Article Text

Abstract
Background Many cancer cells express a major histocompatibility complex class I low/ programmed cell death 1 ligand 1 positive (MHC-Ilo/PD-L1+) cell surface profile. For immunotherapy, there is, thus, an urgent need to restore presentation competence of cancer cells with defects in MHC-I processing/presentation combined with immune interventions that tackle the tumor-initiated PD-L1/PD-1 signaling axis. Using pancreatic ductal adenocarcinoma cells (PDACCs) as a model, we here explored if (and how) expression/processing of tumor antigens via transporters associated with antigen processing (TAP) affects priming of CD8 T cells in PD-1/PD-L1-competent/-deficient mice.
Methods We generated tumor antigen-expressing vectors, immunized TAP-competent/-deficient mice and determined de novo primed CD8 T-cell frequencies by flow cytometry. Similarly, we explored the antigenicity and PD-L1/PD-1 sensitivity of PDACCs versus interferon-γ (IFN-γ)-treated PDACCs in PD-1/PD-L1-competent/deficient mice. The IFN-γ-induced effects on gene and cell surface expression profiles were determined by microarrays and flow cytometry.
Results We identified two antigens (cripto-1 and an endogenous leukemia virus-derived gp70) that were expressed in the Endoplasmic Reticulum (ER) of PDACCs and induced CD8 T-cell responses either independent (Cripto-1:Kb/Cr16-24) or dependent (gp70:Kb/p15E) on TAP by DNA immunization. IFN-γ-treatment of PDACCs in vitro upregulated MHC-I- and TAP- but also PD-L1-expression. Mechanistically, PD-L1/PD-1 signaling was superior to the reconstitution of MHC-I presentation competence, as subcutaneously transplanted IFN-γ-treated PDACCs developed tumors in C57BL/6J and PD-L1-/- but not in PD-1-/- mice. Using PDACCs, irradiated at day 3 post-IFN-γ-treatment or PD-L1 knockout PDACCs as vaccines, we could selectively bypass upregulation of PD-L1, preferentially induce TAP-dependent gp70:Kb/p15E-specific CD8 T cells associated with a weakened PD-1+ exhaustion phenotype and reject consecutively injected tumor transplants in C57BL/6J mice.
Conclusions The IFN-γ-treatment protocol is attractive for cell-based immunotherapies, because it restores TAP-dependent antigen processing in cancer cells, facilitates priming of TAP-dependent effector CD8 T-cell responses without additional check point inhibitors and could be combined with genetic vaccines that complement priming of TAP-independent CD8 T cells.
- CD8-positive T-lymphocytes
- antigen presentation
- immunogenicity
- vaccine
- B7-H1 antigen
- gastrointestinal neoplasms
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- CD8-positive T-lymphocytes
- antigen presentation
- immunogenicity
- vaccine
- B7-H1 antigen
- gastrointestinal neoplasms
Background
CD8 T cells play a crucial role for the control of many human and murine tumors.1 In an optimal scenario, the immune system specifically detects and eliminates aberrant tumor progenitor cells before they develop into clinically manifested tumors.2 This implied that tumor cells per se are immunogenic and can present tumor-associated antigens (TAAs) to the immune system in a manner that allows priming of effector CD8 T-cell responses. However, many tumors developed mechanisms to escape from recognition and elimination by CD8 T cells. Downregulation or loss of molecules involved in MHC-I processing/presentation was evident in many primary human and murine cancer cells, thereby limiting their presentation competence to CD8 T cells.3 Furthermore, PD-L1 expression on tumor cells could function as a molecular shield and directly silence effector T-cell responses through coinhibitory PD-L1/PD-1 interactions.4 On the other hand, there was evidence that interferon-γ (IFN-γ) producing CD8 T cells could upregulate PD-L1 on tumor cells, indicating that a gradual increase in immune-suppressive PD-L1 signaling and/or the efficacy of anti PD-L1/PD-1 therapies may depend on initial CD8 T cell/tumor cell interactions.5–7 However, tumors like pancreatic ductal adenocarcinoma (PDAC) also generate an immune-suppressive microenvironment, composed among others of PD-L1 expressing myeloid-derived suppressor cells and/or regulatory CD4+ Foxp3+ Tregs that may enhance the downregulation of tumor-specific CD8 T-cell responses and/or affect the limited responsiveness of murine and human PDAC to antibody-mediated PD-L1/PD-1 check point inhibition.8–10
The lack of presentation-competent MHC-I molecules on the surface of cancer cells depends on multiple factors, like alterations in MHC-I or beta-2 microglobulin expression and/or in the endogenous antigen processing/presentation machinery, that primarily affect the generation of antigenic peptides in the cytosol by the proteasome complex, or the import of these peptides from the cytosol into the Endoplasmic reticulum (ER) and their loading to nascent MHC-I molecules by transporters associated with antigen processing (TAP).11 In particular, TAP could be a central player for priming tumor-specific CD8 T cells, as some antigens/epitopes (ie, T cell epitopes associated with impaired peptide processing; TEIPPs) were even exclusively presented under MHC-I presentation-prohibiting conditions in TAP-deficient tumors.11 12 A number of TAP-independent pathways have been described for secretory or cell surface associated antigens that are initially targeted to the ER, for example, antigens that contain NH2-terminal ER-targeting signal peptide (SP) sequences.11 13 14 SP and transmembrane helices contain a high density of MHC-I epitopes that could be released in the ER or the trans Golgi network by various processing pathways, for example, involving ER-resident signal peptidase (SPase), SP peptidase, furin or ER aminopeptidases.15 16 In particular, expression and processing of self-antigens in the ER could improve their MHC-I presentation efficacy and antigenicity to prime CD8 T cells, at least, when the antigen processing machinery in the ER generates epitopes with free carboxyl (COOH)-termini that do not require further COOH-terminal trimming before loading to MHC-I molecules.14 17–19 Generation/presentation of these epitopes often was independent of proteasomes and/or TAP.16–18 However, antigens expressed in the ER and translocated into the cytosol, for example, by cross-presentation could also be processed/presented in a TAP-dependent manner.16 20 Little is known if (and how) processing of tumor antigens in the ER and/or in TAP-dependent/-independent pathways affects epitope presentation by MHC-Ilo cancer cells and/or priming of effector CD8 T cells.
Here, we analyzed two central points for CD8 T cell-mediated immunotherapies: (1) processing/presentation requirements of TAAs that allow epitope presentation and CD8 T-cell priming, and (2) immune-suppressive PD-L1-signaling requirements of tumor cells that allow expansion and/or maintenance of functional effector CD8 T cells. To determine if TAP-dependent/-independent antigen processing of two tumor antigens/epitopes expressed in the ER of pancreatic ductal adenocarcinoma cells (PDACCs) affects priming of CD8 T cells, that is, the Cr-1:Kb/cr16-24 epitope from the embryonic antigen Cripto-1 (Cr-1)21 22 and the gp70:Kb/p15E epitope from an endogenous leukemia virus-derived gp70 glycoprotein,23 we immunized TAP-competent/-deficient mice with antigen-expressing vector DNAs.24 Furthermore, we restored MHC-I presentation competence in PDACCs in vitro by IFN-γ-treatment and primed tumor-specific effector CD8 T-cell responses in PD-1/PD-L1-competent/-deficient mice by cell-based immunization. Overall, we established a novel IFN-γ-treatment protocol for PDACCs that restored their MHC-I presentation competence, curtailed the coinhibitory PD-L1/PD-1 signaling axis and allowed priming of TAP-dependent effector CD8 T cells.
Materials and methods
Mice
C57BL/6J (B6) mice were obtained from Janvier (Le Genets-St-Isle; France). RAG1-/- (B6.129S7-Rag1tm1Mom/J; Jackson #002216), TAP1-/- (B6.129S2-Tap1 tm1Arp /J; Jackson #002944), PD-L1-/- 25 and PD-1-/- mice26 were bred and kept under standard pathogen-free conditions in the animal colony of Ulm University. KPC mice were generated by breeding B6.129S2-Trp53tm1Tyj /J (Jackson #002109), LSL-KrasG12D/+ 27 and p48 Cre/+ mice.28
Cell lines
HEK-293 (CRL-1573) and MC38 (CRL-2639) cell lines were obtained from the American Tissue Culture Collection (ATCC, Rockville, Maryland, USA). The murine KPC tumor cell lines were obtained from PDAC developed in KPC mice.29 Briefly, PDAC were digested with collagenase D, trypsinized and passed through a 40 µm cell strainer (passage 0). Five cell lines were generated from individual mice/tumors, expanded in vitro for 3–4 passages, tested for KrasG12D/+ , p53 and Cre expression by PCR and frozen in liquid nitrogen. Cells from frozen bulk stocks were expanded in vitro for about 4–6 weeks and used in the experiments. All cell lines were tested ‘free of mycoplasma’ (PCR Mycoplasma Test Kit; cat. no. A3744, AppliChem, Darmstadt, Germany).
Construction of expression plasmids and characterization of antigen expression
The antigenic sequences of Cr-1 and gp70 were synthesized (codon optimized) by GeneArt (Regensburg, Germany) and cloned into the pCI vector (cat. no. E1731, Promega, Mannheim, Germany) using the NheI and NotI restriction sites. All antigen modifications were carried out using the Q5 Site-Directed Mutagenesis Kit (cat. no. E0554, NEB, Ipswich, USA). Batches of DNA were produced in Escherichia coli using the Qiagen Plasmid Mega Kit (cat. no. 12183; Qiagen, Hilden, Germany). Expression of vector-encoded antigens was tested in transiently transfected HEK-293 cells as described previously.30
Immunization of mice and tumor models
Mice were immunized intramuscularly into both tibialis anterior muscles with 100 µg/mouse DNA or transplanted subcutaneously into the left/right flank with 2.5×105 tumor cells (in 100 µL PBS). For cell-based immunizations, tumor cells were pretreated with recombinant mouse IFN-γ (20 ng/mL, cat. no. 554587, BD Biosciences, Heidelberg, Germany) for 16–20 hours, washed and cultured for indicated times before gamma irradiation (30 Gy). Where indicated, anti-PD-1 (cat. no. BP0146; Bio X Cell), anti-CD8 (cat. no. BE0117; Bio X Cell), anti-CD4 (cat. no. BE0119; Bio X Cell) and rat IgG2a or IgG2b isotype control antibodies (cat. no. BP0089, BE0090; Bio X Cell) were injected intraperitoneally (100 µg/mouse). Tumor growth was monitored by regular palpation with calipers. Mice were sacrificed when the tumor diameter reached 1 cm. Determination of antigen-specific CD8 T-cell frequencies by flow cytometry (FCM) was carried out as described previously.30
Gene expression analyses
Total RNA was isolated from five individual cell lines. The quality was analyzed with a bioanalyzer (Agilent Technologies, Santa Clara, USA). Gene expression analysis was carried out using the SurePrint G3 Mouse Gene Expression 8×60K Microarray (Design ID 028005; Agilent Technologies). Samples were labeled with the Low Input Quick Amp Labeling Kit (Agilent Technologies) according to the manufacturer’s guidelines. Slides were scanned using a G2565CA microarray scanner (Agilent Technologies). Raw data files were extracted using the Feature Extraction software (v.10.7.3.1, Agilent Technologies). Raw microarray data were background corrected and quantile normalized using the R and Bioconductor package limma (https://www.R-project.org).31 32 After filtering control probes and low expressed probes, differential expression analysis was performed using limma.
Determination of MHC-I and PD-L1 expression on cancer cells
Expression of MHC-I (H2-Kb) and PD-L1 on established KPC cell lines and KPC cells isolated ex vivo from subcutaneously growing tumors was characterized by FCM using the following antibodies: PE Rat anti-mouse H2-Kb (cat. no. 116508, BioLegend), PE Rat anti-mouse CD274 (cat. no. 558091, BD Biosciences) and PE Rat IgG2a isotype control (cat. no. 12-4321-83, eBioscience). To exclude contaminations with hematopoietic cells in ex vivo isolated tumor cell populations, CD45.2+ and CD11c+ cells were electronically gated and excluded. This ensured that histograms reflect H2-Kb and PD-L1 levels on tumor cells, satisfying the respective forward/sideward (FSC/SSC) features.33
Statistics
Data were analyzed using PRISM software (GraphPad, San Diego, USA). Unless mentioned otherwise, all column scatter plots show respective frequencies±SD. Where indicated, the statistical significance of differences in the mean cell frequencies between multiple groups was determined by analysis of variance (ANOVA) (with 95% CI) followed by Tukey’s multiple comparisons test. Alternatively, differences between two groups were evaluated using the unpaired student’s t-test. The p<0.05 (*), p<0.01 (**), p<0.001 (***), p<0.0001 (****) values were considered significant.
Results
PDACCs established from highly aggressive tumors in KrasG12D/+/Trp53fl/fl/ p48 C re/+ (KPC) mice29 barely express MHC-I molecules on the cell surface (online supplementary figure S1a). We here asked whether tumor antigens expressed in the ER of these PDACCs and processed in TAP-independent/-dependent pathways are preferentially presented to CD8 T cells under these conditions.
Supplemental material
TAP-dependent/-independent priming of CD8 T cells by TAAs
We initially analyzed the Cr-1 antigen (Cr-1 or TDGF-1; www.uniprot.org/uniprot/P51865) that is expressed during embryonic development in men and mice and re-expressed in KPC-derived PDACCs (online supplementary figure S1b,c) and in about 44% of human PDAC.21 22 This antigen contains a single Cr-1:Kb/cr16-24 epitope located at the extreme COOH-terminus of its ER-targeting SP (SPCr-1),22 which is released during Cr-1 maturation by ER-resident SPase (figure 1A). To determine TAP-dependent/-independent presentation of the Cr-1:Kb/cr16-24 epitope, we used a DNA immunization approach. For this, we generated a Cr-1-encoding vector (pCI/Cr-1; figure 1B) and determined Cr-1 expression in transiently transfected HEK-293 cells. Endoglycosidase H treatment of cell lysates showed that Cr-1 was mainly expressed in an about 20 kDa glycosylated gpCr-1 form (figure 1C), confirming an almost quantitative SPCr-1-mediated translocation of Cr-1 into ER/Golgi compartments. A single injection of pCI/Cr-1 efficiently primed Cr-1:Kb/cr16-24-specific CD8 T cells in B6 and TAP-deficient (TAP1-/-) mice, detectable in liver and spleen by ex vivo staining with Cr-1:Kb/cr16-24 dimers or after peptide-specific stimulation and determination of IFN- γ + Cr-1:Kb/cr16-24-specific CD8 T-cell frequencies by FCM (figure 1D; groups 2). Priming of Cr-1:Kb/cr16-24-specific CD8 T cells thus did not require TAP-mediated antigen processing.

TAP-independent induction of Cr-1:Kb/cr16-24-specific CD8 T cells by DNA immunization. (A) Map of the Cripto-1 (Cr-1) protein. The NH2-terminal signal peptide (SPCr-1), the epidermal growth factor (EGF)-like domain, the Cripto_Frl-1_Cryptic domain (CFC) and the glycosylphosphatidylinositol anchor (GPI) are indicated. The amino acid sequences of SPCr-1, SPCr-1/R26P, the Kb/cr16-24 epitope and the predicted SPase cleavage site are shown. (B) Schematic presentation of pCI/Cr-1, pCI/Cr-1R26P (Cr-1 containing an arginine to proline exchange at position 26) and of pCI/stT77Cr-1 (Cr-1 containing a strep-tag (ST) and Hsp73-capturing SV40 T1-77 (T77) domain). (C) lysates of non-transfected HEK-293 cells and HEK-293 cells transiently transfected with pCI/Cr-1 (lanes 1), pCI/Cr-1R26P (Lane 2) or pCI/stT77Cr-1 (lanes 3) were subjected to SDS-PAGE followed by beta-actin-, Cr-1- or st-specific Western blotting. Where indicated, cell lysates were treated with endoglycosidase H (E-Hf +) or remained untreated (E-Hf -) prior to SDS-PAGE. The positions of glycosylated (gp) and non-glycosylated forms of Cr-1 are indicated. (D) B6 mice were immunized with pCI (group 1, n=6, in two independent experiments), pCI/Cr-1 (group 2, n=6, in two independent experiments), pCI/Cr-1R26P (group 3, n=3, one experiment) or pCI/stT77-Cr-1 (group 4, n=3, one experiment) and TAP1-/- mice (n=6, in two independent experiments) were injected with PCI (group 1) or pCI/Cr-1 (group 2). Frequencies of dimer+ Kb/cr16-24 specific CD8 T cells in liver and spleen of immunized mice were determined ex vivo by FCM at day 12 postinjection. Additionally, antigen-specific IFN-γ+ CD8 T cells were determined by ex vivo stimulation of spleen cells with Kb/cr16-24 (closed circles) and control Kb/ova257-264 peptides (open circles). Graphs show the actual proportions (%) of Kb/cr16-24-dimer+ or IFN-γ+ cells of total CD8+ T cells±SD for statistical evaluation, indicated groups of mice were compared using one-way ANOVA (with 95% CI) followed by Tukey’s multiple comparisons test (B6) or by the unpaired Student’s t-test (TAP-/-). Only statistically significant differences are indicated (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). ANOVA, analysis of variance; Cr-1, Cripto-1; FCM, flow cytometry; IFN-γ, interferon-γ; SPase, signal peptidase; SP, signal peptide; TAP, transporter associated with antigen processing.
It has been reported that introduction of a proline (P) immediately following the SP cleavage sites (position +1) generated non-cleavable antigen variants.34 The SP identification program SignalP V. 5.0 (http://www.cbs.dtu.dk/services/SignalP/data.php)35 predicted a cut at position 26 of the murine SPCr-1, generating an epitope-containing SP1-25 fragment (figure 1A). To confirm the crucial role of ER-resident SPase for priming Cr-1:Kb/cr16-24-specific CD8 T cells, we generated a pCI/Cr-1R26P vector that contained an amino acid exchange at Cr-126 from arginine (R) to proline (P) (figure 1A,B). This vector expressed an about 22 kDa gpCr-1R26P glycoprotein that migrated above the gpCr-1 protein band in SDS-PAGE, demonstrating that the SPCr-1 SP was not cleaved off in this protein (figure 1C, lane 2). As a consequence, we could not induce Kb/cr16-24-specific CD8 T cells in B6 mice by pCI/Cr-1R26P immunization (figure 1D, group 3). Expression/processing of Cr-1 in the ER of antigen-presenting cells (APCs) targeted by DNA immunization was, thus, a prerequisite to induce Cr-1:Kb/cr16-24-specific CD8 T cells. Confirmatory, targeting Cr-1 to the cytosol, using a NH2-terminal, Dna-J homologous SV40/T77 domain that stably binds cytosolic Hsp73 in transfected cells36 37 (pCI/stT77Cr-1) (figure 1B,C; online supplementary figure S2) did not induce Cr-1:Kb/cr16-24-specific CD8 T cells in vaccinated B6 mice (figure 1D; group 4).
Supplemental material
Next, we searched for an epitope-containing tumor antigen that, in contrast to Cr-1, is expressed in the ER of PDACCs but processed for MHC-I presentation and CD8 T cell priming in a TAP-dependent manner. We identified the glycoprotein 70 (gp70), originating from an endogenous murine leukemia virus integrated as a provirus in the mouse germline DNA (www.uniprot.org/uniprot/P03386), which fulfilled these criteria. Gp70 was stably expressed in PDACCs (online supplementary figure S1b,c) but not in somatic cells.23 30 38 In contrast to Cr-1:Kb/cr16-24, the gp70:Kb/p15E epitope is not located in the ER-targeting SP (SPgp70) (figure 2A) and its processing/presentation apparently depends on proteasomal processing39 and TAP.38 Therefore, this antigen/epitope needs retrotranslocation from the ER to the cytosol and/or ‘cross-presentation’ for efficient MHC-I processing/presentation.20 40 Neither full-length gp70 nor epitope-containing gp70 fragments induced CD8 T cells in H-2d BALB/c or H-2b B6 mice.30 41 42 Confirmatory, we here show that an ER-targeted SPIgκstgp70 antigen, composed of a gp70327-615 fragment and a NH2-terminal heterologous Igκ SP sequence (figure 2A,B), did not induce gp70:Kb/p15E-specific CD8 T cells in B6 mice (figure 2C; groups 2). In contrast, a cytosolic Hsp73-bound stT77gp70 fusion antigen (pCI/stT77gp70) (figure 2A,B; online supplementary figure S2), associated with a high cross-priming capacity,37 43 induced high frequencies of gp70:Kb/p15E-specific CD8 T cells in B6 but not in TAP1-/- mice (figure 2C; groups 3). Priming of gp70:Kb/p15E and Cr-1:Kb/cr16-24-specific CD8 T cells thus differed substantially, depending on expression of tumor antigens in the ER versus the cytosol (figures 1 and 2; online supplementary table 1). Furthermore, Cr-1:Kb/cr16-24- and gp70:Kb/p15E-specific CD8 T cells were functional in B6 mice and almost quantitatively and specifically eliminated CFSE-labeled APCs pulsed with Cr-1:Kb/cr16-24 (online supplementary figure S3) or gp70:Kb/p15E peptides30 in in vivo cytotoxicity assays. This suggested that priming of these TAA-specific CD8 T cells in B6 mice was not restricted by central and peripheral tolerance mechanisms.
Supplemental material
Supplemental material
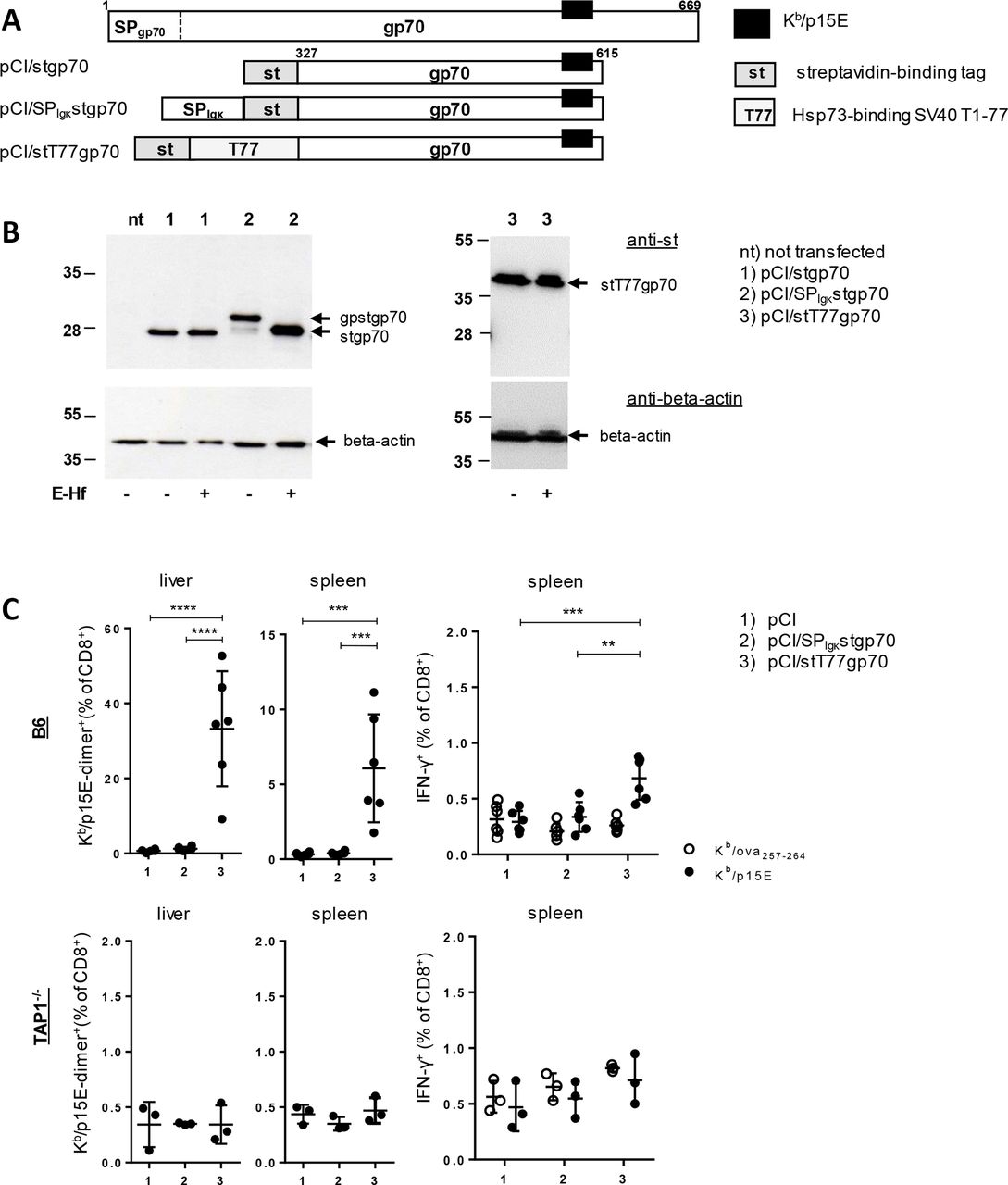
TAP-dependent induction of gp70:Kb/p15E-specific CD8 T cells by DNA immunization. (A) Schematic presentation of the gp70 protein and its variants stgp70 (a 289-residue gp70327-615 fragment with a NH2-terminal strep-tag domain), SPIgκstgp70 (stgp70 with an additional NH2-terminal ER-targeting Igκ signal peptide (SPIgκ)) and stT77gp70 (stgp70 with a 77-residue Hsp73- binding T77 domain between ST and gp70). The position of the Kb/p15E epitope is indicated. (B) Cell lysates of nt HEK-293 cells or cells transiently transfected with pCI/stgp70 (lanes 1), pCI/SPIgκstgp70 (lanes 2) or pCI/stT77gp70 (lanes 3) were subjected to SDS-PAGE followed by anti-st (upper panels) and anti-beta-actin (lower panels) specific Western blotting. Prior to SDS-PAGE, cell lysates were treated with endoglycosidase HF (E-Hf +) or remained untreated (E-Hf -). The positions of glycosylated (gp) and non-glycosylated forms of gp70 are indicated. (C) B6 mice (upper panels, data from two independent experiments) and TAP1-/- mice (lower panels, one experiment) (n=3–6) were immunized with pCI (group 1), pCI/SPIgκstgp70 (group 2) or pCI/stT77gp70 (group 3). Twelve days postimmunization, frequencies of Kb/p15E-dimer+ CD8 T cells in spleen and liver were determined by FCM. Additionally, antigen-specific IFN-γ production of CD8 T cells was determined by ex vivo stimulation of spleen cells with Kb/p15E (closed circles) and control Kb/ova257-264 peptides (open circles). Graphs show the actual proportions (%) of Kb/p15E-dimer+ or -IFN-γ+ CD8 T cells of total CD8 T cells±SD for evaluation of the statistical significance of differences between indicated groups, one-way ANOVA (with 95% CI) followed by Tukey’s multiple comparisons test was used. Only significant differences are depicted (**p<0.01, ***p<0.001, ****p<0.0001). ANOVA, analysis of variance; Cr-1, Cripto-1; ER, endoplasmic reticulum; FCM, flow cytometry; IFN-γ, interferon-γ; TAP, transporter associated with antigen processing.
Manipulation(s) of KPC cells for their usage as cell-based vaccines
A major aim was to explore whether tumor cell vaccines could be used to prime anti-tumor-specific CD8 T-cell responses. We, thus, asked if MHC-I expression can be restored on KPC tumor cells and affect priming of TAP-independent (Cr-1:Kb/cr16-24) or TAP-dependent (gp70:Kb/p15E) CD8 T-cell responses, and if tumor cell-initiated immune-suppressive (eg, PD-L1-mediated) mechanisms interfere with these immune responses.
In line with established data, we could upregulate MHC-I expression on different KPC cells by an in vitro treatment with IFN-γ44 (figure 3A). Microarrays performed on untreated and IFN-γ-treated KPC cells identified 291 significant differentially expressed probesets (Benjamini-Hochberg adjusted p<0.05). Among those probesets, 289 probesets (corresponding to 238 genes) were upregulated and only one gene (two probesets) was downregulated in IFN-γ-treated cells (figure 3B; online supplementary table 2). Unsupervised hierarchical clustering-based heatmap of the differentially expressed genes showed clear segregation of untreated and IFN-γ-treated cells (figure 3B). Furthermore, among 91 genes present in the KEGG pathway ‘Antigen processing and presentation’ (mmu04612) 27 genes were significantly upregulated; for example, TAP1 and TAP2 (figure 3C). In line with these findings, Gene Set Enrichment Analysis based on the REACTOME database showed significant upregulation of 105 gene sets in IFN-γ-treated cells (FDR<0.05) including ‘Antigen processing and presentation’ and ‘IFN-γ signaling’ (FDR q value<0.001; p<0.001) (online supplementary figure S4a). In particular, the upregulation of TAP1 was found within the top three leading-edge genes based on the immunological signatures database (online supplementary figure S4b). However, these analyzes also revealed a significant upregulation of coinhibitory PD-L1 (CD274) in IFN-γ-treated KPC cells (online supplementary table 2), correlating with a prominent upregulation of PD-L1 on the cell surface (figure 3A).
Supplemental material
Supplemental material

IFN-γ-induced gene expression in KPC cell lines. (A) Different KPC cell lines were either untreated or treated with 20 ng/mL recombinant mouse IFN-γ for 20 hours, followed by FCM analysis of H2-Kb and PD-L1 surface expression. One representative histogram per cell line out of three independent experiments is shown. (B) Unsupervised hierarchical clustering based on differentially expressed probesets by comparing five IFN-γ-treated (for 20 hours) KPC cell lines (+IFN-γ) to the respective untreated KPC cell lines (ctrl). Red represents upregulated probesets while blue shows downregulated ones in the IFN-γ-treated (+IFN-γ) group. The samples clearly clustered based on the experimental arms (pink=ctrl; light blue=+IFN-γ). (c) KEGG ‘Antigen processing and presentation’ (mmu04612) gene expression profile in IFN-γ treated KPCs (+IFN-γ) to untreated KPCs (ctrl). We identified 27 genes that showed significant differential expression (BH adjusted p<0.05) and are listed to the right of the heatmap. FCM, flow cytometry; IFN-γ, interferon-γ.
KPC7735 cells and IFN-γ-pretreated KPC7735 cells efficiently grafted as solid tumors in transplanted B6 mice (figure 4A). This suggested that upregulation of co-inhibitory PD-L1 on IFN-γ-treated KPC7735 cells was sufficient to prevent CD8 T cell-mediated rejection of transplanted tumors through direct activation of the PD-L1-signaling axis with PD-1+ CD8 T cells. To confirm this assumption, we transplanted KPC7735 cells into B6 mice and treated them with anti-PD-1 antibody. Anti-PD-1 but not isotype control antibodies inhibited the outgrowth of KPC7735 tumors in three out of six animals (figure 4B), indicating that transplanted KPC7735 cells per se induced CD8 T cells that unleashed their effector functions only after blocking PD-L1/PD-1 interactions.45 Confirmatory, untreated KPC7735 cells and, with a higher robustness, IFN-γ-treated KPC7735 cells induced an antitumor immunity that rejected tumor transplants in PD-1-/- mice (figure 4C). Comparable to B6 mice, neither non-treated nor IFN-γ-treated KPC7735 cells were rejected in PD-L1-/- mice (figure 4A–D). We, thus, concluded that PD-L1 molecules on the surface of KPCs function as a dominant gatekeepers that suppressed effector CD8 T-cell responses in B6 mice via the PD-L1/PD-1 signaling axis.

PD-1/PD-L1-dependent growth of KPC7735 tumors in transplanted hosts. (A) 2.5×105 untreated (left panel, n=9) or IFN-γ treated (+IFN-γ) (right panel, n=8) KPC7735 cells were transplanted subcutaneously into the left flank of B6 mice. Data shown were collected from two independent experiments. (B) B6 mice were transplanted with untreated KPC7735 cells and injected intraperitoneally with anti-PD-1 (n=6) or isotype control antibodies (n=5) on days 3 and 6 post tumor inoculation. (C, D) Untreated (left panels) or IFN-γ treated KPC7735 cells (right panels) were transplanted subcutaneously into PD-1-/- (C) (n=6 per group) or PD-L1-/- (D) mice (n=4 per group). Tumor rejection rates are indicated. Data shown were collected from two independent experiments. (E) (Upper panel) in two independent experiments, RAG1-/- mice (n=8) were transplanted subcutaneously with 2.5×105 naïve KPC7735 cells and tumor development was monitored. (Lower panels) surface expression of MHC class I molecules (H2–Kb) and PD-L1 was assessed on ex vivo isolated KPC7735 tumors at day 12 post-transplantation into B6 or RAG1-/- mice. One representative histogram (representing an individual explanted tumor cell suspension out of four B6 and RAG1-/- mice, respectively) is shown. IFN-γ, interferon-γ.
KPC7735 cells transplanted subcutaneously into B6 mice showed a prominent upregulation of both, MHC-I and PD-L1 molecules on the cell surface, resembling the effects of the in vitro IFN-γ-treatment (figure 4E). The upregulation of MHC-I and PD-L1 cell surface expression was weakened in subcutaneous KPC7735 tumors developing in T- and B cell-deficient syngeneic RAG1-/- mice (figure 4E). This indicated that transplanted KPC7735 cells stimulated the adaptive immune system in immune-competent mice, which in turn induced both, MHC-1 and PD-L1 expression on these tumor cells,5 and thereby established tumor-eradicating effector CD8 T-cell responses when the PD-L1/PD-1 signaling axis was silenced/blocked in anti-PD-1-treated B6 or in PD-1-/- mice.
To proof that tumor cells induce PD-L1/PD-1-sensitive effector CD8 T cells, we generated PD-L1-deficient KPC7735PD-L1KO cells using the CRISPR/Cas9 system. KPC7735PD-L1KO cells upregulated expression of MHC-I but not PD-L1, Cr-1 or gp70 on IFN-γ-treatment (online supplementary figure S5a‒c) and were quantitatively rejected in transplanted B6 mice (figure 5A). An additional challenge of these KPC7735PD-L1KO-immune, tumor-free B6 mice at day 28 with KPC7735 cells resulted in a quantitative rejection of these transplanted cells (figure 5A) along with a prominent CD8 T-cell response against the TAP-dependent gp70:Kb/p15E but not the TAP-independent Cr-1:Kb/cr16-24 epitope (figure 5B). Similarly, a single immunization of B6 mice with KPC7735PD-L1KO cells induced high frequencies of gp70:Kb/p15E-specific CD8 T cells and low frequencies of Cr-1:Kb/cr16-24-specific CD8 T cells (figure 5C). Interestingly, also KPC7735 cells primed gp70:Kb/p15E- and Cr-1:Kb/cr16-24-specific CD8 T cells in B6 mice, but the frequencies were lower than in KPC7735PD-L1KO-immune mice (figure 5C). It was striking that a significant higher proportion of gp70:Kb/p15E-specific CD8 T cells primed in KPC7735-immune B6 mice expressed the PD-1 exhaustion marker (figure 5D), which could limit their effector phenotype and explain the rejection of KPC7735PD-L1KO but not of KPC7735 tumor cells in these mice.
Supplemental material

Testing the antigenicity of KPC7735PD-L1KO cells. (A) 2.5×105 KPC7735PD-L1KO cells were transplanted subcutaneously into the left flank of B6 mice (n=6, from two independent experiments) and tumor growth was monitored. At day 28, mice were challenged with 2.5×105 KPC7735 cells and rejection rates are indicated. (B) Non-treated B6 mice (group 1, n=3) and three tumor-free mice shown in (A) were sacrificed at day 45 (group 2) and Cr-1:Kb/cr16-24- and gp70:Kb/p15E-specific CD8 T-cell frequencies were determined in the liver and spleen. Graphs show the actual proportions (%) of Kb/p15E-dimer+ and Kb/cr16-24-dimer+ cells among all CD8 T cells±SD. Statistical significant differences were determined using the unpaired Student’s t-test. (C) B6 mice were either non-treated (group 1, n=4) or immunized with 2.5×105 KPC7735 (group 2, n=4) and KPC7735PD-L1KO cells (group 3, n=5). Twelve days post cell injection Cr-1:Kb/cr16-24- and gp70:Kb/p15E-specific CD8 T-cell frequencies were determined as described in (A). ANOVA, followed by Tukey’s multiple comparisons test, was used to compare T-cell frequencies between all groups. (D) Expression of PD-1 was assessed on gp70:Kb/p15E-dimer+ CD8 T cells and statistically evaluated using the unpaired Student’s t-test. In the right panels one representative dot blot, showing the staining of PD-1 on gated Kb/p15E-dimer+ CD8 T cells is depicted. Data shown in (C, D) were obtained from one experiment. (*p<0.05, **p<0.01). ANOVA, analysis of variance; Cr-1, Cripto-1; ns, not significant.
Development of a novel IFN-γ-treatment protocol to selectively restore MHC-I presentation-competence on PDACCs
Considering, that the deletion of PD-L1 in tumor cells with the CRISPR/Cas9 system is an attractive but laborious and time-consuming way to generate effective cell-based tumor vaccines for PD-1-competent B6 mice, we explored conditions to manipulate KPC tumor cells for selectively restoring their MHC-I presentation competence. To determine whether the kinetics of IFN-γ-inducible MHC-I and PD-L1 surface expression differ, and could be exploited to manipulate the upregulation of PD-L1, tumor cells were treated for 20 hours with IFN-γ, washed and further cultured in vitro (figure 6A,B). This showed that upregulation of PD-L1 in different KPC cells was less sustained as compared with MHC-I and declined toward basal levels around day 3 (d3) post IFN-γ-treatment start, whereas MHC-I expression still was prominent (figure 6B). In contrast, the expression of gp70 and Cr-1 tumor antigens was not (or rarely) affected by the IFN-γ-treatment protocol (online supplementary figure S6).
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Development of a tumor cell-based immunization protocol. (A) Schematic presentation of the IFN-γ-treatment protocol: KPC7735 cells were treated with IFN-γ for 16–20 hours, washed and further cultured up to day 3 post treatment start. Subsequently, cells were irradiated and subcutaneously transplanted (2.5×105 cells per mouse) into the left flank (L) of mice. After 14–19 days, mice were challenged with 2.5×105 naïve KPC7735 cells into the opposite flank (R). (B) Five different KPC cell lines were treated with 20 ng/mL IFN-γ for 20 hours, washed and further cultured up to 7 days. Surface expression of MHC-I molecules (H2-Kb, open circles) and PD-L1 (closed squares) was assessed at the indicated time points by FCM. (C) B6 mice were injected with irradiated KPC7735 cells (KPC7735-ir, n=5), IFN-γ-treated KPC7735 cells irradiated immediately after the IFN-γ treatment (KPC7735-I-ir, n=5) or IFN-γ-treated KPC7735 cells irradiated at day 3 post-IFN-γ treatment start (KPC7735-Id3-ir, n=4) and challenged at day 19 post immunization with KPC7735 cells. Parallel groups of KPC7735-Id3-ir-immune mice (n=4) were additionally treated with anti-CD4 (n=4) or anti-CD8 antibodies (n=5) on days 3 and 6 post tumor challenge (indicated by an arrow). Graphs show tumor growth on the left (L) and right flank (R) and tumor rejection rates are indicated. Data shown were obtained from one experiment. (D) Similarly, PD-1-/- mice were injected with KPC7735-ir (n=4) or KPC7735-Id3-ir cells (n=4), challenged at day 19 with KPC7735 cells and tumor growth was monitored. (E) B6 (n=7) and PD-1-/- mice (n=6) were injected with 2.5×105 IFN-γ-treated KPC-Id3-ir cells in two independent experiments. On day 12 postimmunization, Cr-1:Kb/cr16-24- and gp70:Kb/p15E-specific CD8 T-cell frequencies were determined in the liver and spleen. Graphs show the actual proportions (%) of Kb/p15E-dimer+ and Kb/cr16-24-dimer+ cells among all CD8 T cells±SD, corrected for the frequencies detected in non-treated mice. The two mouse groups were compared using the unpaired Student’s t-test. FCM, flow cytometry; IFN-γ, interferon-γ; ns, not significant.
For immunization studies, we additionally irradiated tumor cells and transplanted either non-treated irradiated KPC7735 cells (KPC7735-ir), KPC7735 cells irradiated immediately after IFN-γ treatment (KPC7735-I-ir) and KPC7735 cells irradiated at d3 post IFN-γ treatment (KPC7735-Id3-ir) into the left flank of B6 mice (figure 6A–C). We did not observe outgrowth of tumors in these mice, confirming the efficacy of our irradiation conditions (figure 6C). At d19 postvaccination, mice were challenged with 2.5×105 naïve KPC7735 cells into the opposite right flank (figure 6A–C). KPC7735-ir and KPC7735-I-ir cells did not induce protective immune responses and consecutively transplanted KPC7735 cells formed solid tumors (figure 6C). In contrast, KPC7735-Id3-ir cells induced a tumor-specific immunity that efficiently suppressed tumor development of transplanted KPC7735 cells (figure 6C). Suppression of tumor growth in this group was reverted by the injection of anti-CD8 but not anti-CD4 mAbs (figure 6C), demonstrating that CD8 T cells played a crucial role in the rejection of KPC7735 tumor transplants. Similarly, KPC7735-Id3-ir cells primed a tumor-protective immunity in PD-1-/- mice (figure 6D). However, in contrast to B6 mice, vaccination of PD-1-/- mice with KPC7735-ir cells was also sufficient to suppress the outgrowth of consecutively transplanted KPC7735 cells (figure 6C,D). This suggested that the immune-mediated upregulation of MHC-I and PD-L1 (figure 4E) was, at least transiently active in transplanted KPC7735-ir cells and allowed priming of an anti-tumor response in PD-1-/- (lacking the PD-L1/PD-1 signaling axis) but not in B6 mice. Confirmatory, irradiation of KPC cells resulted in a prominent cell death (>90%) during a subsequent in vitro culture for 48 hours, but did not significantly affect the IFN-γ-induced upregulation of MHC-I and PD-L1 molecules on the small population of live cells (online supplementary figure S7).
Supplemental material
KPC7735-Id3-ir cells primed a prominent gp70:Kb/p15E-specific CD8 T-cell response and a weak Cr-1:Kb/cr16-24-specific CD8 T-cell response in B6 mice (figure 6E; online supplementary table 1). Interestingly, gp70:Kb/p15E- and Cr-1:Kb/cr16-24-specific CD8 T-cell frequencies were comparable in KPC7735-Id3-ir-immune B6 and PD-1-/- mice (figure 6E), indicating that a missing PD-1 expression did not affect de novo priming of CD8 T cells. However, as compared with KPC7735-ir cells that did not elicit a tumor-protective immunity in B6 mice (figure 4C), KPC7735-Id3-ir cells induced higher frequencies of gp70:Kb/p15E-specific CD8 T cells associated with a decreased single positive (PD-1+ or LAG-3+) and double positive PD-1+/LAG-3+ exhaustion marker profile (online supplementary figure S8) which could preserve their expansion and/or functional effector phenotype.46 The IFN-γ-treatment protocol could also be used in B6 mice to prime a tumor-specific immunity against different PD-L1+/MHClo PDACCs (KPC7612-Id3-ir) (online supplementary figure S9). Interestingly, upregulation of PD-L1 in IFN-γ-treated PD-L1+/MHCint MC38 colon adenocarcinoma cells was less sustained as compared with the inducible MHC-I expression and MC38-Id3-ir but not MC38 or MC38-ir cell-based vaccination induced a protective immunity in B6 mice that rejected a subsequent transplant of MC38 cells (online supplementary figure S10).
Supplemental material
Supplemental material
Supplemental material
Above analyzes revealed clear differences in the priming of Cr-1:Kb/cr16-24- and gp70:Kb/p15E-specific CD8 T-cell responses by DNA-based and cell-based immunization (online supplementary table 1). We, thus, tested whether DNA immunization could be used to complement the TAP-dependent gp70:Kb/p15E-specific CD8 T-cell immunity induced by KPC tumor cell-vaccines. Indeed, coimmunization of B6 mice with pCI/Cr-1 and KPC7735PD-L1KO cells efficiently primed both, Cr-1:Kb/cr16-24- and gp70:Kb/p15E-specific CD8 T-cell responses and the respective CD8 T-cell frequencies were even comparable to those primed by individual vaccines (online supplementary figure S11). This strongly suggested that de novo primed Cr-1:Kb/cr16-24- and gp70:Kb/p15E-specific CD8 T cells did not interfere.
Supplemental material
Discussion
Here, we used PDACCs as a model to explore if (and how) expression and processing of tumor antigens in the ER and/or in TAP-dependent/-independent pathways influences the MHC-I epitope presentation and priming of effector CD8 T cells by cell-based immunization.47 48 We showed that subcutaneous transplantation of KPC cells into immune-competent B6 but not into T- and B-cell deficient RAG1-/- mice upregulated MHC-I and PD-L1 expression on the cell surface. This indicated that cells of the adaptive immune system in immune-competent B6 mice directly interfered with transplanted KPC tumor cells and triggered a balanced MHC-I presentation-competent, immune-suppressive cell surface expression profile.5 We could largely mimic this in vivo upregulation of MHC-I and PD-L1 molecules by treating in vitro cultured KPC cells with IFN-γ. Microarrays performed on untreated and IFN-γ-treated KPC cells confirmed the upregulation of genes involved in MHC-1 expression (eg, beta-2 microglobulin), the conventional MHC-I antigen processing/presentation pathway (eg, TAP1)49 but also of PD-L1 molecules. The effects of IFN-γ-initiated upregulation of PD-L1 were superior to the reconstitution of MHC-I presentation-competence, as IFN-γ-treated KPC7735 cells grew out to tumors in B6 and PD-L1-/- mice, but not in PD-1-/- mice. In PD-L1-/- mice, PD-L1 molecules were exclusively expressed on transplanted tumor cells, demonstrating that the engagement of PD-1 by PD-L1 on tumor cells was sufficient to suppress the anti-tumor responses to KPC7735 cells. When PD-L1/PD-1 signaling was blocked in PD-1-/- mice or anti-PD-1 treated B6 mice, subcutaneously transplanted KPC7735 cells induced an antitumor immunity against themselves, pointing to a dominant negative PD-L1-mediated mechanism. Similarly, the depletion of PD-L1 in KPC7735PD-L1KO tumor cells was sufficient to prevent the PD-L1/PD-1 signaling axis and preferentially induce TAP-dependent gp70:Kb/p15E-specific CD8 T cells. Overall, the KPC7735PD-L1KO-induced immunity led to their own rejection and protected these B6 mice from consecutively transplanted KPC7735 tumors. Based on these observations, we established an in vitro IFN-γ treatment protocol for KPC tumor cells that enhanced their antigenic potential to prime CD8 T cells in B6 mice in an antigen specific manner. This method reconstituted TAP-dependent MHC-I presentation-competence and concomitantly bypassed the IFN-γ-mediated upregulation of PD-L1 in KPC cells. As a consequence, cell-based immunization with KPC7735-Id3-ir cells, but neither KPC7735-ir nor KPC7735-I-ir cells, induced a tumor-protective immunity in B6 mice that rejected a subsequent inoculum of naïve KPC7735 cells. An interesting aspect of this method was that codelivery of anti-PD-1 or anti-PD-L1 antibodies that provided potent tools for immune checkpoint control but also induced serious side effects, like the induction of autoimmune diabetes,45 50 were not necessary to induce this protective CD8 T-cell immunity by cell-based immunization.
Cr-1 and gp70 antigens expressed in their natural compartment (the ER) or as Hsp73-bound chimeric antigens in the cytosol elicited a complete opposite pattern of CD8 T-cell responses by DNA immunization (online supplementary table 1). Expression and processing of Cr-1 in the ER was a prerequisite to generate the Cr-1:Kb/cr16-24 epitope and induce Cr-1:Kb/cr16-24-specific CD8 T cells in a TAP-independent manner. Confirmatory, a cytosolic Hsp73-complexed stT77Cr-1 antigen did not induce CD8 T cells, though Hsp73 facilitated cross-priming of CD8 T cells by DNA immunization.20 40 43 It is unknown if and which cross-presentation/-priming mechanism(s) (ie, antigen transfer from antigen-expressing myocytes targeted by vector DNA injection to professional B7.1+ APCs, like dendritic cells 20) were involved in the priming of Cr-1:Kb/cr16-24-specific CD8 T cells by SPase-sensitive Cr-1 but not SPase-resistant Cr-1R26P antigens. Alternatively, Cr-1 could be directly expressed and processed in the ER of professional APCs targeted by intramuscular DNA injection.24 The expression/processing requirements of Cr-1 for priming effector CD8 T cells by DNA immunization strongly resembled the preproinsulin (ppins) self-antigen. Ppins is targeted to the ER by a SPppins domain and contains a Kb/A12-21 epitope in the insulin A-chain localized at the extreme COOH-terminus that does not require further COOH-terminal trimming before binding to Kb.19 Kb/A12-21-specific CD8 T cells and autoimmune diabetes were induced in PD-1-/- and PD-L1-/- by injection of ppins DNA, as well as in ppins-immune B6 mice that replenished their diabetogenic potential after anti PD-L1 antibody treatment.45 Comparable to Cr-1, the expression of ppins in the ER was a prerequisite to generate a low-affine Kb/A12-21 epitope and induce autoreactive CD8 T cells by DNA immunization, most likely because this epitope must not compete for MHC-I binding with the bulk of antigenic peptides generated in the conventional endogenous antigen presentation pathway for TAP-dependent transport into the ER.19 45 A primary effect of ER-expression/processing of self-antigens thus could be that epitopes with a relative weak avidity for MHC-I can directly bind to a substantial number of MHC-I molecules in the ER, necessary to prime/activate autoreactive CD8 T cells. In contrast, expression of gp70 antigens in the ER was not sufficient to directly stimulate gp70:Kb/p15E-specific CD8 T cells in B6 and TAP-deficient mice by DNA immunization. This showed that the gp70 antigen does not belong to the group of TEIPPs, which are exclusively immunogenic under TAP-deficient conditions.11 12 In line with previous reports, indicating that presentation of the gp70:Kb/p15E epitope depends on proteasomal processing and TAP,38 39 we showed that cytosolic Hsp73-bound stT77gp70 complexes induced gp70:Kb/p15E-specific CD8 T cells in B6 but not TAP-deficient mice. A stgp70 antigen lacking the Hsp73-binding domain did not induce gp70:Kb/p15E-specific CD8 T cells.30 This suggested that priming of gp70:Kb/p15E-specific CD8 T cells by cytosolic gp70 antigens critically depend on ‘help’ from Hsp73. Confirmatory, RNA-binding stgp70 designer antigens (eg, stgp70tat, a fusion antigen composed of stgp70 and a cationic HIV-tat domain) induced TLR-7-dependent innate ‘help’ and primed gp70:Kb/p15E-specific CD8 T cells by DNA immunization.30 Furthermore, considering that a proline (P) at position 3 of the Kb/p15E epitope (KSPWFTTL) greatly hindered its TAP-dependent transport and efficient presentation by Kb molecules,39 we think that these adjuvant-like immune-stimulatory immune responses might improve epitope presentation.
It is generally assumed that cross-presentation of tumor cells or tumor cell debris by professional APCs is a prerequisite to prime functional effector CD8 T-cell responses.20 In our transplantation model, KPC7735-Id3-ir cells induced high frequencies of gp70:Kb/p15E- but low frequencies of Cr-1:Kb/cr16-24-specific CD8 T cells in both, B6 and PD-1-/- mice. A comparable effector phenotype was induced by KPC7735PD-L1-KO cells. Therefore, at least for the two antigens tested, the IFN-γ-treatment protocol selectively enhanced priming/expansion of TAP-dependent gp70:Kb/p15E-specific CD8 T cells that correlated with the rejection of consecutively transplanted KPC7735 cells. However, it is obvious that additional multi-specific CD8 T-cell responses against yet unknown antigens/epitopes47 were coprimed by KPC7735-Id3-ir cells and facilitated the almost quantitative rejection of transplanted KPC7735 tumor cells.
In conclusion, we here showed a promising strategy to overcome two initial problems in the development of CD8 T-cell-mediated immunotherapies against cancer: (1) the identification of expression/processing conditions for tumor-associated self-antigens that stimulate effector CD8 T-cell responses by cell-based vaccines and (2) the combination of CD8 T-cell priming with immune interventions that limit CD8 T-cell exhaustion by silencing the tumor-mediated PD-L1/PD-1 signaling axis.
Acknowledgments
We appreciate the expert technical assistance of Katrin Schwengle and Ralf Köhntop. We thank Lieping Chen (Department of Oncology, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA) for PD-L1-/- (B7-H1-/-) mice and Tasuku Honjo (Department of Immunology and Genomic Medicine, Kyoto University, Graduate School of Medicine, Kyoto, Japan) for PD-1−/− mice.
References
Footnotes
Contributors KS, JK, LR, JG, MW and AL designed and performed the experiments, researched and interpreted data. AK, MW, TS and RS conceived the experiments, secured funding, discussed and interpreted the data. KS and RS wrote manuscript. All authors edited and approved the final manuscript.
Funding This work was supported by grants from the Deutsche Forschungsgemeinschaft: DFG SCHI-505/6-1 to R.S. and a BIU Phase II TP I3 grant from the Boehringer Ingelheim Ulm University Biocenter to MW, RS and TS.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Except of the established human cell line HEK-293 used for detection of vector-expressed antigens, this study does not contain human materials. All mouse immunization studies were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the German Federal Animal Protection Law. The protocols were approved by the Committee on the Ethics of Animal Experiments of the University of Ulm (Tierforschungszentrum Ulm, Oberberghof) and the Regierungspräsidium Tübingen (Permit Numbers: 1199 and 1384 to RS and 1273 to AK).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as supplementary information. The datasets generated in this study are now publicly available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE140397.