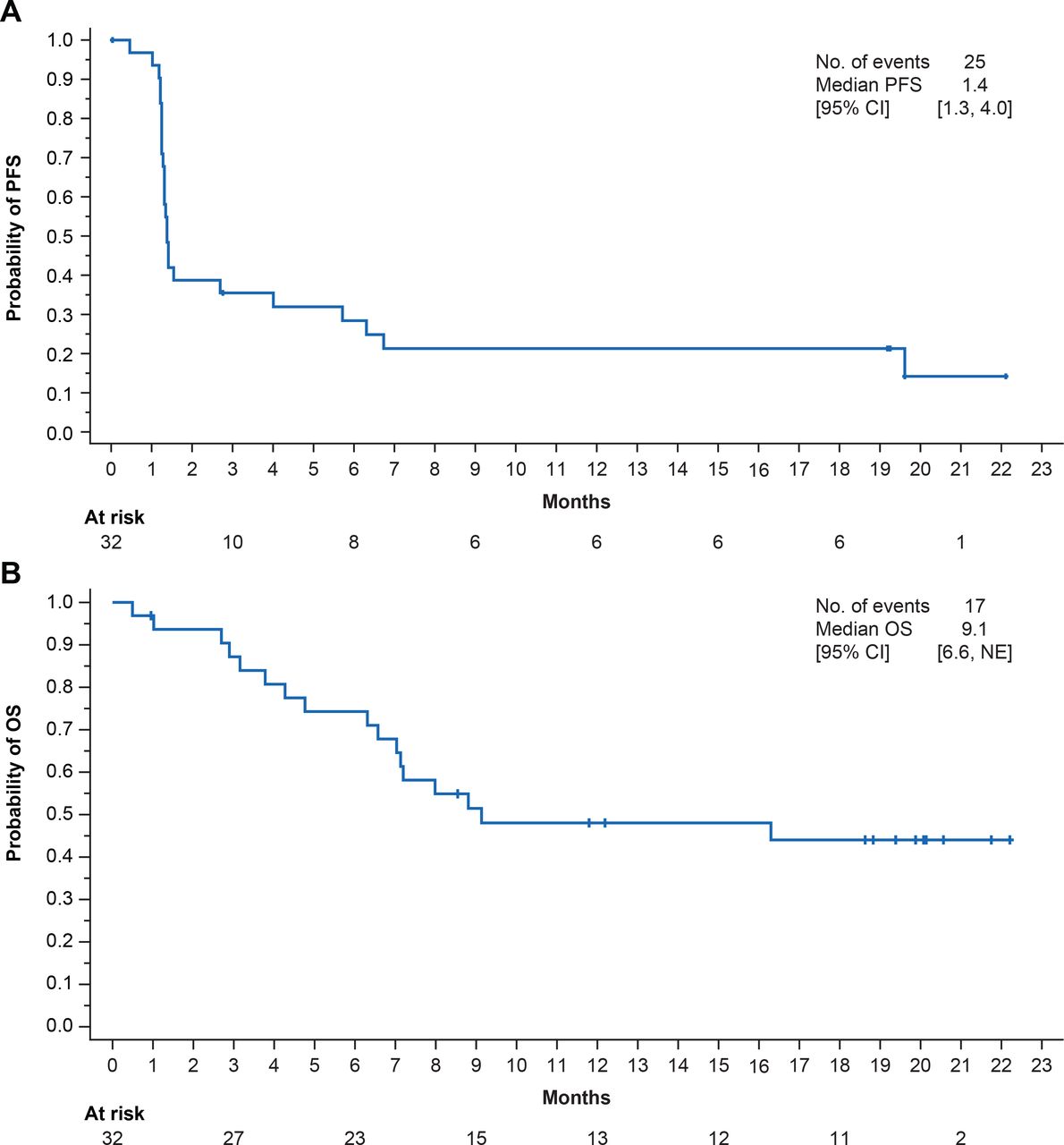
The confirmed ORR by IRC was 13% (95% CI 4% to 29%; 4 PR), and an additional 4 patients had SD, resulting in a DCR of 34% (95% CI 19% to 53%) (table 2 and figure 1A,B). The median DOR was 18.1 months (range 6–20+ months). Two of 4 patients with PR had an ongoing response of 16.5+ and 19.6+ months as of data cut-off (figure 1C). Median PFS was 1.4 months (95% CI 1.3 to 4.0 months) and the 6-month and 12-month PFS rates were 28% (95% CI 14% to 45%) and 21% (95% CI 9% to 37%), respectively; the 18-month PFS rate was stable at 21% (table 2 and figure 2A). Median OS was 9.1 months (95% CI 6.6 months to not estimable (NE)) and the 12-month and 18-month OS rates were 48% (95% CI 30% to 64%) and 44% (95% CI 26% to 61%), respectively (figure 2B).
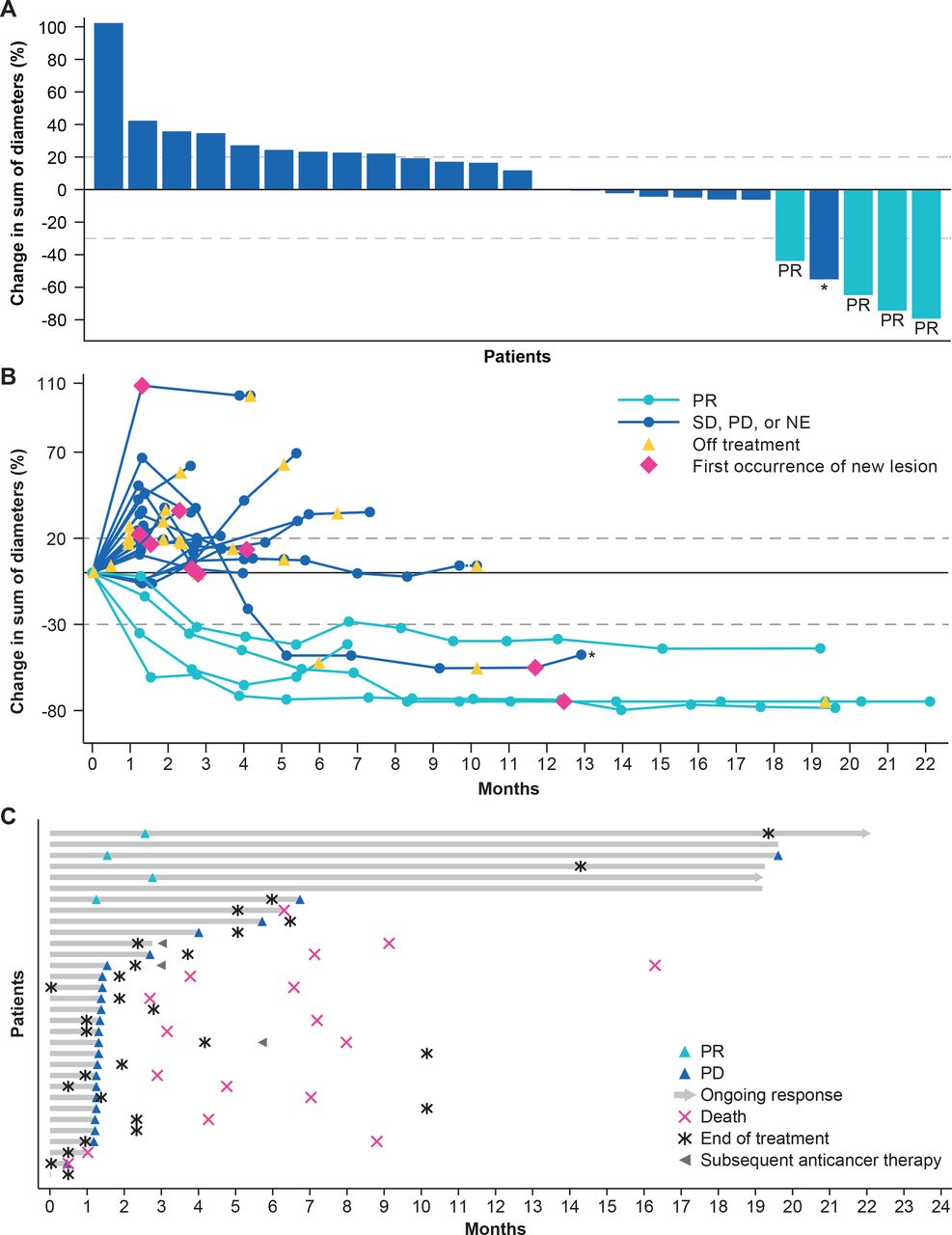
Tumor regression from baseline (A and B) and time to and duration of response (C) in the full analysis set . CR, complete response; NE, not estimable; PD, progressive disease; PR, partial response; SD, stable disease. *Delayed PR after initial PD.

PFS (A) and OS (B) in all patients. NE, not estimable; OS, overall survival; PFS, progression-free survival.
Clinical activity of bintrafusp alfa (N=32)
By investigator assessment, ORR and DCR were 16% and 34%, respectively (table 2). Two patients developed delayed PR after initial PD (time to PR 5.1 and 8.5 months); the patient with time to PR of 8.5 months by investigator assessment did not have target lesions and was therefore assessed as non-CR/non-PD by IRC based on the non-target lesions. The total clinical response rate by investigator assessment was 22%.
Safety and tolerability
Thirty-one patients (97%) experienced a treatment-emergent AE (TEAE) and 24 (75%) experienced a grade ≥3 TEAE. Treatment was discontinued in 8 patients (25%) due to TEAE. Twenty-two patients (69%) experienced treatment-related AEs (TRAEs); of these, 11 (34%) experienced grade 3 TRAEs, which included rash maculopapular (n=2 (6%)) and increased liver enzymes, anemia, diabetic ketoacidosis, colitis, hyperglycemia and hyperthyroidism (n=1 each (3%)) (table 3). No grade 4 or 5 TRAEs were observed. TRAEs led to treatment discontinuation in 2 patients (colitis and SCC of the skin n=1 each (6%)). Keratoacanthomas (n=3 (9%)) and SCC of the skin (n=2 (6%)) were managed with excision and observation. No patient experienced an infusion-related reaction and no infusion rate reductions were necessary. Immune-related AEs were observed in 13 patients (41%), and 4 experienced grade 3 immune-related AEs (colitis and increased alanine and aspartate aminotransferases (n=1 each) and rash maculopapular (n=2)); no grade 4 or 5 immune-related AEs were observed (online supplementary table S4). The occurrence of immune-related colitis led to treatment discontinuation in 1 patient (3%).
Supplemental material
TRAEs of any grade in ≥2 patients or any grade 3 events in any patient (N=32)
Biomarker analysis
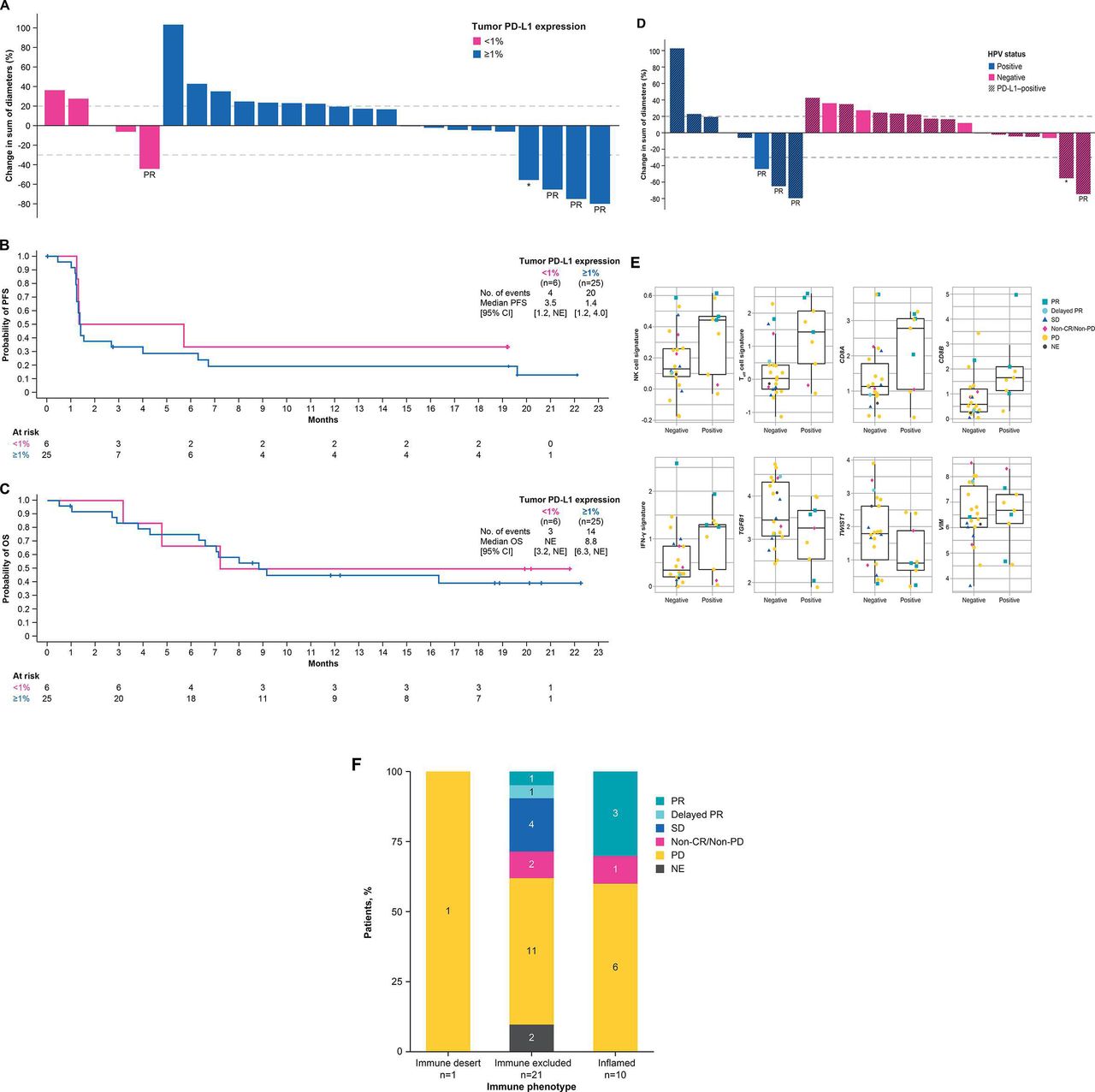
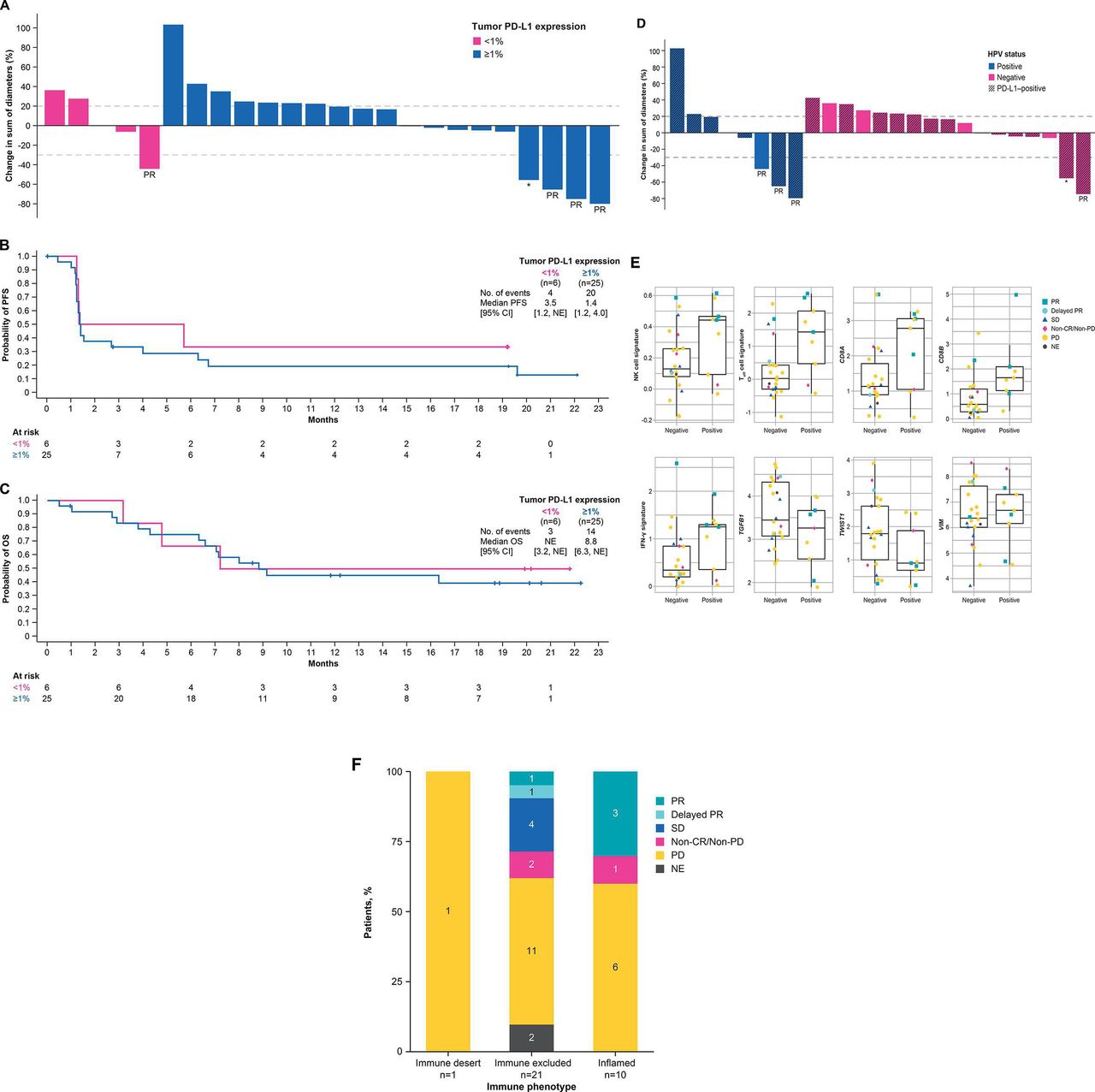
Tumor PD-L1 expression status was available in 31 of 32 patients. Of 25 patients with PD-L1 expression on ≥1% of tumor cells (PD-L1-positive), 3 achieved PR and 3 had SD, resulting in an ORR of 12% (95% CI 3% to 31%) and DCR of 24% (95% CI 9% to 45%). One of 6 patients with PD-L1 expression on <1% of tumor cells (PD-L1-negative) achieved PR (figure 3A and online supplementary table S5). Median PFS in patients with PD-L1-positive tumors was 1.4 months (95% CI 1.2 to 4.0 months) and 3.5 months (95% CI 1.2 months to NE) in patients with PD-L1-negative tumors (figure 3B and online supplementary table S5). Median OS in patients with PD-L1-positive tumors was 8.8 months (95% CI 6.3 months to NE) and NE (95% CI 3.2 months to NE) in patients with PD-L1-negative tumors (figure 3C and online supplementary table S5).
Supplemental material
{kind=link}
{kind=link}
{kind=link}
Tumor regression from baseline (A, D), PFS (B) and OS (C) according to tumor PD-L1 expression and HPV status, respectively, and gene expression profiling status (E) and immune phenotype (F). BOR, best overall response; CR, complete response; HPV, human papillomavirus; IFN-γ, interferon-γ; NE, not estimable; NK, natural killer; OS, overall survival; PD, progressive disease; PD-L1, programmed death-ligand 1; PFS, progression-free survival; PR, partial response; SD, stable disease; Teff, effector T cell; VIM, vimentin. *Delayed PR after initial PD.
Tumor HPV status was evaluable in 31 of 32 patients. Objective responses were observed in 3 of 9 patients with HPV-positive tumors (33% (95% CI 5% to 70%)) and in 1 of 22 patients with HPV-negative tumors (5% (95% CI 0% to 23%)) (figure 3D, online supplementary table S1 and online supplementary figure S1). Median PFS in patients with HPV-positive tumors was 2.7 months (95% CI 1.2 to 19.6 months) and 1.3 months (95% CI 1.2 to 4.0 months) in patients with HPV-negative tumors. Median OS in patients with HPV-positive tumors was 8.0 months (95% CI 2.7 months to NE) and 9.1 months (95% CI 6.3 months to NE) in patients with HPV-negative tumors (online supplementary table S1 and online supplementary figure S1).
Supplemental material
Supplemental material
A total of 30 patients had both HPV and PD-L1 data available. Among 9 patients with HPV-positive tumors, 7 (78%) were PD-L1-positive, and among 21 patients with HPV-negative tumors, 17 (81%) were PD-L1-positive, suggesting no association between HPV status and PD-L1 expression.
RNAseq data were analyzed to evaluate prespecified genes and gene signatures related to the TGF-β pathway and the immune-related pathways versus response to bintrafusp alfa. An extensive exploratory analysis was also conducted to identify genes or gene signatures associated with response to bintrafusp alfa in a biology/pathway agnostic manner. Several genes and signatures that reflect immune activity had higher levels in HPV-positive tumors compared with HPV-negative tumors, although the differences were not statistically significant after correcting for the size of the set tested (figure 3E).
Evaluable patient tumors were characterized according to immune phenotype as either inflamed (tumor infiltration of both CD4+ and CD8+ T cells and other immune cells), immune-excluded (immune cells are present but do not infiltrate the tumor) or immune-desert (lack of immune cells in and around the tumor). Of the 10 patients with a tumor of the inflamed immune phenotype, 3 had responses, and of 21 patients with a tumor of the immune-excluded phenotype, 1 had a response. A response following pseudoprogression was observed in a patient with a tumor of the immune-excluded phenotype (figure 3F).
Discussion
In this pretreated population of patients with advanced SCCHN, bintrafusp alfa monotherapy showed clinical activity, with an ORR of 16% and DCR of 34% per investigator, a 44% OS rate at 18 months and a median DOR (mDOR) of 18.1 months. These preliminary results are comparable to those in similar patient populations treated with the anti-PD-1 immune checkpoint inhibitors pembrolizumab and nivolumab, with ORRs of 13% to 18% and mDORs of 8–18.4 months in previously treated patients with PD-L1-unselected tumors.4 7 17 37 38 Bintrafusp alfa also had a manageable safety profile. Potential TGF-β-related AEs (eg, keratoacanthoma) were well managed with simple excision and observation and did not result in trial discontinuation. No recurrence of keratoacanthoma or SCC has been reported. The incidence, severity and type of immune-related AEs observed with bintrafusp alfa were comparable to those seen with anti-PD-L1 agents.4 7 17 37
Bintrafusp alfa monotherapy showed activity irrespective of tumor PD-L1 expression. DCRs in patients with PD-L1-positive and PD-L1-negative tumors were 24% and 33% and 18-month OS rates were 40% and 50%, respectively. We also observed numerically higher ORR in patients with HPV-positive tumors (33%) compared with patients with HPV-negative tumors (5%). A separate study found that bintrafusp alfa stimulated an HPV-specific response,31 suggesting that bintrafusp alfa may have additional activity in HPV-positive disease. This observation may be due to the link between TGF-β signaling and HPV-positive tumors and to the greater inflammatory immune response generally observed in virus-associated cancers.23–25 27 Indeed, a retrospective subgroup analysis of the dose-finding cohort of this phase I trial revealed an ORR of 46% in 11 patients with HPV-positive cancers (mixed cohort of patients with cervical, anal and SCCHN tumors) receiving bintrafusp alfa monotherapy administered at doses of 0.3–30 mg/kg, with durable and ongoing responses.31 Furthermore, while responses to PD-L1 agents are generally observed in inflamed immune microenvironments,39 patients who achieved objective response or disease control in this study had tumors with either an inflamed or immune-excluded immune phenotype.
Thus, bifunctional targeting of the TGF-β and PD-L1 pathways with bintrafusp alfa may be an encouraging approach for patients with advanced SCCHN and in HPV-associated cancers. Bintrafusp alfa is being evaluated in phase I and II trials, including the phase II NCT03427411 study, which is specifically enrolling patients with HPV-associated malignancies, such as cervical cancer, p16+ oropharyngeal SCCHN and anogenital cancers, including vaginal, vulvar, anal and penile carcinomas.
A preclinical study investigated the link between TGF-β and PD-L1 and found correlations between the expression of several genes and antitumor activity.30 Genes related to both pathways targeted by bintrafusp alfa (natural killer cell signature, effector T-cell signature, CD8A, CD8B, interferon-γ signature, TGFB1, TWIST1 and VIM) were examined in archival tumor samples using RNASeq; these data were found to be consistent with HPV-positive tumors having more pre-existing immune response.
The single-arm design of this study is a potential limitation, making it difficult to compare these results with those in patients treated with chemotherapy or established immune checkpoint inhibitors. Furthermore, the small sample sizes of subgroups (eg, for the HPV-positive cohort (n=9) and PD-L1-negative cohort (n=6)) limit conclusions. Nevertheless, the results in this expansion cohort warrant further investigation of bintrafusp alfa in advanced SCCHN.
Conclusions
In summary, bintrafusp alfa monotherapy showed clinical activity and had a manageable safety profile in this phase I cohort of patients with heavily pretreated, advanced SCCHN with limited or no available therapeutic options. Further investigation of bintrafusp alfa in SCCHN is warranted and ongoing.
Acknowledgments
The authors would like to thank the patients and their families, investigators, co-investigators and study teams at each of the participating centers and at Merck KGaA, Darmstadt, Germany, and EMD Serono Research & Development Institute, Billerica, Massachusetts, USA (a business of Merck KGaA, Darmstadt, Germany). The authors would also like to thank Christian Ihling, of Merck KGaA, Darmstadt, Germany, for his substantial contribution to the immune phenotype analysis. Medical writing support was provided by Shaun Rosebeck, PhD, of ClinicalThinking, Hamilton, New Jersey, USA, which was also funded by Merck KGaA and GlaxoSmithKline in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @gulleyj1
Correction notice This article has been corrected since online publication. The disease control rate in the abstract, efficacy results and Table 2 has been updated from 25% to 34%. Additionally, the number of responses by immune phenotype quoted in the text was updated to match the data shown in Figure 3.
Contributors BCC, AD, AR, SS, NI, EM, AA, CB, JLG and NP collected and assembled the data. LSO, CH and PAR analyzed and interpreted the data. All authors were involved in writing the report and approved the final version of the report.
Funding Merck KGaA provided the study drug and worked with investigators on the trial design and plan, collection and analysis of data and interpretation of results. This work was supported by Merck KGaA, Darmstadt, Germany, and is part of an alliance between Merck KGaA and GlaxoSmithKline. Funding for a professional medical writer with access to the data was provided by Merck KGaA and GlaxoSmithKline. No grant number is applicable.
Competing interests BCC reports research funding from Novartis, Bayer, AstraZeneca, MOGAM Institute, Dong-A ST, Champions Oncology, Janssen, Yuhan, Ono, Dizal Pharma, MSD; consulting role for Novartis, AstraZeneca, Boehringer Ingelheim, Roche, BMS, Ono, Yuhan, Pfizer, Eli Lilly, Janssen, Takeda, MSD; stock ownership in TheraCanVac; royalties from Champions Oncology. CH is an employee of Merck KGaA, Darmstadt, Germany. LSO and PAR are employees of EMD Serono, Billerica, Massachusetts, USA, a business of Merck KGaA, Darmstadt, Germany. JLG reports that the National Cancer Institute (NCI) has a Cooperative Research and Development Agreement (CRADA) with EMD Serono. Resources are provided by this CRADA to the NCI. JLG gets no personal funding from this CRADA but is the co-primary investigator of the CRADA.
Patient consent for publication Not required.
Ethics approval Patients were enrolled in accordance with a protocol approved by the principal and coordinating investigators of the trial and relevant regulatory authorities. International standards of Good Clinical Practice and the Declaration of Helsinki were followed. Written informed consent was provided by patients or their representatives. Ethics committees at all participating institutions (online supplementary table S1) approved the protocol.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. Any requests for data by qualified scientific and medical researchers for legitimate research purposes will be subject to Merck’s Data Sharing Policy. All requests should be submitted in writing to Merck’s data sharing portal (https://www.merckgroup.com/en/research/our-approach-to-research-and-development/healthcare/clinical-trials/commitment-responsible-data-sharing.html). When Merck has a co-research, co-development, or co-marketing or co-promotion agreement, or when the product has been out-licensed, the responsibility for disclosure might be dependent on the agreement between parties. Under these circumstances, Merck will endeavor to gain agreement to share data in response to requests.