Article Text

Abstract
Background Dendritic cells (DCs) are crucial for the efficacy of cancer vaccines, but current vaccines do not harness the key cDC1 subtype required for effective CD8+ T-cell-mediated tumor immune responses. Vaccine immunogenicity could be enhanced by specific delivery of immunogenic tumor antigens to CD141+ DCs, the human cDC1 equivalent. CD141+ DCs exclusively express the C-type-lectin-like receptor CLEC9A, which is important for the regulation of CD8+ T cell responses. This study developed a new vaccine that harnesses a human anti-CLEC9A antibody to specifically deliver the immunogenic tumor antigen, NY-ESO-1 (New York esophageal squamous cell carcinoma 1), to human CD141+ DCs. The ability of the CLEC9A-NY-ESO-1 antibody to activate NY-ESO-1-specific naïve and memory CD8+ T cells was examined and compared with a vaccine comprised of a human DEC-205-NY-ESO-1 antibody that targets all human DCs.
Methods Human anti-CLEC9A, anti-DEC-205 and isotype control IgG4 antibodies were genetically fused to NY-ESO-1 polypeptide. Cross-presentation to NY-ESO-1-epitope-specific CD8+ T cells and reactivity of T cell responses in patients with melanoma were assessed by interferon γ (IFNγ) production following incubation of CD141+ DCs and patient peripheral blood mononuclear cells with targeting antibodies. Humanized mice containing human DC subsets and a repertoire of naïve NY-ESO-1-specific CD8+ T cells were used to investigate naïve T cell priming. T cell effector function was measured by expression of IFNγ, MIP-1β, tumor necrosis factor and CD107a and by lysis of target tumor cells.
Results CLEC9A-NY-ESO-1 antibodies (Abs) were effective at mediating delivery and cross-presentation of multiple NY-ESO-1 epitopes by CD141+ DCs for activation of NY-ESO-1-specific CD8+ T cells. When benchmarked to NY-ESO-1 conjugated to an untargeted control antibody or to anti-human DEC-205, CLEC9A-NY-ESO-1 was superior at ex vivo reactivation of NY-ESO-1-specific T cell responses in patients with melanoma. Moreover, CLEC9A-NY-ESO-1 induced priming of naïve NY-ESO-1-specific CD8+ T cells with polyclonal effector function and potent tumor killing capacity in vitro.
Conclusions These data advocate human CLEC9A-NY-ESO-1 Ab as an attractive strategy for specific targeting of CD141+ DCs to enhance tumor immunogenicity in NY-ESO-1-expressing malignancies.
- melanoma
- immunogenicity, vaccine
- dendritic cells
- immunotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
As the initiators and orchestrators of adaptive immune responses, dendritic cells (DCs) are essential for the induction of tumor-specific CD8+ T cell responses and are crucial for the efficacy of cancer vaccines.1 2 DC vaccines are safe and immunogenic when administered to patients with cancer and in some cases have demonstrated clinical efficacy. Advances in DC biology has revealed that current DC vaccine formulations may not be optimal for inducing antitumor CD8+ T cells. New, more potent vaccines are thus required in order to improve clinical response rates.
DCs are comprised of distinct cell subsets that are highly specialized in driving specific immune responses.3 In mice, the cDC1 DC subset (also known as Batf3-dependent, CD103+ tissue resident and CD8α+ lymphoid resident DC) is responsible for driving CD8+ T-cell-mediated control in a variety of tumor models and for responses to radiotherapy, checkpoint inhibitors and adoptive T cell therapies.4 5 The human cDC1 equivalents, also known as CD141 (BDCA-3)+ DCs, share a high degree of homology in their transcriptomes, phenotype and function with their mouse counterparts.6–9 They are more effective than other DC subtypes at cross-presentation of cellular antigen (Ag), the process by which tumor-specific CD8+ T cell responses are generated.6 10 The presence of CD141+ DC in many solid human tumors is associated with increased survival and responses to anti-PD-1 checkpoint inhibition in melanoma,11–14 highlighting the importance of this immune subset in human tumor immunity. Clinical response rates may be improved by more immunogenic vaccines that specifically harness human CD141+ DC.
Antibodies (Abs) specific to Ag uptake receptors expressed by DCs are attractive candidates for delivering vaccine cargo directly to DCs in vivo.15 16 Abs specific to the C-type-lectin receptor, DEC-205, highly expressed by mouse cDC1 but more broadly expressed by human leukocytes, can deliver Ag to DC for priming CD4+ and CD8+ T cell responses in vivo, and when combined with adjuvant, are more effective therapeutic cancer vaccines than peptide, protein or cellular DC vaccines in mice.17–19 For specific delivery of vaccine cargo to human CD141+ DC, the C-type-lectin-like receptor, CLEC9A (also called DNGR-1), is an appealing candidate given its exclusive expression by human CD141+ DC and its critical role in regulation of CD8+ T cell responses.20–24 Anti-mouse Clec9A Abs are equally effective as DEC-205 Ab at delivering Ag to mouse cDC1 for priming CD8+ T cell responses in vivo,25 and induce CD4+ T cell responses, humoral responses and protective antitumor immunity.20 21 Moreover, anti-human CLEC9A Abs are more effective at mediating cross-presentation by CD141+ DC and activation of cytomegalovirus (CMV) pp65-specific memory CD8+ T cells compared with DEC-205 Ab.26
New York esophageal squamous cell carcinoma 1 (NY-ESO-1) is an immunogenic cancer-testis antigen of unknown function that is widely expressed by a variety of cancer types with limited expression in normal tissues.27 Broad humoral, CD4+ and CD8+ T cell immune responses against NY-ESO-1 spanning different HLA haplotypes are detectable in many patients with cancer, making NY-ESO-1 an attractive target for immunotherapy. There are currently dozens of clinical trials harnessing the immunogenicity of NY-ESO-1, including adoptively transferred NY-ESO-1-specific T cells, vaccines and combination therapies (clinicaltrials.gov and Thomas et al 27). NY-ESO-1 vaccines are safe, well tolerated, have elicited both humoral and cellular immune responses, and are associated with longer recurrence-free and overall survival relative to historical controls in melanoma.28 Vaccination with a human DEC-205 Ab genetically conjugated to NY-ESO-1 (CDX-1401) combined with polyICLC adjuvant was feasible and well tolerated in patients with advanced solid malignancies and myelodysplastic syndrome, with enhanced NY-ESO-1-specific CD4+ and CD8+ T cell responses observed in some patients.29 30 Moreover, DEC-205 Ab vaccines could be safely combined with standard treatments including doxorubicin and or decitabine30 and may also benefit patients receiving checkpoint inhibitor therapies.29
In this study, we developed a new vaccine for specific targeting of NY-ESO-1 to CD141+ DCs. This vaccine is comprised of a human anti-CLEC9A IgG4 Ab genetically fused to immunogenic NY-ESO-1 epitopes. Our data demonstrate that CLEC9A-NY-ESO-1 Abs are effective at delivery and cross-presentation of multiple NY-ESO-1 epitopes by CD141+ DCs for activation of NY-ESO-1-specific CD8+ T cells. CLEC9A-NY-ESO-1 Ab activated both naïve and memory NY-ESO-1-specific T cell responses and were more effective than DEC-205-NY-ESO-1 and untargeted control NY-ESO-1 Ab. CLEC9A-NY-ESO-1 Ab is therefore a promising new vaccine candidate for NY-ESO-1-expressing malignancies.
Methods
Human samples
Blood was obtained from healthy adult volunteers and umbilical cord blood (UCB) was obtained from the Queensland Cord Blood Bank. Cryopreserved peripheral blood mononuclear cells (PBMCs) from patients with melanoma that had been vaccinated with NY-ESO-1 protein and ISCOMATRIX adjuvant31 or monocyte-derived DC (MoDC) pulsed with autologous tumor32 were used for some experiments. CD34+ hematopoeitic stem cells (HSCs) were isolated from UCB by Ficoll-Paque (GE Healthcare) density centrifugation and using a CD34+ isolation kit (Miltenyi Biotec) and cryopreserved. HLA typing was performed by BGI Health (Hong Kong) and or in-house PCR.33 CD34+ HSCs were expanded and differentiated into CD141+ DC using a modification of a previously described protocol34 to include 1 µM StemRegenin-1 (SR-1, Stemcell Technologies) during both expansion and differentiation stages.
Production and validation of recombinant Ab
Expression constructs were generated in pcDNA3.1+ plasmids encoding human IgG4 and κ constant regions and variable regions of anti-human CLEC9A clone 4C6, anti-human DEC-205 clone MMRI-7 or isotype control anti-β-galactosidase (β-gal) clone GL117.26 The antibody heavy chain sequence was fused to cDNA encoding an alanine linker and NY-ESO-1 antigenic sequence (AAAALLEFYLAMPFATPMEAELARRSLAQDAPPLPVPGVLLKEFTVSGNILTIRLTAADHRQLQLSISSCLQQLSLLMWITQCFLPVFLAQPPSGQRR) containing the HLA-A*0201-restricted 157-165epitope SLLMWITQC (SLL) and the HLA-Cw*030392-100-restricted epitope LAMPFATPM (LAM) (GeneArt). The resulting chimeric Abs, carrying two NY-ESO-1 antigenic sequences per Ab, were expressed in Freestyle 293F cells (Invitrogen) using 293Fectin (Invitrogen) and purified from the supernatant using Protein A sepharose (GE Healthcare). Ab specificity was confirmed by binding to 293F cells transiently transfected with full-length recombinant CLEC9A or DEC-205 as previously described.26
Flow cytometry
Binding specificity to human PBMC subsets was assessed by blocking with combined rat and mouse serum followed by incubation with chimeric Ab followed by anti-human IgG4-biotin (Life Technologies) then Streptavidin-PE (SA-PE, Life Technologies). Lymphocyte populations were identified by staining with Ab cocktails comprised of Live/Dead (Zombie) Aqua (Biolegend), anti-human CD3-BV711 or Pacific Blue (clone OTK3), CD19-Pacific Blue (HIB19), CD20-Pacific Blue (2H7), CD56-FITC or Pacific Blue (HCD56), CD14-APC or Pacific Blue (HCD14), CD16-BV785 or Pacific Blue (3G8), HLA-DR-PerCPCy5.5 or PE-Cy7 (L243), CD141-PE-Cy7 (M80), CD1c-Alexa Fluor 700 (L161), CLEC9A-PE (8F9), DEC-205-FITC (all from Biolegend), CD11c-PE-CF594 (B-LY6), CD141-BV711 (1A4) and CD123-BUV395 (7G3) (all from BD), and CADM1-Alexa Fluor 647 (3E1, Jomar/MBL). Samples were acquired on a Becton Dickinson LSR Fortessa X-20 or Beckman Coulter CytoFLEX-S flow cytometer or sorted on a MoFlo Astrios (Beckman Coulter) and analyzed using Flowjo V.9 or 10 software (Treestar).
Antigen presentation assays
CD8+ T cell lines specific for HLA-A*0201 restricted SLL and HLA-Cw*03:03 LAM NY-ESO-1 epitopes were generated and maintained as previously described.35 Purified CD141+ DCs were incubated with 5 µg/mL chimeric Ab, an equivalent amount of untargeted NY-ESO-1 Ag (0.7 µg/mL), SLL or LAM peptides or no Ag for 2 hours at 37°C, then washed and incubated with NY-ESO-1-specific T cell lines. Interferon-γ (IFN-γ) production was detected after 16–20 hours by ELISPOT (Mabtech). For some experiments, PBMCs from melanoma patients with known NY-ESO-1 responses were incubated with chimeric Ab or no antigen for 16 hours and IFN-γ production detected by ELISPOT.
Generation and vaccination of humanized mice expressing NY-ESO-1 SLL T cell receptor
NOD.Cg-Prkdcscid IL2rg tm1Wjl Tg (HLA-A/H2-D/B2M) 1Dvs/SzJ transgenic for human HLA-A*0201 (NSG-A2) mice were purchased from The Jackson Laboratory mice (stock no: 014570). Humanized mice were generated following reconstitution with human CD34+ HSC transduced with lentivirus encoding the HLA-A*0201-restricted NY-ESO-1 SLL T cell receptor (TCR) according to previously published protocols.36 37
Following human CD45+ reconstitution, humanized mice received 2×50 µg subcutaneous injections of Flt3L 4 days apart to expand DC followed by vaccination with 10 µg of chimeric Ab or no antigen with 50 µg poly I:C (InVivogen) and mice were harvested 1 week post vaccination. Spleens were digested in collagenase IV (Worthington Biochemical) and DNase I (Roche/Sigma) followed by Percoll density gradient as previously described36 and enriched for human leukocytes using a Mouse/Human Chimera EasySep Kit (Stemcell). Expression of the NY-ESO-1 SLL TCR was confirmed by staining with NY-ESO-1 SLL dextramer-APC (Immudex), anti-mouse CD45-V500 (30-F11, BD), anti-human CD45-BUV395 (HI30, BD), CD3-Pacific Blue or BV711, CD8-PE-Cy7 (RPA-T8), CD197-BV711 (3D12, BD) and CD45RA-PE (H130, Biolegend).
In vitro expansion and effector function of NY-ESO-1-specific T cells
For priming of naïve T cells in vitro, splenocytes from non-immunized humanized mice expressing the NY-ESO-1 SLL TCR were stimulated with SLL peptide or control-pulsed HLA-A*0201+ allogeneic irradiated lymphoblastoid cell lines (LCLs). IFNγ was measured in the supernatants after 3 days by ELISA (Thermo Fisher) and cultures expanded in media containing 100 U/mL IL-2, 10 ng/mL IL-7 and 20 ng/mL IL-15 for 20 days. For reactivation of in vivo-primed NY-ESO-1-specific T cells, PBMCs from vaccinated patients with melanoma or splenocytes from immunized humanized mice were incubated with 10 µg/mL chimeric Abs, SLL peptide or no Ag in the presence of poly I:C and R848 (InvivoGen) for 2 hours at 37°C, then washed and expanded in media containing IL-2, IL-7 and IL-15 for 9–14 days. Expansion of NY-ESO-1 SLL-specific CD8+ T cells was measured by SLL dextramer staining as described above.
Cytokine secretion was assessed by restimulation of the cultures for 6 hours in the presence or absence of SLL peptide, Brefeldin A, Monensin and CD107a-BV785, followed by staining with Live/dead Aqua, CD8-PerCpCy5.5 and CD3-BUV737. Cells were fixed and permeabilized then stained with MIP1β-PE, IFNγ-FITC, TNFα-PE-Cy7 and IL-2-APC or isotype controls for flow cytometry analysis. Cytotoxic activity of the T cells was assessed against SLL or control peptide (HLA-A2 restricted CMV pp65 NLVPMVATV) pulsed T2 targets, and melanoma cell lines LM-MEL 44 (HLA–A*0201+, NY-ESO-1+) or SK-MEL 28 (HLA-A*0201-, NY-ESO-1-) at an effector:target ratio of 10:1 using a Cytotox 96 Kit (Promega). Specific lysis of target cells was calculated as:
(Experimental-EffectorSpontaneous–TargetSpontaneous)/(TargetMaximum–TargetSpontaneous)×100.
Statistical analysis
Data sets were tested for normal distribution using the Kolmogorov-Smirnoff test. Multigroup comparisons were performed by using repeated measures one-way analysis of variance (ANOVA) or non-parametric equivalent (Freidmann’s) followed by appropriate post multiple comparison post-tests (Tukey’s/Dunn’s). Paired comparisons were performed using a paired t-test or non-parametric Wilcoxon’s signed rank test. Statistical significance was defined as a p value equal to or less than 0.05. GraphPad Prism V.7 was used for graphical representation and statistical analysis.
Results
Generation of human CLEC9A-NY-ESO-1 and DEC-205-NY-ESO-1 Ab
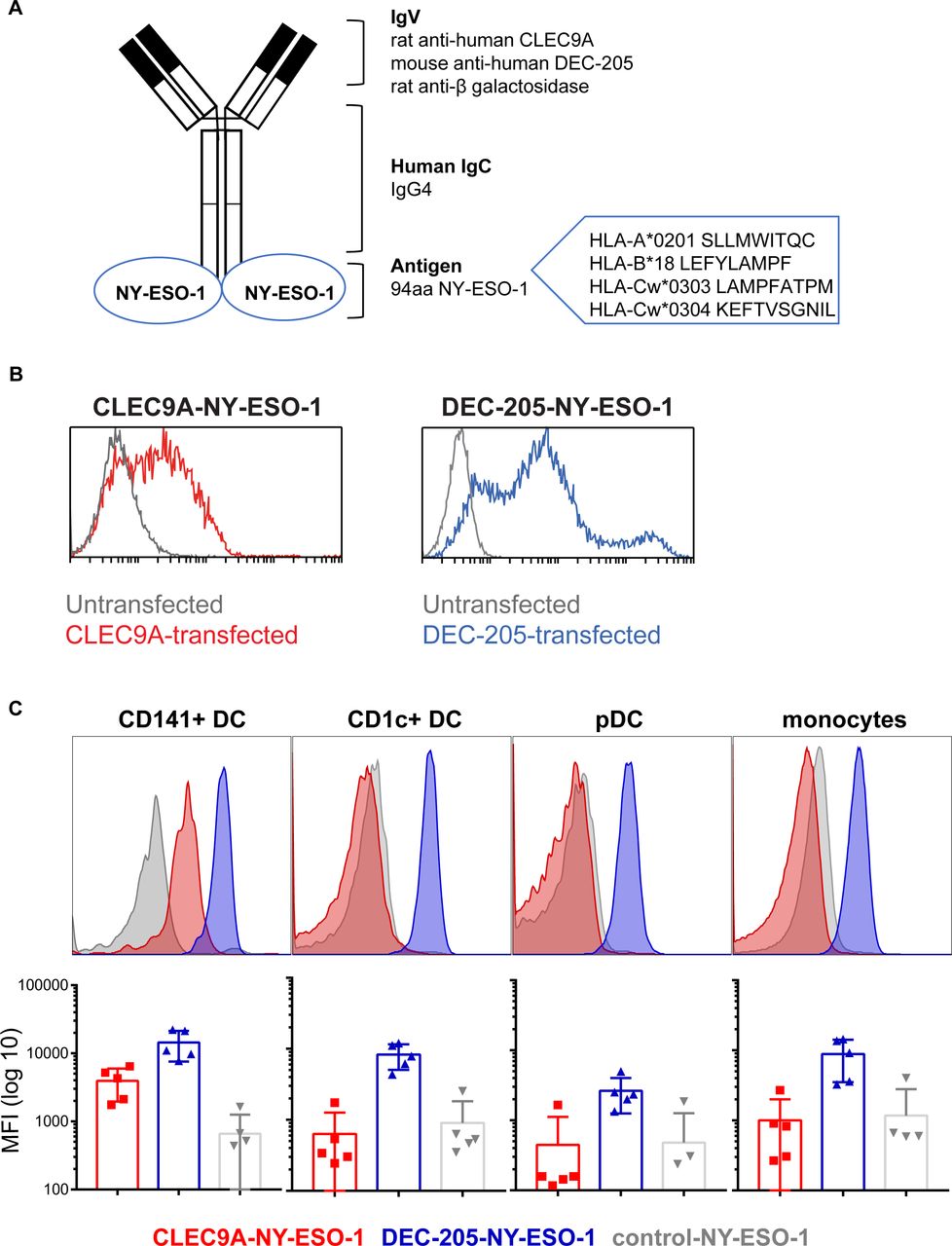
We previously developed constructs encoding human chimeric IgG4 Ab specific for CLEC9A, DEC-205 and β-gal (untargeted isotype control). The NY-ESO-1 sequence, encoding epitopes across multiple HLA alleles, was genetically fused to the heavy chain of each Ab via an alanine linker so that each Ab contained two NY-ESO-1 antigenic sequences (figure 1A). Following expression in 293F cells and purification by affinity chromatography, Ab binding specificity to target receptors was confirmed on CLEC9A or DEC-205 transfected cell lines and PBMC and by ELISA (figure 1B,C and online supplementary figure 1). As expected, the CLEC9A-NY-ESO-1 bound specifically to CD141+ DC, whereas the DEC-205-NY-ESO-1 bound to most human leukocyte subsets, with the highest staining intensity observed on CD1c+ DC and CD141+ DCs. The β-gal control Ab did not bind human leukocytes, with the exception of some non-specific binding on B cells.
Supplemental material
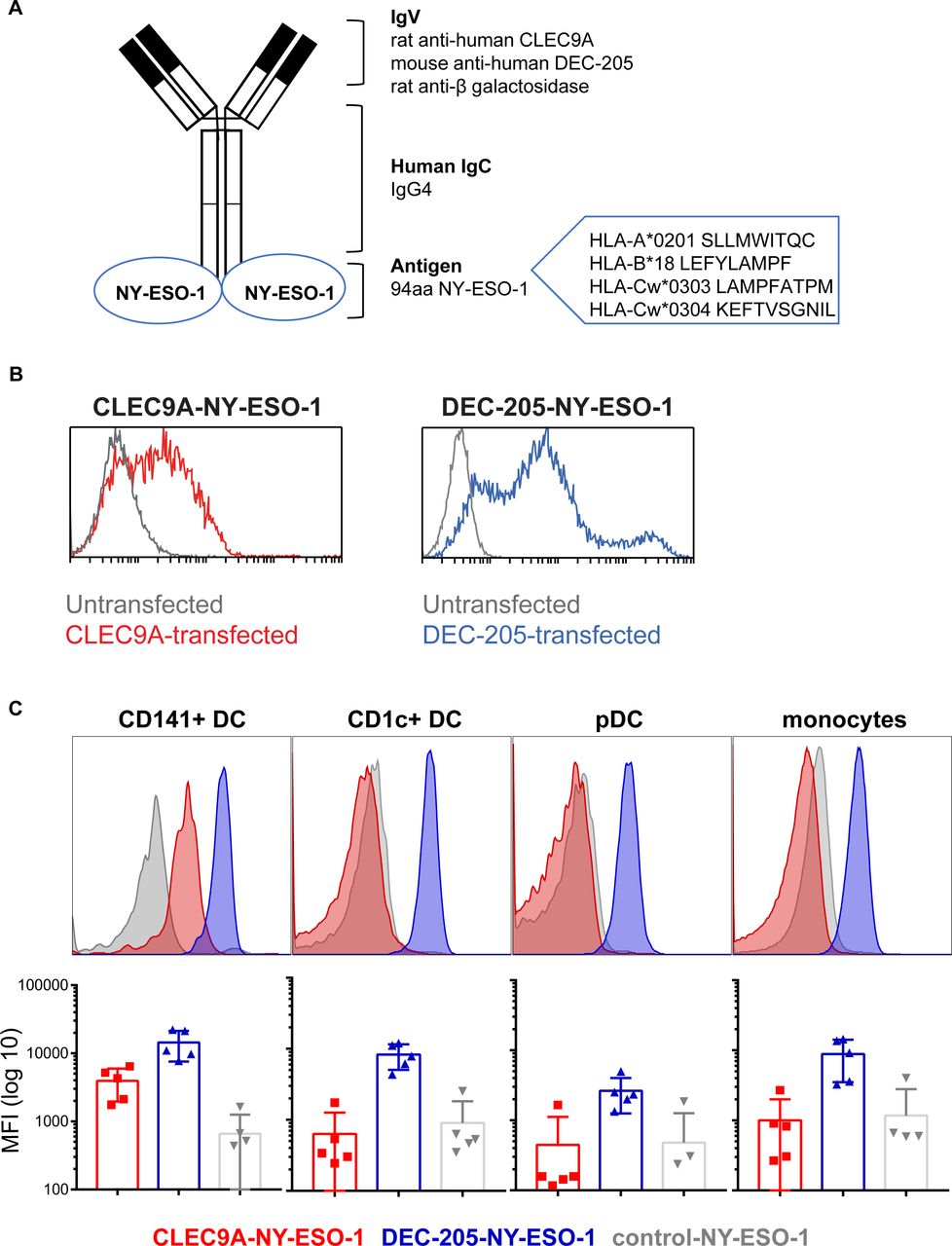
Generation and binding of human CLEC9A-NY-ESO-1, DEC-205-NY-ESO-1 and control-NY-ESO-1 chimeric Ab. (A) Diagram of chimeric Ab consisting of rat or mouse variable regions specific for human CLEC9A, DEC-205, or β-galactosidase (control), and human IgG4 κ constant regions genetically fused to an antigenic sequence of NY-ESO-1. (B) Binding of CLEC9A-NY-ESO-1 (blue, left panel), DEC-205 (red,right panel) to 293 F cells transfected with CLEC9A (left) or DEC-205 (right) and untransfected controls (gray). (C) Binding of chimeric Abs to human Ag presenting cells, shown as representative histograms from one donor and the median fluorescence intensity±SD compiled from five individual healthy donors. Ab, antibody; Ag, antigen; MFI, median fluorescence intensity; NY-ESO-1, New York esophageal squamous cell carcinoma 1.
Cross-presentation of multiple NY-ESO-1 epitopes to CD8+ T cells by CLEC9A-NY-ESO-1 Ab
As CD141+ DCs are extremely rare in human blood, we derived these cells from human cord blood CD34+ HSC using a previously validated in vitro culture system34 (online supplementary figure 2). The transcriptome, phenotype and function of the in vitro derived CD141+ DCs closely resembled those of their human blood counterparts34 and this was confirmed in our hands (data not shown). None of the targeting Ab induced activation of DC measured by expression of costimulatory molecules CD83 and CD86 (online supplementary figure 2B). CLEC9A-NY-ESO-1 and DEC-205-NY-ESO-1 Ab, but not control-NY-ESO-1, bound to in vitro-derived CD141+ DCs (figure 2A). Notably, the staining intensity of DEC-205-NY-ESO-1 was higher than that of CLEC9A-NY-ESO-1. Following uptake by HLA-A*0201+ CD141+ DC, only CLEC9A-NY-ESO-1 mediated cross-presentation of the HLA-A*0201-restricted SLL epitope to specific CD8+ T cells (figure 2B and online supplementary figure 2C). By contrast, both CLEC9A-NY-ESO-1 and DEC-205-NY-ESO-1 mediated cross-presentation of the HLA-Cw*0303-restricted NY-ESO-1 LAM epitope (figure 2C and online supplementary figure 2C). Thus, CLEC9A-NY-ESO-1 Abs are effective delivery vehicles to CD141+ DCs for cross-presentation of multiple NY-ESO-1-specific CD8+ T cell epitopes.

Cross-presentation of NY-ESO-1 epitopes following uptake of CLEC9A-NY-ESO-1 by CD141+ DC. (A) Binding of chimeric Abs to in vitro cultured CD141+ DC shown as representative histograms (left) and the median fluorescence intensity (MFI) collated from three independent donors (right); *p=0.04. (B) Cross-presentation of the HLA-A*0201-restricted NY-ESO-1 SLL epitope following uptake of CLEC9A-NY-ESO-1, DEC-205-NY-ESO-1, control-NY-ESO-1 chimeric Abs or no Ag by in vitro cultured HLA-A*0201+ CD141+ DC. Cross-presentation was measured by interferon γ (IFNγ) production by NY-ESO-1 SLL-specific CD8+ T cells by ELISPOT (spot forming units (SFU) per 10,000 T cells). (C) Cross-presentation of the HLA-Cw3-restricted NY-ESO-1 LAM epitope following uptake of chimeric Ab. Cross-presentation was measured by IFNγ production by NY-ESO-1 LAM-specific CD8+ T cells by ELISPOT (SFU per 10,000 T cells). Data shown are the mean±SD collated from replicates of four independent donors. *p<0.0001 by one-way analysis of variance and Tukey’s multiple comparison test. Ab, antibody; Ag, antigen; DC, dendritic cell; NY-ESO-1, New York esophageal squamous cell carcinoma 1.
CLEC9A-NY-ESO-1 Ab reactivate NY-ESO-1 memory responses in patients with melanoma
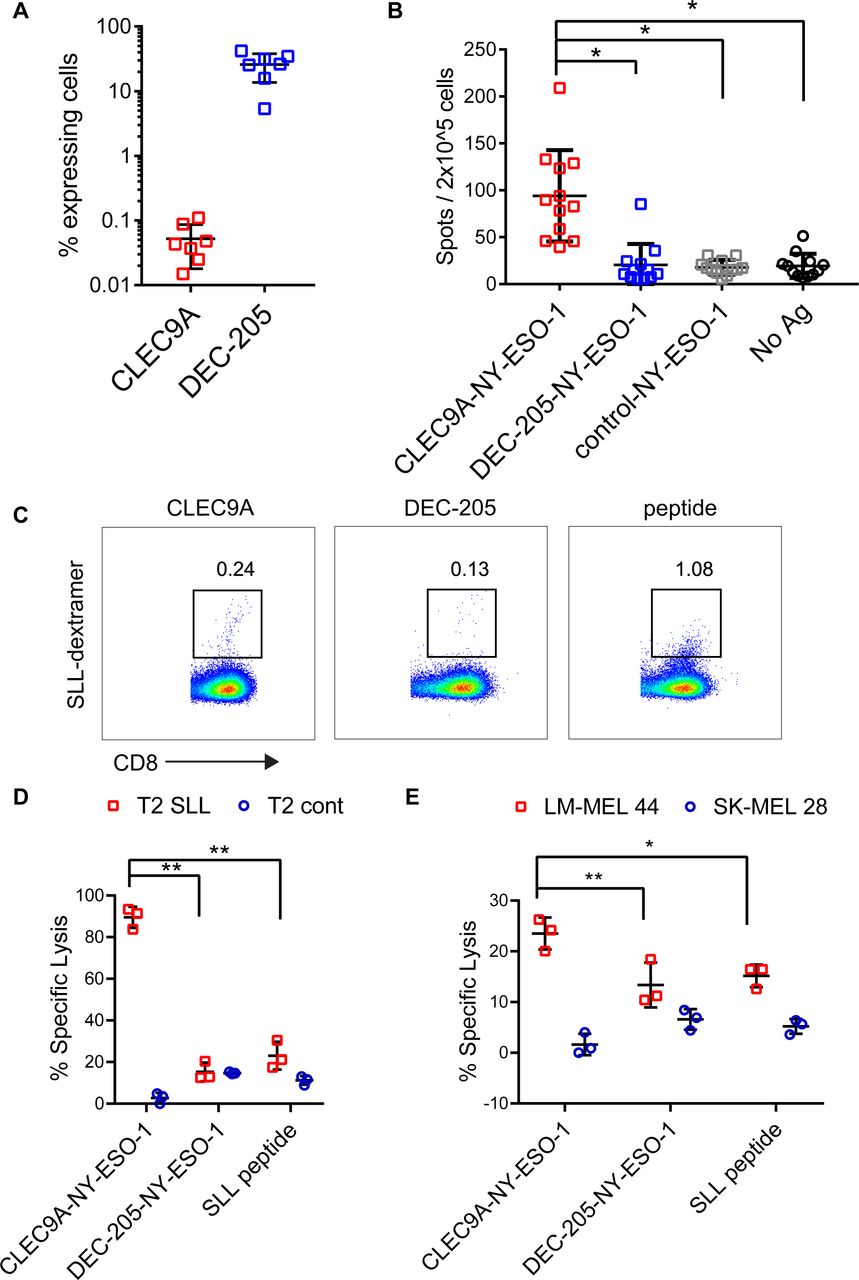
To investigate the ability of CLEC9A-NY-ESO-1 Ab to reactivate NY-ESO-1-specific T cell responses, we used PBMC from melanoma patients that had been previously vaccinated with NY-ESO-1 protein and ISCOMATRIX adjuvant31 or MoDC pulsed with autologous tumor32 and had detectable responses to NY-ESO-1. The percentage of CLEC9A-expressing cells ranged from 0.015% to 0.11% (mean 0.05%) of PBMC and was restricted to the CD141+ DC population (figure 3A), consistent with healthy donors.26 By contrast, DEC-205 was expressed by 5.35%–41.9% (mean 25.8%) of melanoma patient PBMC, with the highest expression seen on monocytes, CD141+ DC and CD1c+ DC, consistent with healthy donors. Despite only a small percentage of PBMC expressing CLEC9A, these were potent at reactivating NY-ESO-1-specific T cell responses following a short stimulation with CLEC9A-NY-ESO-1 Ab ex vivo (figure 3B). In contrast, DEC-205-NY-ESO-1, control-NY-ESO-1 and unconjugated CLEC9A Ab did not induce responses (figure 3B and online supplementary figure 3). Consistent with previous findings,31 responses to the HLA-A*0201-restricted SLL epitope were confirmed in only a few (2/8) of the HLA-A*0201+ patients. Three of the donors had previously identified responses to a very broad range of MHC I and MHC II restricted epitopes (patients 7, 9, 2131). In another donor, the NY-ESO-1-specific response was mapped to a novel HLA-B*5601 -restricted NY-ESO-193-102 AMPFATPMEA epitope (Dr Volker Lennerz and Dr Thomas Wölfel, University Medical Center, Mainz, Germany, personal communication). Responses to known Cw3-restricted epitopes were not detectable in any of the 7 HLA-Cw*03+ patients (online supplementary figure 3 and not shown). Thus, the majority of the responses appeared to be restricted to other, potentially as yet undescribed CD8 and/or CD4 epitopes. For one patient that had pre-existing responses to the HLA-A*0201-restricted SLL epitope, we examined whether the chimeric Ab could also expand NY-ESO-1-specific responses with effector function. For these experiments, patient PBMC were incubated with chimeric Ab or no antigen and then expanded for 2 weeks in the presence of T cell cytokines. Both CLEC9A-NY-ESO-1 and DEC-205-NY-ESO-1 Ab could expand a small percentage of NY-ESO-1 SLL-specific CD8+ T cells as observed by NY-ESO-1 SLL dextramer staining (figure 3C). Although the control-NY-ESO-1 Ab was also able to expand NY-ESO-1 SLL-specific CD8+ T cells, there were insufficient cell numbers from this and the no antigen control cultures to assess in killing assays. T cells expanded by CLEC9A-NY-ESO-1 Ab, but not DEC-205-NY-ESO-1 Ab, could kill SLL peptide coated T2 tumor targets (figure 3D). T cells expanded by CLEC9A-NY-ESO-1 Ab were also more effective at lysis of HLA-A*0201+NY-ESO-1+ tumor targets (figure 3E).
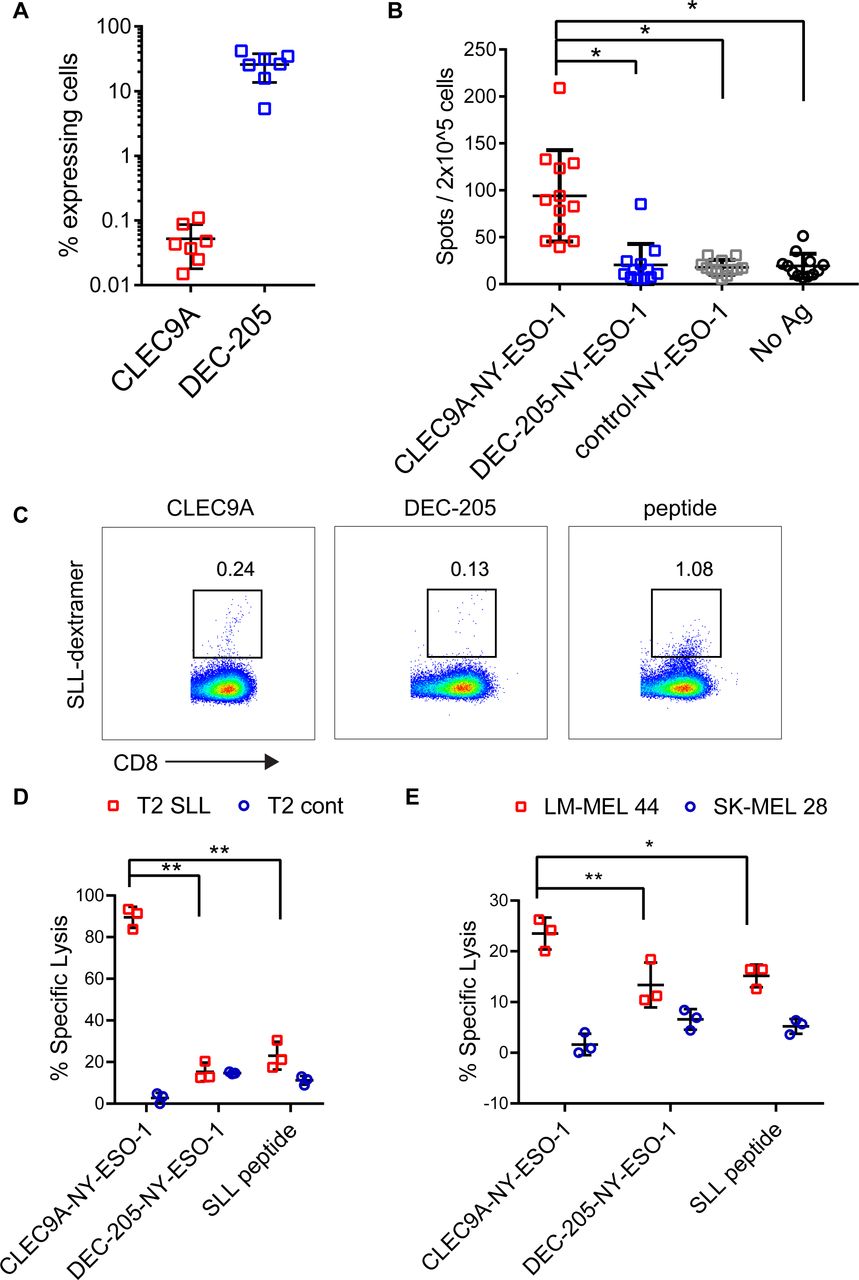
CLEC9A-NY-ESO-1 targeting Ab reactivates NY-ESO-1-specific responses in patients with cancer. (A) Percentage of viable patient peripheral blood mononuclear cells (PBMC) expressing CLEC9A or DEC-205. (B.) PBMCs from cancer patients with known NY-ESO-1-specific responses were stimulated with chimeric Ab or no Ag and interferon γ (IFNγ) production was measured by ELISPOT 18 hours later. Data shown are the mean±SD from 12 patients. (C–E) PBMC from a patient with known responses to the HLA-A2-restricted NY-ESO-1-SLL epitope were restimulated with chimeric Ab or SLL peptide. (C) Percentage of SLL-dextramer+ cells in the cultures. (D) Killing of cognate SLL peptide-pulsed T2 tumor target cells and irrelevant peptide (NLVPMVATV)-pulsed control T2 cells by chimeric Ab- or peptide-expanded T cells. (E) Killing of LM-MEL 44 (HLA-A*0201+, NY-ESO-1+) and SK-MEL 28 (HLA-A*0201- NY-ESO-1-) tumor cell lines by chimeric Ab- or peptide-expanded T cells. *p<0.001; **p<0.005 by one-way analysis of variance and Tukey’s multiple comparison test. Ab, antibody; Ag, antigen; NY-ESO-1, New York esophageal squamous cell carcinoma 1.
CLEC9A-NY-ESO-1 Ab prime naïve NY-ESO-1-specific CD8+ T cells in humanized mice
Humanized mice were generated following reconstitution with human CD34+ HSC transduced with lentivirus encoding NY-ESO-1 SLL TCR (figure 4A). Introduction of the TCR transgene into HSC inhibits endogenous TCR gene rearrangement in recombinant T cells, resulting in exclusive expression of the transgenic TCR on the cell surface via a process known as allelic exclusion.38 Engraftment in neonatal NSG-A2 mice enabled thymic selection of NY-ESO-1 SLL-specific CD8+ T cells on human HLA-A*0201. The mean percentage of human CD45+ cell engraftment in blood ranged from 0.04% to 94% (mean 17.53%±23.56 SD, n=66). Engrafted mice developed human CD141+ DC, CD1c+ DC, plasmacytoid DC, monocytes and T cells in similar proportions to those previously described in NSG-A2 mice36 37 (figure 4B,C). No SLL-specific (dextramer+) CD8+ T cells were detectable in mice engrafted with mock transduced CD34+ HSC, suggesting an absence or very low frequency of NY-ESO-1 SLL-specific CD8+ T cell precursors (not shown). In mice engrafted with TCR lentiviral transduced CD34+ HSC, the NY-ESO-1 SLL TCR was restricted to 0%–47.1% of splenic CD8+ T cells (mean 5.932%±11.05 SD, figure 4D). Importantly, all of the CD8+ T cells expressing the SLL TCR coexpressed CD45RA and CCR7, indicative of a naïve phenotype (figure 4D). Ex vivo coculture of splenocytes from these mice with SLL peptide-pulsed HLA-A*0201+ allogeneic irradiated LCLs led to the expansion of NY-ESO-1 SLL-specific CD8+ T cells (figure 4E), and induced IFNγ production on restimulation with cognate SLL peptide (figure 4F). The expanded NY-ESO-1 SLL-specific CD8+ T cells were capable of lysing T2 target cells pulsed with cognate SLL peptide, and HLA-A*0201+NY-ESO-1+ (LM-MEL 44), but not HLA-A*0201-NYESO-1- (SK-MEL 28) tumor cell lines (figure 4G). Collectively, these data demonstrate successful generation of humanized mice comprised of both human DC subsets and a detectable repertoire of naïve NYESO-1-specific CD8+ T cells that can be primed to proliferate and develop effector function.
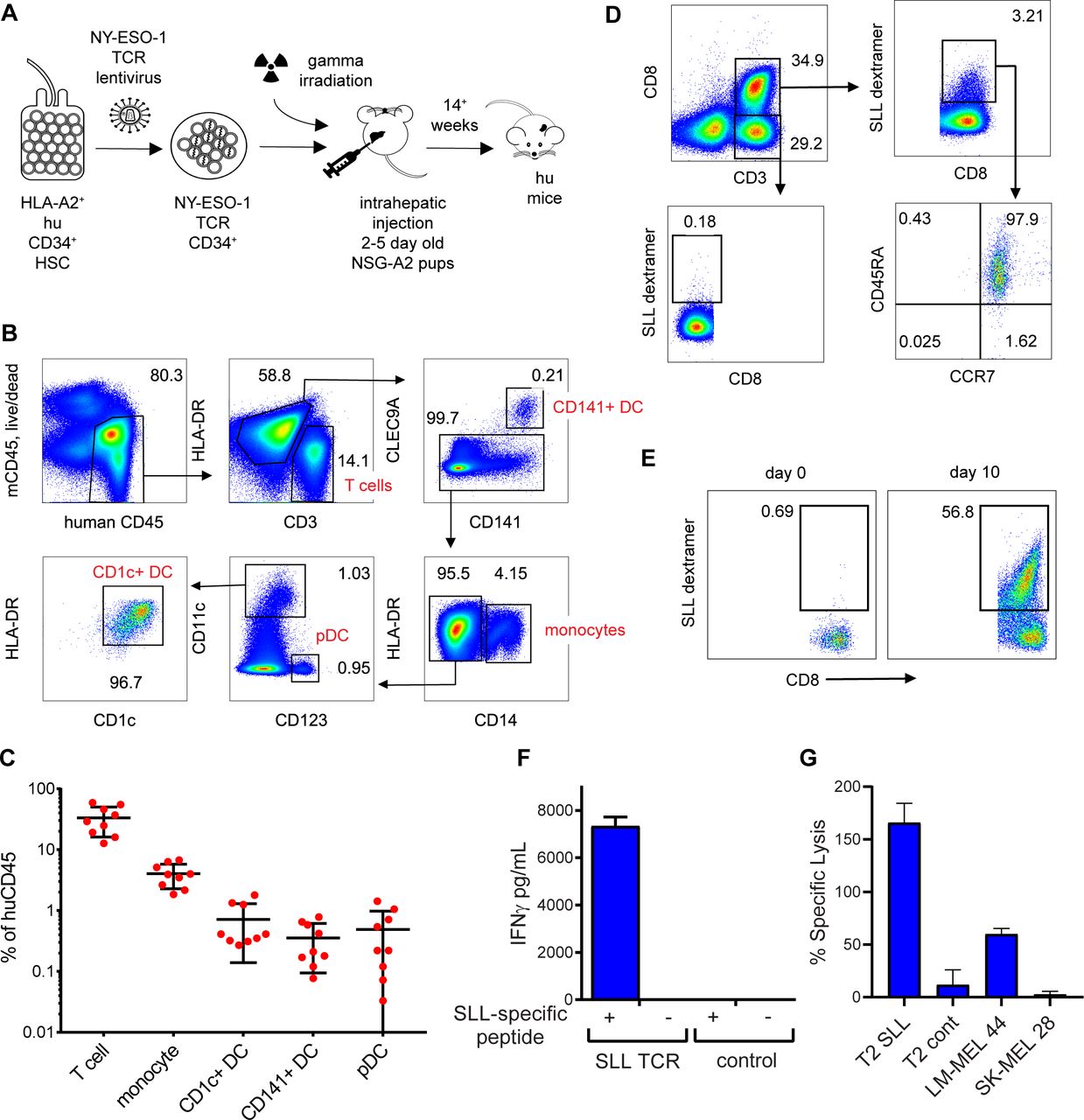
Development of naïve NY-ESO-1 SLL-specific CD8+ T cells in humanized mice. (A) Schematic showing generation of humanized mice following lentiviral transduction of human CD34+ progenitors. (B) Representative flow cytometry plots showing development of human T cell and antigen-presenting cell subsets in the spleens of humanized mice. (C) Frequencies of human hematopoietic subsets expressed as a percentage of human CD45+ cells. (D) Representative flow cytometry plots showing NY-ESO-1 SLL-specific CD8+ T cells (SLL dextramer+) in the spleens of humanized mice, the majority of which express a naïve phenotype (CD45RA+CCR7+). (E) Expansion NY-ESO-1 SLL-specific (SLL dextramer+) CD8+ T cells following 9 days culture of splenocytes from T cell receptor (TCR) lentivirus-transduced mice with SLL peptide-pulsed allogeneic irradiated lymphoblastoid cell lines (LCLs). (F) Interferon γ (IFNγ) production by splenocytes from humanized mice expressing NY-ESO-1 SLL TCR or not (control) following 3 days activation with cognate SLL peptide-pulsed allogeneic irradiated LCLs (+) or no antigen (-). (G) Lysis of SLL or control peptide pulsed T2 targets HLA-A*0201+NY-ESO-1+ (LM-MEL 44) or HLA-A*0201-NY-ESO-1- (SK-MEL 28) target cancer cells by expanded NY-ESO-1 SLL-specific CD8+ T cells. Data are the mean±SD from three replicates. DC, dendritic cell; NY-ESO-1, New York esophageal squamous cell carcinoma 1.
Using this model, we examined the capacity of CLEC9A-NY-ESO-1 to prime naïve NY-ESO-1-specific CD8+ T cells. For these experiments, humanized mice were injected with Flt3L to expand DC subsets, then mice were immunized with chimeric Ab (or HBSS no Ag) along with poly I:C adjuvant (figure 5A). Increased percentages of NY-ESO-1 SLL-specific CD8+ T cells were observed in some of the CLEC9A-NY-ESO-1- and DEC-205-NY-ESO-1-immunized animals compared with the control-NY-ESO-1 vaccinated mice but this was not statistically significant (figure 5B). However, splenocytes from mice immunized with CLEC9A-NY-ESO-1 demonstrated significantly higher capacity to produce IFNγ compared with DEC-205-NY-ESO-1 and control-NY-ESO-1 immunized mice following a short ex vivo reactivation with cognate SLL peptide (figure 5C). This suggested that CLEC9A-NY-ESO-1 could prime naïve CD8+ T cells in vivo.

CLEC9A-NY-ESO-1 primes naïve NY-ESO-1-specific T cells in humanized mice. (A) Experiment schematic. (B) Percentage of NY-ESO-1 SLL-specific CD8+ T cells measured by SLL dextramer 1 week after priming in vivo. Each point represents splenocytes from individual humanized mice. (C) Interferon γ (IFNγ) production by splenocytes from vaccinated mice following ex vivo stimulation with SLL peptide. Data are the combined replicates from three independent experiments, shown as IFNγ spot forming units (SFU) per 50,000 human CD45+ splenocytes. *p=0.005, **p=0.003, ***p=0.002 (two-way analysis of variance and Tukey’s multiple comparison test). Ab, antibody; NY-ESO-1, New York esophageal squamous cell carcinoma 1; poly I:C, polyinosinic:polycytidylic acid.
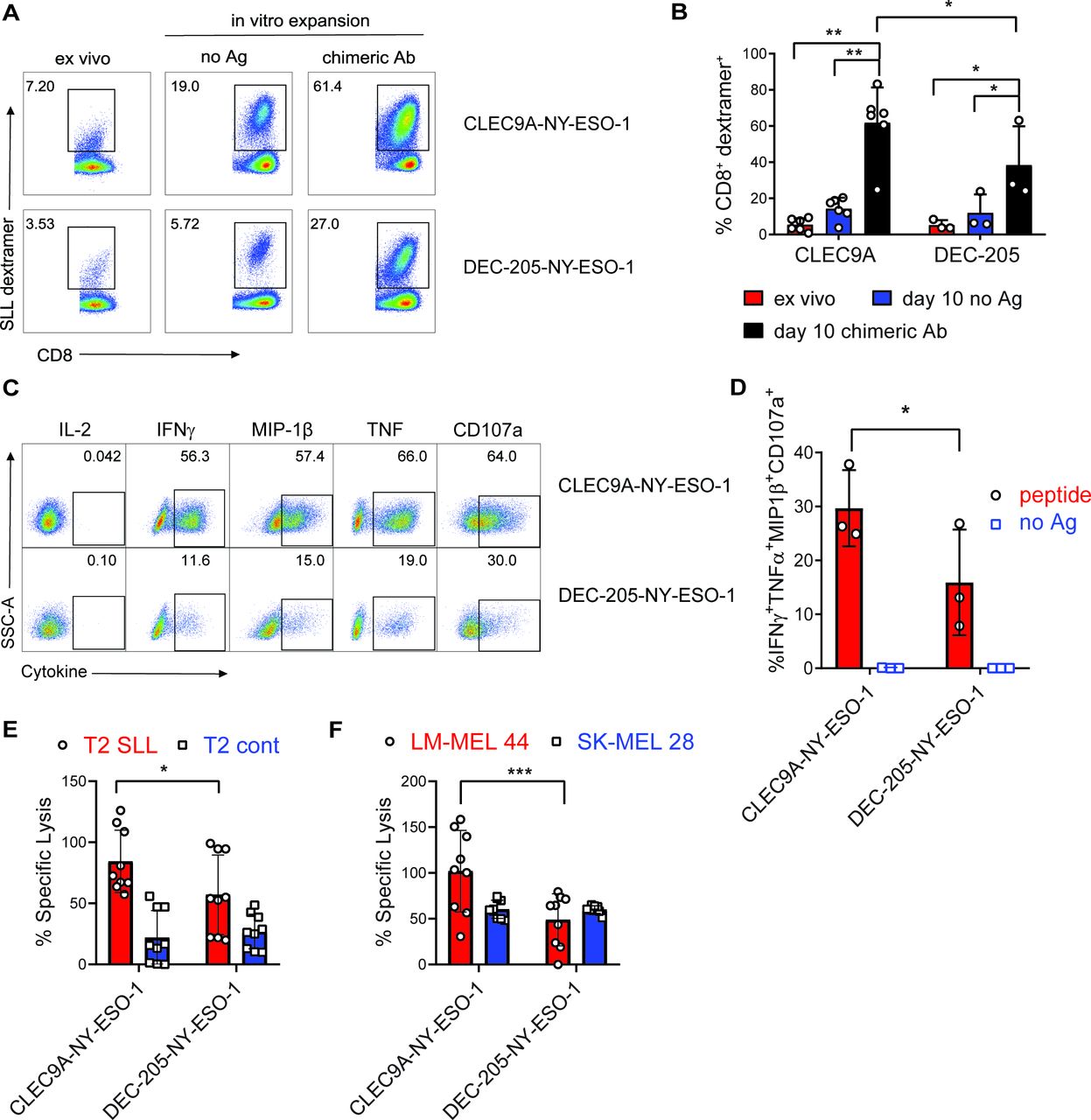
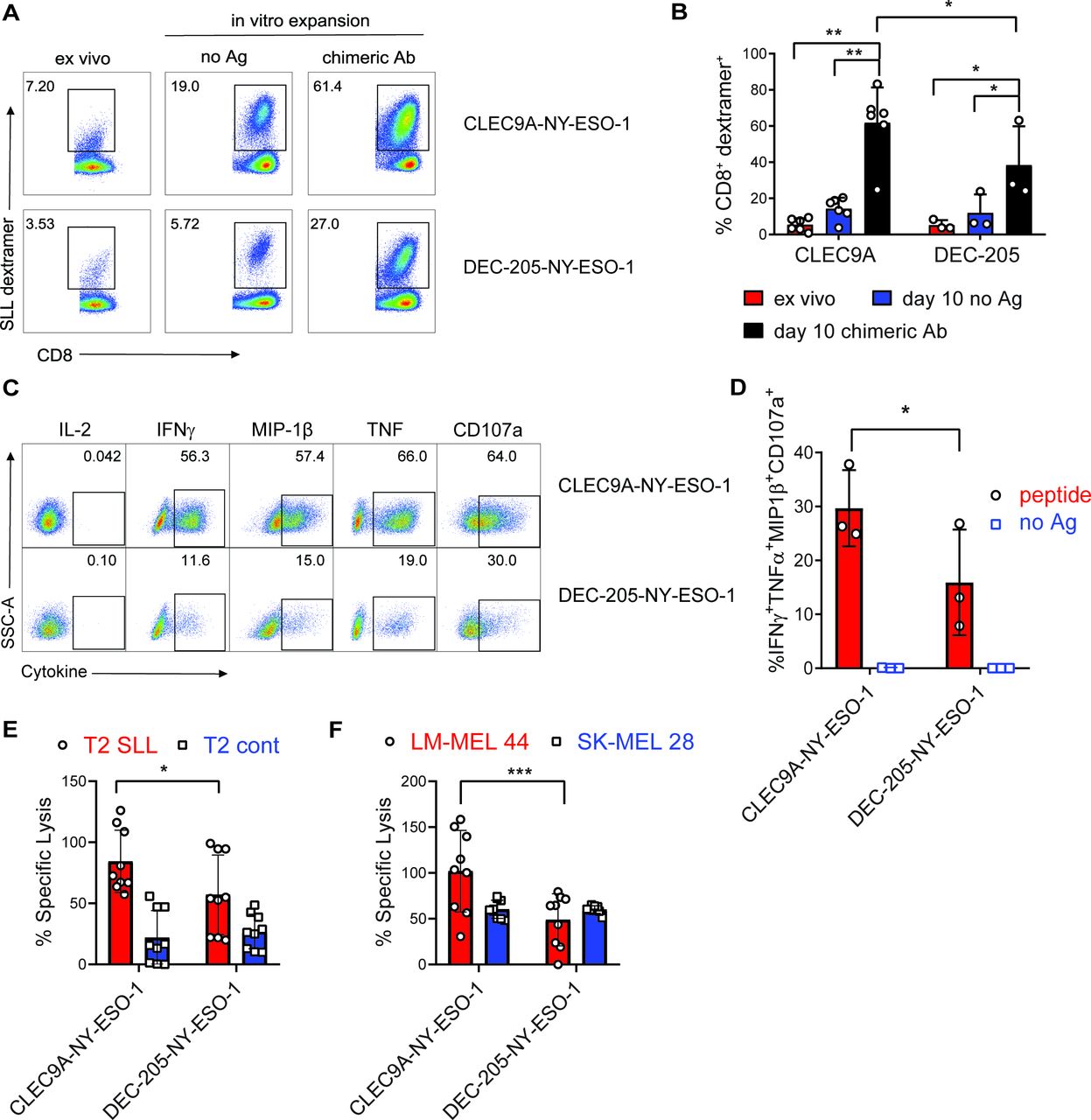
To investigate the effector function of CLEC9A-NYESO-1-primed CD8+ T cells, splenocytes were harvested 1 week after vaccination and restimulated ex vivo with no Ag or the same chimeric Ab they were vaccinated with. NY-ESO-1 SLL-specific CD8+ T cells were expanded from the splenocytes of CLEC9A-NY-ESO-1 or DEC-205-NY-ESO-1 vaccinated mice, with a significantly higher degree of expansion after CLEC9A-NY-ESO-1 immunization (figure 6A,B). Splenocytes from non-immunized control mice failed to expand in vitro and minimal expansion was observed following restimulation with recombinant NY-ESO-1 protein (not shown). CD8+ T cells primed by either CLEC9A-NY-ESO-1 or DEC-205-NY-ESO-1 did not produce IL-2, but expressed cytotoxic degranulation marker CD107a, and produced IFNγ, MIP-1β and TNF following reactivation with SLL cognate peptide (figure 6C, online supplementary figure 3). Boolean gating analysis showed that CD8+ T cells primed with CLEC9A-NY-ESO-1 had a significantly higher proportion of polyfunctional Ag-specific CD8+ T cells than those primed with DEC-205-NY-ESO-1, defined as the percentage of CD8+ T cells coexpressing IFNγ, MIP-1β, TNF and CD107a (figure 6D). Effector function of the CLEC9A-NYESO-1 primed CD8+ T cells was further demonstrated by their capacity to lyse T2 target cells pulsed with cognate SLL peptide and HLA-A*0201+ NY-ESO-1+ melanoma cells (LM-MEL 44), but not T2 cells pulsed with irrelevant peptide (CMV pp65) or HLA-A*0201-NYESO-1- melanoma cells (figure 6E,F). CD8+ T cells primed by DEC-205-NY-ESO-1 were also capable of lysing target cells but were less when compared with those primed with CLEC9A-NY-ESO-1. The percentage of cytokine-producing effector cells and target cell lysis correlated with the percentage of SLL-specific T cells in the cultures, suggesting that CLEC9A-NY-ESO-1 priming stimulates increased expansion rather than T cell quality on a per cell basis. These data demonstrate that CLEC9A-NY-ESO-1 Ab can prime naïve CD8+ T cells to expand and develop into polyclonal effectors.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CLEC9A-NYESO-1 primes naïve NY-ESO-1-specific CD8+ T cells for effector function. Humanized mice were vaccinated with CLEC9A-NY-ESO-1 or DEC-205-NYESO-1 in the presence of poly I:C. (A) Percentage of NY-ESO-1 SLL dextramer+ CD8+ T cells in splenocytes 1 week after vaccination (ex vivo) and again after 10 days in vitro expansion in the presence of no Ag or the same chimeric Ab used for immunization shown as representative plots. (B) Mean percentage of NY-ESO-1 SLL-specific CD8+ T cells±SD from individual mice. (C) Cytokine secretion, from CD8+ T cells that were primed and expanded with CLEC9A-NY-ESO-1 or DEC-205-NY-ESO-1, following a 6-hour restimulation with SLL peptide or no Ag as representative flow cytometry plots for each cytokine. (D) Boolean gating analysis of the percentage of CD8+ T cells producing all four effector molecules (IFNγ, MIP-1β, TNF and CD107a) as the compiled mean±SD from individual mice. (E) Lysis of T2 target cells pulsed with NY-ESO-1 SLL peptide or irrelevant control peptide (NLVPMVATV) pulsed T2. (F) Lysis of HLA-A*0201+ NY-ESO-1+ (LM-MEL-44) and HLA-A*0201-NY-ESO-1- (SK-MEL-28) melanoma cell lines by CD8+ T cells primed and expanded with CLEC9A-NY-ESO-1 or DEC-205-NYESO-1. Compiled replicate means±SD from groups of n=3 individual mice are shown. *p<0.05, **p<0.0001, ***p<0.0005 by two-way analysis of variance and Tukey’s multiple comparison test. Ab, antibody; Ag, antigen; IFNγ, interferon γ; NY-ESO-1, New York esophageal squamous cell carcinoma 1; poly I:C, polyinosinic:polycytidylic acid; TNF, tumor necrosis factor.
Discussion
We report the development of a human CLEC9A-NY-ESO-1 Ab vaccine that specifically delivers NY-ESO-1 to CD141+ DCs for cross-presentation of multiple MHC Class I epitopes. Targeting CD141+ DCs with CLEC9A-NY-ESO-1 Ab resulted in more effective reactivation of NY-ESO-1-specific responses in melanoma patient PBMC compared with either an untargeted control-NY-ESO-1 or DEC-205-NY-ESO-1 Abs. Moreover, we showed that the CLEC9A-NY-ESO-1 Ab primed naïve NY-ESO-1-specific CD8+ T cells leading to expansion of CD8+ T cells with polyclonal effector function that could lyse NY-ESO-1 expressing tumor cells. Collectively, these data demonstrate the potential of CLEC9A-NY-ESO-1 Ab as a vaccine for malignancies that express NY-ESO-1.
CLEC9A-NY-ESO-1 offers advantages over cancer vaccines currently being trialed in the clinic. First, it can be produced as an ‘off-the-shelf’ clinical-grade formulation, thereby circumventing the financial and logistical issues associated with patient-specific vaccines. Second, this vaccine specifically targets the key DC subset required for initiation of tumor-specific immune responses, thereby reducing off-target effects while maximizing potential efficacy.15 16 The human IgG4 isotype, combined with the specificity of CLEC9A-NY-ESO-1 for CD141+ DCs favors retention in the bloodstream, enhancing the longevity of exposure and the potential for T cell activation. Third, when compared with untargeted NY-ESO-1 or targeting more broadly to other Ag presenting cells via DEC-205 Ab, CLEC9A-NY-ESO-1 is superior at reactivating T cell responses across multiple HLA haplotypes, making it widely applicable to many patients.
The generation of CLEC9A-NY-ESO-1 and DEC-205-NY-ESO-1 Abs on the same IgG4 construct enabled a direct comparison of their ability to deliver NY-ESO-1 epitopes for cross-presentation. The immunodominant HLA-A*0201-restricted NY-ESO-1 SLL epitope was cross-presented more efficiently by CD141+ DC following uptake of CLEC9A-NY-ESO-1 compared with DEC-205-NY-ESO-1 or control-NY-ESO-1 Ab. This is consistent with our previous findings demonstrating superior cross-presentation of an immunodominant HLA-A*0201-restricted epitope from CMV pp65 by CLEC9A Ab compared with DEC-205 despite comparable rates of internalization.26 However, the HLA-Cw*03-restricted NY-ESO-1 LAM epitope was cross-presented by CD141+ DC following uptake of either CLEC9A-NY-ESO-1 or DEC-205-NY-ESO-1 Ab. There are several possibilities that might explain this phenomenon. DEC-205 is known to traffic to lysosomes, whereas CLEC9A localizes to early endosomes where cross-presentation is most efficient and prolonged due to higher pH and slower antigen degradation kinetics.16 39 40 The efficiency of cross-presentation from lysosomes following DEC-205 delivery may be related to the positioning of the epitopes relative to the IgG4 heavy chain, with the HLA-Cw*03 LAM epitope, which is proximal to the Ag heavy chain, likely being degraded at a slower rate than the more distal HLA-A*0201 SLL epitope. Second, there may be redundancy in the circumstances under which the HLA-Cw*03 LAM epitope can be cross-presented. In support of this, the HLA-Cw*03 LAM epitope can be cross-presented by MoDC when NY-ESO-1 is formulated with ISCOMATRIX adjuvant but not when delivered as naked protein or conjugated to DEC-205 Ab.35 41 Furthermore, this epitope can be processed by both proteasome dependent and independent mechanisms depending on the mode of delivery.35 Unlike other DC, CD141+ DC are superior at cross-presentation from lysosomes39 and use proteasome-dependent mechanisms.10 Thus, at least for the HLA-Cw*03 LAM epitope and potentially others, there may be flexibility and redundancy in cross-presentation efficiency depending on the DC type, mode of delivery and intracellular trafficking. Others, such as HLA-A*0201 SLL epitope, may be more efficiently processed via the classical cross-presentation pathway in early endosomes, which is maximized by targeting CD141+ DC with CLEC9A Ab.
Regardless of the mechanisms, CLEC9A-NY-ESO-1 Ab was able to efficiently deliver antigen to CD141+ DC for cross-presentation of both HLA-A*0201 SLL and HLA-Cw*03 LAM epitopes. Moreover, in all cancer patients tested, CLEC9A-NY-ESO-1 was more effective than DEC-205-NY-ESO-1 and control-NY-ESO-1 at reactivating NY-ESO-1-specific memory T cells after a short ex vivo stimulation. In most cases, the low frequency of NY-ESO-1-specific memory T cells in patient PBMC, even after vaccination, necessitates in vitro expansion for their detection.42 Thus, CLEC9A targeting may be a highly sensitive approach for the detection of Ag-specific memory T cells ex vivo. Responses to the HLA-A*0201 SLL epitope were detected in only two of the eight HLA-A*0201+ patient samples tested, while none of the 7 HLA-Cw*03+ patient samples had detectable responses to the HLA-Cw*03 LAM epitope after direct ex vivo stimulation. This is consistent with previous findings where responses to NY-ESO-1 vaccines are known to be broad and span multiple epitopes and HLA haplotypes, many being as yet undefined.29 31 Our data therefore suggest that CLEC9A-NYESO-1 Ab can reactivate broad NY-ESO-1-specific memory responses to multiple, potentially unknown NY-ESO-1 epitopes. In contrast, weak responses induced by DEC-205-NY-ESO-1 were detectable in only a few patients, suggesting that cross-presentation of the HLA-Cw*03 LAM epitope by DEC-205-NY-ESO-1 Ab is unlikely to be a general feature of other epitopes. Furthermore, our data suggest that specific targeting of the rare CD141+ DC subset via CLEC9A Ab may be sufficient and more effective than targeting CD141+ DC as well as other more abundant Ag presenting cells via DEC-205 Ab.
The generation of humanized mice reconstituted with human immune cell subsets, including DC subsets and a detectable repertoire of naïve NY-ESO-1 SLL-specific CD8+ T cells allowed us to investigate naïve CD8+ T cell priming. We demonstrated that CLEC9A-NY-ESO-1 could induce priming and expansion of naïve human NY-ESO-1-specific CD8+ T cells, which developed polyclonal effector function and lysed tumor cells in an Ag-specific manner. Although DEC-205-NY-ESO-1 Abs could also prime naïve NY-ESO-1-specific CD8+ T cells with effector function, they were less efficient at CD8+ T cell expansion. These data support the notion that effective CD8+ T cell priming in humans requires cross-presentation by CD141+ DCs.
A potential role for human Ag-presenting cells other than CD141+ DCs in tumor immune responses may not be fully apparent following targeting with DEC-205-NY-ESO-1 Ab. Human CD1c+ DCs, which are more abundant than CD141+ DCs and equivalent to mouse cDC2s, do not efficiently cross-present Ags after delivery via DEC-205 but can cross-present when Ag is delivered to early endosomes, such as via CD40.16 39 43 However, DEC-205 and CD40 Abs conjugated to influenza matrix protein M1 epitopes were equally capable of reactivating M1-specific memory CD8+ T cells in a humanized mouse model, suggesting that cross-presentation by both CD1c+ DCs and CD141+ DCs is unlikely to enhance CD8+ T cell responses in vivo.43 Although we cannot yet exclude a role for CD1c+ DC in the enhancement of tumor-specific CD4+ T cell responses, as was recently shown in mice,44 we have shown that CLEC9A and DEC-205 Abs are similar in their capacity to promote CD4+ T cell responses after uptake by human CD141+ DCs,26 and also to prime HIV-specific CD4+ T cell responses to a similar extent in mice.25
The requirement for adjuvant to enhance T cell priming by vaccines is well established and an important consideration for maximizing CD8+ T cell responses. Priming of naïve NY-ESO-1-specific CD8+ T cells was successful when the CLEC9A-NY-ESO-1 was administered in combination with Flt3L and poly I:C, which we have previously shown to enhance CD141+ DC numbers and activation respectively in humanized mice.45 46 Poly-ICLC (Hiltonol, a stabilized poly I:C derivative) is safe to administer in combination with DEC-205 targeting Ab (CDX-1401) and can induce humoral and cellular immunity in patients with cancer, making this adjuvant a logical choice to combine with CLEC9A-NY-ESO-1 in the clinic.29 30 Although poly I:C combined with a TLR8 agonist can lead to even more potent activation of CD141+ DC in vivo,46 this did not appear to confer any advantage to DEC-205 targeting in the clinical setting.29 Whether Flt3L can enhance responses to vaccination with DEC-205 targeting and poly-ICLC is currently being evaluated in a phase II clinical trial in melanoma (NCT02129075) with early results demonstrating safety and tolerability.47
Adoptive transfer of T cells genetically engineered to express NY-ESO-1-specific TCR induce objective tumor regression in a large majority of patients with melanoma and sarcoma, but few remissions are durable.48 49 Animal models show that T cell expansion and persistence after adoptive transfer can be enhanced when combined with DC vaccines. This is under investigation in the clinic (NCT01946373, NCT01697527, NCT02775292, NCT02070406) with early results demonstrating safety and feasibility.50 Trafficking to the tumor site is another limitation of adoptive T cell therapies in solid tumors, and in mice is mediated by cDC1 DCs.11 12 Thus, CLEC9A-NY-ESO-1 Ab may be an attractive and practical strategy to enhance and sustain responses to adoptive T cell therapies. The key role of mouse cDC1s in responses to radiation therapy, viral therapies and immune checkpoint inhibitors suggests that CLEC9A-NYESO-1 Ab may also enhance immune responses in these settings. Indeed, the induction of humoral and cellular NY-ESO-1 specific responses in patients treated with the CTLA-4 checkpoint inhibitor ipilimumab (NCT01176474, NCT01176461) provides a strong rationale to investigate NY-ESO-1 vaccines in combination with checkpoint inhibitor therapies. Moreover, the triple combination of NY-ESO-1 DC vaccines with checkpoint inhibitors and adoptive T cell therapy is also feasible and well tolerated50 (NCT02775292, NCT02070406). Together, these highlight the potential of CLEC9A-NY-ESO-1 vaccines to enhance immunogenicity and response rates when combined with a variety of immunotherapies and standard treatments in patients with cancers that express NY-ESO-1.
The heterogeneity of NY-ESO-1 expression within tumors and its loss due to promoter hypomethylation is a potential limitation of this Ag as a therapeutic target. One approach to overcoming this is the use of demethylating agent, decitabine, to induce expression of NY-ESO-1, which can be safely used in combination with DEC-205 targeting in patients with high-risk myelodysplastic syndrome.30 Interestingly, this study reported increases in CD141+ DC associated with vaccine responses, providing a further rationale for specifically targeting them via CLEC9A-NY-ESO-1 and a rationale to explore combinatorial therapies with decitabine. CLEC9A Ab can also be readily fused to other Ag to maximize CD8+ T cell enhancing potential.
Our data advocate the potential of human CLEC9A-NY-ESO-1 as an attractive new vaccine, that will be broadly applicable to many cancer types and appropriate to combine with a variety of other therapies to enhance tumor immunogenicity.
Acknowledgments
We thank Robyn Rodwell and staff at the Queensland Cord Blood Bank (Mater Hospitals, Brisbane, Australia), and Teoni Hale (Mater Research Institute, Brisbane, Australia) for consenting donors and collecting UCB samples, and staff at the Translational Research Institute Flow Core and Biological Research Facilities (Mater Research Institute, Brisbane, Australia). Dr Volker Lennerz and Dr Thomas Wölfel (University Medical Center Langenbeckstr. 1, D-55131 Mainz, Germany) mapped the novel HLA-B*5601-restricted NY-ESO-1 93-102 epitope.
References
Footnotes
K-AM and OLH contributed equally.
Contributors K-AM, OLH and IML-R designed and performed experiments, analyzed data and drafted the manuscript. KMT and MHL designed and generated the chimeric antibodies. FEP, CW, LO, NR and GD contributed to data acquisition and analysis. DBK, RPH and EG generated the TCR-encoding lentivirus and provided protocols and expertise for human CD34+ HSC transduction. JC and CS provided T cell and tumor cell lines, patient PBMC and expertise on NY-ESO-1. KJ conceived, designed and performed experiments, analyzed and interpreted data, and wrote the manuscript. All authors read and approved the final manuscript.
Funding This work was supported by the Assistant Secretary of Defense for Health Affairs through the Prostate Cancer Research Program under Award No. W81XWH-15-1-0451. Opinions, interpretations, conclusions and recommendations are those of the author and are not necessarily endorsed by the Department of Defense. KJR is supported by the Mater Foundation, Brisbane, Australia and project grants from the National Health and Medical Research Council of Australia (NHMRC). FEP was supported by Worldwide Cancer Research UK (grant 15-0181). KMT was supported by a Cancer Council Australia Postdoctoral Fellowship. MHL and IC are supported by project grants from the NHMRC. This work was made possible through Victorian State Government Operational Infrastructure Support and Australian Government NHMRC Independent Research Institute Infrastructure Support Scheme. The Translational Research Institute is supported by a grant from the Australian Government.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Written informed consent was obtained for human sample acquisition in line with standards established by the Declaration of Helsinki. Study approval was granted by the Mater Human Research Ethics Committee (HREC13/MHS/83 and HREC13/MHS/86) and The US Army Medical Research and Materiel Command (USAMRMC) Office of Research Protections, Human Research Protection Office (HRPO; A-18738.1, A-18738.2, A-18738.3). All animal experiments were approved by the University of Queensland Animal Ethics Committee and conducted in accordance with the Australian Code for the Care and Use of Animals for Scientific Purposes in addition to the laws of the USA and regulations of the Department of Agriculture.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.