Article Text

Abstract
Background Tumor-infiltrating lymphocyte (TIL) therapy is a personalized cancer treatment which involves generating ex vivo cultures of tumor-reactive T cells from surgically resected tumors and administering the expanded TILs as a therapeutic infusion. Phase 1 of many TIL production protocols use aldesleukin (IL-2) alone to establish TIL cultures (termed “PreREP” (Pre-Rapid Expansion Protocol)); however, this fails to consistently produce TIL cultures from renal cell carcinoma (RCC) in a timely manner. Adding mitogenic stimulation via anti-CD3/anti-CD28 beads along with IL-2 to the fresh tumor digest (FTD) during TIL generation (termed “FTD+ beads”) increases successful TIL culture rates; however, T cells produced by this method may be suboptimal for adoptive transfer. We hypothesize that adherent cell depletion (ACD) before TIL expansion will produce a superior TIL product by removing the immunosuppressive signals originating from adherent tumor and stromal cells. Here we investigate if “panning,” a technique for ACD prior to TIL expansion, will impact the phenotype, functionality and/or clonality of ex vivo expanded RCC TILs.
Methods Tumor specimens from 55 patients who underwent radical or partial nephrectomy at the University of Kansas Medical Center (KUMC) were used to develop the panning method and an additional 19 specimens were used to validate the protocol. Next-generation sequencing, immunohistochemistry/immunocytochemistry and flow cytometry were used during method development. The phenotype, functionality and clonality of autologous TILs generated in parallel by panning, PreREP, and FTD+ beads were assessed by flow cytometry, in vitro co-culture assays, and TCRB CDR3 sequencing.
Results TIL cultures were successfully generated using the panning protocol from 15/16 clear cell, 0/1 chromophobe, and 0/2 papillary RCC samples. Significantly fewer regulatory (CD4+/CD25+/FOXP3+) (p=0.049, p=0.005), tissue-resident memory (CD8+/CD103+) (p=0.027, p=0.009), PD-1+/TIM-3+ double-positive (p=0.009, p=0.011) and TIGIT+ T cells (p=0.049, p=0.026) are generated by panning relative to PreREP and FTD+ beads respectively. Critically, a subset of TILs generated by panning were able to degranulate and/or produce interferon gamma in response to autologous tumor cells and the average tumor-reactive TIL yield was greatest when using the panning protocol.
Conclusions Removing immunosuppressive adherent cells within an RCC digest prior to TIL expansion allow for the rapid production of tumor-reactive T cells with optimal characteristics for adoptive transfer.
- lymphocytes
- tumor infiltrating
- T lymphocytes
- kidney neoplasms
- therapies
- investigational
- immunotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Tumor-infiltrating lymphocyte (TIL) therapy is an individualized cancer treatment which involves generating tumor-reactive T cells ex vivo from a patient’s own surgically resected tumor and administering these cells as a therapeutic infusion. TIL therapy has produced durable responses in numerous forms of cancer with recent clinical trials exploring efficacy for the treatment of melanoma, cervical cancer, ovarian cancer, breast cancer, non-small cell lung cancer, hepatocellular carcinoma, head and neck squamous cell carcinoma, glioblastoma, gastric cancer, colorectal cancer, neuroendocrine tumors, osteosarcoma, soft-tissue sarcomas, pancreatic ductal adenocarcinoma, and cholangiocarcinoma (ClinicalTrials.gov).1–6 Clear cell renal cell carcinoma (ccRCC) has many of the hallmarks of tumor types that respond well to TIL therapy: it is a highly immunogenic neoplasm with a reactive T-cell infiltrate and is known to respond to other immunotherapies including aldesleukin (IL-2), interferon alpha (IFN-α), and checkpoint inhibitors.7–13 However, RCC has been labeled a difficult target for TIL-based therapies and at present there is only one ongoing clinical trial exploring TIL therapy for RCC (NCT02926053).14 This may be due to the results of the only randomized, phase III trial exploring TIL treatment for RCC, which was terminated early in 1997 due to the lack of efficacy because 41% of patients randomized to the experimental group did not receive a TIL infusion due to an insufficient number of viable T cells being generated during in vitro TIL manufacturing.15 Since the termination of this trial, little work has been done to explore why in vitro culture failed for such a large proportion of patients with RCC leaving an unmet need to develop novel methods for TIL expansion from RCC.
Although RCC is an immunogenic tumor with a consistent T-cell infiltrate, the conundrum remains regarding why it is so challenging to clinically generate TIL cultures. TIL expansion in the phase III trial was attempted using a two-stage protocol. IL-2 (1200 IU/mL) alone was used to establish TIL cultures followed by an early version of a rapid expansion protocol involving phytohemagglutinin stimulation after CD8 selection. TIL expansion protocols have changed considerably since this trial, and Andersen and colleagues have shown that with an improved Pre-Rapid Expansion Protocol (PreREP) protocol and pooling of the bulk “young TIL”, a TIL generation success rate of 92% can be achieved from RCC; however, it required up to 2 months of culture for this first phase of the TIL expansion protocol to be complete.14 This prolonged duration required to establish TIL cultures from RCC may hinder the application of this therapy for patients with aggressive disease. This limitation was partially addressed by Baldan et al who showed that optimized tumor digestion and immediate addition of mitogenic stimulation via anti-CD3/anti-CD28 paramagnetic beads to the FTD increased successful TIL generation rates from RCC in a 15-day time frame.16
We hypothesize that TIL generation from RCC can be further improved by using an additional technique: removal of the immunosuppressive tumor and stromal cells that are present in the surgical specimen by adherence-based separation. During efforts to optimize TIL generation from RCC, we first experimented with ways to enrich TILs from FTDs including fluorescent activated cell sorting and magnetic bead-based sorting to remove TILs from their immunosuppressive environment prior to expansion. These methods added an additional time and resource requirement to the already labor-intensive and resource-intensive TIL production process. Labeling and additional manipulation were also required which subjected the limited tumor digests to cell loss prior to expansion. However, the immunosuppressive cell types within the tumor microenvironment which we aimed to exclude, including tumor cells, cancer-associated fibroblasts, and some myeloid derived suppressor cells, all share the in vitro characteristic of adherence. Therefore, we developed and evaluated a technique to promote the expansion of RCC TIL (referred to as “panning”), which involves an overnight ACD step following tumor dissociation prior to TIL stimulation. We report that this strategy, which requires minimal time, resources, and manipulation, increases average TIL yield in a 14-day time frame and creates fewer regulatory T cells, tissue resident memory T cells, and T cells expressing multiple immune checkpoints all of which are phenotypes expected to contribute to the robustness of TIL function for use in antitumor clinical applications.
Materials and methods
Patients and samples
Deidentified clinical samples were provided from the Biospecimen Repository Core Facility (BRCF) at the University of Kansas Medical Center (KUMC) along with relevant clinical information. Tissue specimens were obtained from patients enrolled under the repository’s institutional review board approved protocol (HSC no: 5929) and following the US Common Rule. Tissue from 74 patients who underwent radical or partial nephrectomy within the University of Kansas Health System for suspected RCC was obtained between August 2017 and January 2020. Online supplemental table 1 provides clinical information and basic research notes on how each tumor tissue sample was used. Tissues were delivered from the operating room to surgical pathology where the BRCF staff obtained a portion of the discarded tissues, deidentified the samples, and immediately transported the specimens at room temperature in RPMI-1640 to the basic research laboratory for TIL and tumor cell line (TCL) generation.
Supplemental material
Supplemental material
Creation of fresh tumor digests (FTDs)
Using aseptic techniques in a laminar flow hood, fibrotic, necrotic, and normal adjacent tissues were grossly dissected from viable tumor. Viable tumor was mechanically and enzymatically digested in a cocktail containing RPMI-1640 (without serum), 1 mg/mL type IV collagenase (Sigma), 100 units/mL deoxyribonuclease I (MP Bio), 1× penicillin, streptomycin, and amphotericin B (Antibiotic-Antimycotic; Gibco). Viable tumor and digestion cocktail were added to a Miltenyi C Tube and disrupted using a gentleMACS Dissociator followed by a 30 min incubation at 37°C with gentle rocking; mechanical disruption and 30 min incubations were continued until a slurry was obtained (approximately 1–1.5 hours). The slurry was passed through a 100 µm cell strainer, pelleted, subjected to ammonium chloride–potassium (ACK)-mediated RBC lysis (BioLegend) and washed in phosphate-buffered saline (PBS). This FTD was either used immediately for culture or cryopreserved for future use. Digests of normal adjacent kidney tissue were performed on selected samples using the same protocol.
Generation of TIL and TCL cultures by panning, PreREP, and FTD+ beads
Panning
Total viable cells of 4–6×106 from an FTD were plated in a BioLite 175 cm2 vented flask (Thermo Fisher Scientific) and incubated overnight at 37°C and 5% CO2 in T-cell media consisting of RPMI-1640 with 2.05 mM L-glutamine, 10% fetal bovine serum (FBS), 25 mM HEPES (Gibco), 1 mM sodium pyruvate (HyClone), 1× non-essential amino acids (HyClone), and 1× penicillin/streptomycin (HyClone). Non-adherent cells were then pooled by washing five to eight times with PBS until no non-adherent cells were observed in the flask. The non-adherent cells were pelleted and resuspended in 2 mL of fresh T-cell media and transferred to a single well of a 24-well plate. 6000 IU/mL of IL-2 (teceleukin) and 4×105 anti-CD3/anti-CD28 Dynabeads (Thermo Fisher Scientific) were then added. TILs were split three times (1 well to 8 wells of a 24-well plate) by resuspending the T cells and transferring 1 mL into a fresh well and then adding 1 mL of fresh media (FM) to each well. Splitting of TILs occurred when clusters of expanding T cells were grossly visible in the well. Eight wells of TILs were then pooled and transferred to an upright 175 cm2 flask with 50 mL of media (16 mL of TILs+34 mL FM). 50 mL of additional media was added 24–48 hours later and subsequent 1:1 splitting to multiple upright T175 flasks occurred when a carpet of expanding T cells was grossly visible in the flask. Media was never removed from the TIL culture using the panning protocol because splitting was always indicated before media acidified and 6000 IU/mL of IL-2 was maintained by bolus addition after each splitting. Beads were removed at the completion of TIL expansion (day 14). Adherent cells were grown in parallel in DMEM (Dulbecco’s Modified Eagle Medium) with high glucose (HyClone) supplemented with 10% FBS, 1× penicillin/streptomycin (HyClone), and 1× Insulin-Transferrin-Selenium (Gibco) to establish TCLs.
PreREP
Total viable cells of 4–6×106 of an FTD were plated in 2 wells of a 24-well plate (2–3×106/well) with 2 mL of T-cell media per well and 6000 IU/mL of IL-2. Splitting occurred as described in the panning protocol; however, half of the media was regularly required to be removed (slowly pipetting from the top of the well) and replaced with FM and IL-2 before splitting was indicated. 6000 IU/mL of IL-2 was maintained by bolus addition after each splitting or media replacement.
FTD+ beads
Total viable cells of 4–6×106 of an FTD were plated in 2 wells of a 24-well plate (2–3×106/well) with 2 mL of T-cell media per well, 4×105 anti-CD3/anti-CD28 Dynabeads and 6000 IU/mL of IL-2. Splitting occurred as described in the panning protocol; however, half of the media was regularly required to be removed (slowly pipetting from the top of the well) and replaced with FM and IL-2 before splitting was indicated. 6000 IU/mL of IL-2 was maintained by bolus addition after each splitting or media replacement. Beads were removed at the completion TIL expansion (day 14).
All cell counts in this study were performed using a Muse Cell Analyzer (MilliporeSigma) and the Muse Cell Count and Viability kit.
Immunohistochemistry, immunocytochemistry, and histology
Prior to digestion, a portion of the RCC sample and normal adjacent tissue were fixed in 10% neutral buffered formalin at room temperature for 48 hours. Tissues were then dehydrated in 70% ethanol for 24 hours. The samples were then embedded in paraffin and processed by the KUMC BRCF staff using a Tissue-Tek Processor (Sakura). Microtomy was performed on a Leica RM2255 Microtome (Leica Biosystems). Automated staining for pan-cytokeratin and the paired box 8 protein (PAX-8) was performed using an intelliPATH instrument (Biocare). Immunocytochemistry was performed using the same protocol with cell pellets of the primary TCLs established from the tumor sample. Images were acquired using MetaMorph Microscopy Image Analysis Software (Molecular Devices) on a Nikon Eclipse 80i Microscope.
Flow cytometry
Flow cytometry was performed using a BD LSR II in the KUMC Flow Cytometry Core Laboratory. Cell surface staining was performed in 2% FBS in PBS, and BD Horizon Brilliant Stain Buffer (BD Biosciences) was added when multiple BV dyes were used in the same panel. Cells were subsequently washed in protein-free PBS and stained for viability using Ghost Dye 780 (Tonbo). Cells were resuspended in 2% FBS in PBS for analysis. Transcription factor staining was performed using the True Nuclear Transcription Factor Buffer Set (BioLegend), and cytokine staining was performed using the eBioscience IC Fixation Buffer and Permeabilization Buffer (Invitrogen). The flow cytometry panels used in this study can be found in the online supplemental methods. Analyses were performed using FlowJo (V.10.6.1 and V.10.6.2; FlowJo). Analyses included debris exclusion (Forward Scatter-Area vs Side Scatter-Area (FSC-A vs SSC-A)), single events (Forward Scatter-Area vs Forward Scatter-Height (FSC-A vs FSC-H)), and live cell gates before examining other parameters (online supplemental figure 1).
Lymphocyte function assay
Expanded TILs rested overnight in the absence of CD3/CD28 stimulation and/or cytokines were either left unstimulated, stimulated with 10 µg/mL of OKT3 or added to autologous tumor cells in T-cell media at an effector:target ratio of 5:1 (5×105 TIL:1×105 tumor cells). The cells were cultured in the presence of 0.7 µL/mL monensin (BD GolgiStop) and anti-CD107a-BV421 in a final volume of 100 µL/condition at 37°C and 5% CO2 for 4 hours after being briefly centrifuged to promote immediate effector/target contact. Cells were then stained with the remainder of the panel using the indicated antibodies, viability dye, and fix/perm buffer set.
Next-generation sequencing (NGS) of primary RCC cell lines and parent tissue
DNA was extracted from primary TCLs which were established from three consecutive RCC samples using the QIAmp DNA Mini Kit. DNA was quantified using a Tecan NanoQuant Plate on a Tecan Infinite M200 Pro and then underwent a multiplex PCR reaction targeting exons of 275 solid tumor-related genes using a commercially available kit (Qiagen). The prepared libraries were then subjected to NGS on a NextSeq 500 instrument (Illumina) to generate fastQ files (text-based format for storing nucleotide sequences). The reads were mapped to the Homo_sapiens_sequence hg19 reference using QIAGEN Biomedical Genomics Workbench pipeline. Metrics for reported variants such as depth of coverage, variant allele frequency, and average quality cut-offs were evaluated individually for pathogenic and likely pathogenic hits compared with uncertain significance variants. The Variant Call Format files were generated and loaded into QIAGEN Clinical Insight to assess individual calls for pathogenicity and quality control parameters.
TCRB CDR3 sequencing
DNA was extracted from TILs expanded in each condition from three specimens using the QIAmp DNA Mini Kit. Samples were provided to Adaptive Biotechnologies for TCRB CDR3 sequencing. Adaptive Biotechnologies performed quality checks and data normalization using their established pipeline and uploaded the data for analysis using the ImmunoSEQ Analyzer 3.0 platform.
Statistical analysis
Statistical significance was considered a p-value less than 0.05 determined by using a ratio paired t-test on Graph Pad Prism 8.2.1.
Results
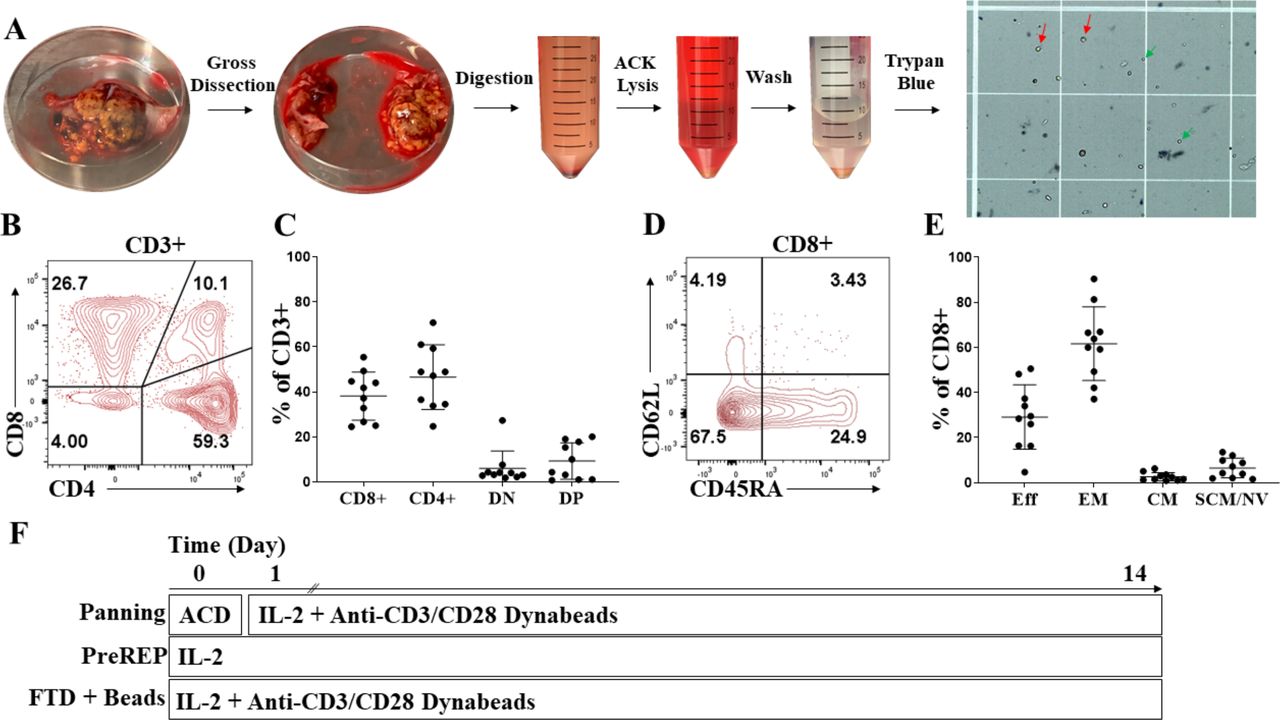
Consistent liberation of viable CD8+ effector memory T cells from ccRCC
Using our RCC tumor digestion protocol involving gross tumor dissection, mechanical and enzymatic digestion, and ACK RBC lysis, we observe both viable presumptive lymphocytes (green arrows) and viable presumptive tumor cells (red arrows) in the preparation using trypan blue exclusion (figure 1A). We confirmed that there was both a viable CD4+ and CD8+ T-cell infiltrate liberated within tumor digests from the 10 ccRCC samples which pre-expansion phenotypes were analyzed and found the average CD4/CD8 ratio to be 1.24 (figure 1B,C). We also found that 50% of the samples had greater than 10% of double-positive T cells (CD3+/CD4+/CD8+) prior to expansion (figure 1B,C). Double-positive T cells within the tumor microenvironment have been proposed to be a clonal/oligoclonal population of dysfunctional, tumor-specific T cells capable of being reactivated by checkpoint inhibitors; however, the significance of these double-positive T cells outside of the thymus is largely unknown.17 18 We next characterized the differentiation state of pre-expansion TILs and showed that an average of 62% of ccRCC-infiltrating CD8+ T cells have an effector memory (Tem=CD45RA-/CD62L-) phenotype prior to expansion, a subset which is known to have proliferative potential, in contrast to the average 29% with an effector phenotype (Teff=CD45RA+/CD62L-, AKA Temra) which have a minimal ability to divide and are prone to apoptosis (figure 1D,E). In summary, we consistently liberated viable CD8+ Tem cells from RCC tumor specimens which should have the capacity for in vitro expansion. Therefore, in subsequent experiments we directly compared PreREP to other methods of TIL expansion to determine if the reported high failure rates for expanding TILs from whole RCC FTDs with IL-2 alone could be overcome with alternative protocols. Figure 1F depicts the schema for the three TIL expansion protocols which were explored.

Consistent liberation of viable CD8+ effector memory T cells from clear cell renal cell carcinoma (ccRCC). (A) Creation of a fresh tumor digest (FTD) from ccRCC. Green arrows indicate viable presumptive lymphocytes, and red arrows indicate viable presumptive tumor cells after trypan blue exclusion. (B) Representative CD4+ and CD8+ staining of pre-expansion CD3+ RCC tumor-infiltrating lymphocytes (TILs). (C) Frequencies of pre-expansion CD8+, CD4+, double-negative (DN), and double-positive (DP) T cells from 10 ccRCC samples. (D) Representative effector (eff), effector memory (em), central memory (cm) and stem-cell like memory/naive (scm/nv) staining of pre-expansion CD3+/CD8+ RCC TILs. (E) Frequencies of pre-expansion CD3+/CD8+ with an eff, em, cm, and scm/nv phenotype from 10 ccRCC samples. (F) Outline of the three TIL expansion methods used. ACD, adherent cell depletion; ACK, ammonium chloride–potassium; IL-2, aldesleukin; PreREP, Pre-Rapid Expansion Protocol.
Tumor cells predominate in the adherent cell population within ccRCC and express immunosuppressive ligands
We hypothesized that inhibitory signals from tumor and/or stromal cell populations within the FTD could be suppressing mitogenic stimulation from tumor antigens and exogenous IL-2 and that this could be responsible for the reported failures for generating TIL cultures from whole RCC FTDs with IL-2 supplementation alone. First, we observed that the adherent cultured cells from the ccRCC samples have a striking morphology which differs from adjacent normal renal cortical tissue (figure 2A). There are prominent vacuoles within the cytoplasm of the adherent cells, likely filled with lipids and glycogen, which give the parent tissue its characteristic “clear cell” morphology. This was our first clue that tumor cell growth dominated the adherent population as opposed to other stromal populations. Also, primary adherent cell cultures from ccRCC uniformly express cytokeratin reflecting the epithelial origin of these tumors as well as the transcription factor PAX-8, which is expressed in the proximal tubules of the kidney that is thought to be the cell of origin of ccRCC (figure 2B). Next, to further characterize the adherent cells, we evaluated gene mutations using a NGS gene panel (275 genes) on DNA isolated from three consecutive adherent ccRCC cultures. We detected pathogenic VHL mutations, the most commonly mutated gene in ccRCC,19 in all three of the adherent primary cultures. Mutant PBRM1 (2 of 3), SETD2 (1 of 3), and TP53 (1 of 3) were also detected in the adherent cell cultures, all with greater than 98% mutant allele frequency, confirming a near pure population of clonal tumor cells grows out after five or more passages (online supplemental table 2). Next, we asked if these adherent tumor cells expressed immunosuppressive ligands in vitro, which could be responsible for failed TIL generation from whole FTDs. We found that adherent cells in culture are able to express programmed cell death ligand 1 (PD-L1), a well-characterized inhibitory immune checkpoint ligand. Two of three samples displayed at least a 10-fold increase in mean fluorescence intensity (MFI) relative to the isotype when cultured in FM; however, in response to autologous stimulated TIL-conditioned media (TIL CM) for 24 hours all three samples massively upregulated PD-L1 with an average 80-fold increase in MFI relative to the isotype control (figure 2C). This is likely due to IFN-ɣ present in the TIL CM (online supplemental figure 2) because PD-L1 has been shown to be an IFN-inducible ligand in many cell types.20 21 Galectin-9 (Gal-9) is a ligand for the T-cell immunoglobulin and mucin-domain containing-3 (TIM-3) receptor and it has been shown to be a negative prognostic factor in ccRCC likely due to the suppression of type 1 T-cell responses by linking the inhibitory TIM-3 receptor to the supramolecular activation cluster on the surface of T cells.22–24 We found that adherent RCC cells during in vitro culture express Gal-9, as shown by the average fivefold increase (n=3) in MFI relative to the isotype control (figure 2D). IFN-ɣ is known to upregulate Gal-9 in certain cell types25; however, we observed a slight decrease in MFI when RCC cells were cultured in TIL CM. This could be due to complex interplay of Gal-9 expression and secretion because Gal-9 is known to be secreted by non-classical pathways involving multivesicular bodies which mediate exosome biogenesis; however, little is known about the regulation of this secretion.26 IFN-ɣ is also known to increase expression of the antigen-presenting major histocompatibility complexes (MHCs/human leukocyte antigen (HLA)). We show that RCC cells uniformly express conventional class I HLA molecules; however, interestingly only two of three RCC cultures upregulated HLA-A/B/C in response to autologous TIL CM and the other showed a slight decrease in expression (online supplemental figure 3). Structurally related to the conventional class I MHC molecules is the non-classical immunosuppressive HLA-G, which under normal physiological conditions is known to be expressed in the placenta and promote maternal immune tolerance to paternal antigens expressed by the developing fetus.27 However, RCC is known to ectopically express this molecule to evade the immune system.28 Although expression levels were considerably lower than conventional class I HLA molecules and the other immunosuppressive ligands evaluated, HLA-G was detectable on the surface of the adherent RCC cells and slightly upregulated in response to TIL CM with an average 1.7-fold increase in MFI relative to the isotype (figure 2E).

Tumor cells predominate in the adherent cell population within clear cell renal cell carcinoma (ccRCC) and express immunosuppressive ligands. (A) H&E staining of adjacent normal renal cortex and autologous ccRCC (top panel) and corresponding passage 0 adherent cell lines established from those tissues (bottom panel). (B) Pan-cytokeratin (PanCK) and PAX-8 staining of RCC tissue (top panel) and the autologous, adherent cells at passage 0 derived from the same sample (bottom panel). (C) PD-L1 expression on the surface of adherent primary RCC cells cultured in fresh media (FM) or conditioned media from autologous TILs (TIL CM). (D) Galectin-9 (Gal-9) expression within adherent primary RCC cells. (E) Human leukocyte antigen G (HLA-G) expression on adherent primary RCC cells. (F) 40× photomicrographs of TILs expanding in 24-well plates using the panning, Pre-Rapid Expansion Protocol (PreREP), and fresh tumor digest (FTD)+ beads protocols at 18 hours, day 4, and day 6 post-surgery. PE, phycoerythrin; PD-L1, programmed cell death ligand-1.
In addition to the immunosuppressive properties of the adherent RCC cells themselves, myeloid derived suppressor cells (MDSCs: CD33+/HLA-DR-) are known to impact T-cell expansion and function. Therefore, we examined if monocytic MDSCs (mMDSCs: CD33+/HLA-DR-/CD14+), granulocytic MDSCs (pmnMDSCs: CD33+/HLA-DR-/CD15+), and/or early MDSCs (CD33+/HLA-DR-/CD14-/CD15-) were depleted by removing adherent cells prior to expansion. We found that MDSCs were a rare subset within three RCC FTDs and that CD33+ myeloid cells in general were depleted during the ACD step involving overnight culture in T-cell media (ie, rare in both the adherent and non-adherent population relative to day 0); however, some of the persisting MDSCs were detected in the adherent population indicating that panning does reduce T-cell exposure to this non-desirable cell type during TIL expansion (online supplemental figures 4 and 5 ).
Together, these data suggest that adherent cells left in culture during TIL expansion will express multiple inhibitory ligands that have the potential to counteract the stimulatory signals from exogenous IL-2. This can be visualized by comparing cultures of autologous TILs expanded by panning, PreREP, and FTD+ beads over the first 6 days of culture, in which TILs expanded after ACD rapidly form proliferating clusters of T cells requiring splitting at day 4 and again at day 6, relative to both other protocols in which adherent cells are left in culture and splitting was not indicated in the first 6 days (figure 2F).
TILs generated by panning have an optimal phenotype for adoptive transfer
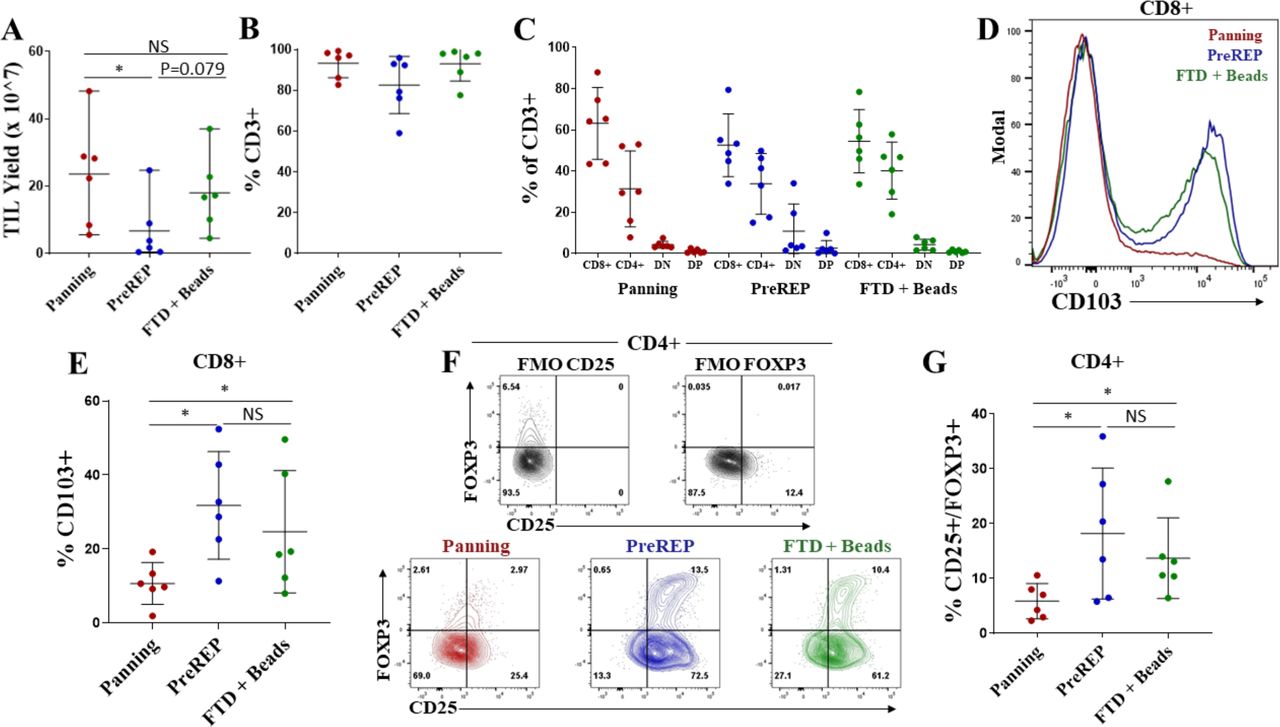
Within the validation cohort we observed a 94% success rate (15/16 consecutive samples) for establishing TIL cultures from histologically confirmed ccRCC using our panning protocol and a 0% success rate for establishing TIL cultures from chromophobe (0/1) and papillary (0/2) RCC. Digests of six of the ccRCC samples in the validation cohort contained at least 1.2×107 total viable cells (samples designated by “*” in online supplemental table 1), which was the minimum number of total cells within the FTD needed to compare autologous TIL expansion using all three protocols (4×106 cells/protocol). No more than 6×106 total viable cells were used to initiate TIL cultures using each protocol to improve comparability between samples. Importantly, five of the six samples used to compare protocols were from late-stage (TNM (tumor, node, metastases) stage 3) ccRCC making this cohort clinically relevant because early-stage RCC is often cured by nephrectomy,29 whereas late-stage disease has a poor prognosis and is the main focus of efforts to improve outcomes using immunotherapies.
Successful TIL generation was considered to be a cell culture which required splitting at least four times (1 well of a 24-well plate to an upright 175 cm2 flask) using the panning protocol or three times (2 wells of a 24-well plate to an upright 175 cm2) using the PreREP or FTD+ beads protocols in the 14-day time frame. Average TIL yield was 23.6×107 (range=5.5×107–4.8×108) using the panning protocol, 6.6×107 (range=4.6×106–2.5×108) using the PreREP protocol, and 18.1×107 (range=4.5×107–3.7×108) using the FTD+ beads protocol (figure 3A). Using the described criteria for successful TIL generation, of the six samples used for comparing the protocols, 6/6 panning, 3/6 PreREP, and 6/6 FTD+ beads were successful; however, there were enough viable cells in the three failed PreREP cultures to perform immunophenotyping and functional assays—which was the rationale for using a minimum of 4×106 total cells to initiate each culture. An average of 93.2%, 82.6%, and 93.0% of cells were CD3+ after expansion using the panning, PreREP, and FTD+ beads protocols, respectively (figure 3B), and there were no statistically significant differences in the percentage of CD4+ or CD8+ T cells generated by each protocol (figure 3C). However, TILs expanded from two donors via PreREP had greater than 20% double-negative (DN) T cells which parallels Markel et al’s report using the PreREP protocol for generating TILs from RCC,30 but the biological significance of these postexpansion DN TILs is largely unknown.

Tumor-infiltrating lymphocytes (TILs) generated by panning have an optimal phenotype for adoptive transfer. (A) TIL yields from six clear cell renal cell carcinoma samples using the panning, Pre-Rapid Expansion Protocol (PreREP), and fresh tumor digest (FTD)+ beads protocols to expand autologous TILs. (B) Percentage of CD3+ cells in the final TIL products. (C) Frequencies of CD8+, CD4+, double-negative (DN), and double-positive (DP) T cells in the final TIL products. (D) Representative CD103 staining of CD3+/CD8+ T cells in the final TIL products. (E) Frequency of CD103+/CD8+ T cells in the final TIL products. (F) Representative regulatory T cells (Treg) staining and fluorescence minus one (FMO) controls. (G) Frequencies of CD25+/FOXP3+ T cells within the CD4+ subset in the final TIL products. NS, not significant.
Significantly fewer CD103+/CD8+ T cells were observed when TILs were expanded by panning relative to both other protocols with an average of 10.6%, 31.8%, and 24.6% of CD8+ T cells expressing CD103 in the TIL products expanded by panning, PreREP, and FTD+ beads, respectively (figure 3D,E). CD103 is a marker of tissue-resident memory T cells (TRM) and binds to E-cadherin, playing a role in retaining these T cells in epithelial tissues.31 32 Although TRM cells may play an important role in the immune response to epithelia-derived tumors, we believe they are not optimal for adoptive transfer because they are not known to recirculate33 and therefore are unlikely to contribute to systemic responses after adoptive transfer. Another T-cell subset that we believe is not optimal for adoptive transfer is regulatory T cells (Tregs), which are known to suppress antitumor effector functions. We show that panning produces a significantly smaller proportion of Tregs (Tregs=CD4+/CD25+/FOXP3+) with an average of 5.9%, 18.2%, and 13.7% of CD4+ T cells expressing CD25 and FOXP3 after expansion by panning, PreREP, and FTD+ beads, respectively (figure 3F,G). The high levels of Tregs produced by both other protocols may be a result of simultaneous activation and PD-1/PD-L1 signaling that occurs as a result of leaving adherent cells in the culture. This combination of stimulatory and inhibitory signaling has been implicated in Treg induction.34 There were no statistically significant differences in the expression of CD28, CD27, KLRG1, CD62L, or IL-7Ra as a result of expansion method; however, a slight (average 2.1% vs 3.8%), but statistically significant increase in CCR7 expression was observed when TILs were expanded by FTD+ beads relative to panning (online supplemental figure 6). Although there is minimal significance when evaluating each of these markers individually, when we evaluate expression of all of these markers simultaneously using t-Distributed Stochastic Neighbor Embedding, we see differential clustering (online supplemental figures 7‒12).
There is strong evidence that coexpression of multiple inhibitory immune checkpoints correlates with a state of decreased function and “exhaustion”35 36; therefore, we also characterized the presence and relative abundance of immune checkpoint molecules on TILs following expansion using each of the three protocols. We report that panning, compared with PreREP and FTD+ beads, generates significantly fewer PD-1+ T cells (an average of 7.2%, 31.2%, and 15.4%, respectively) (figure 4A,B). We show that on average over 75% of RCC TILs expanded in all conditions express TIM-3 (figure 4A,C) with no significant differences in the percentage of TIM-3+ T cells between methods. However, the percentage of PD-1+/TIM-3+ double-positive T cells is also significantly lower in the TIL products expanded by panning relative to PreREP and FTD+ beads with an average of 6.0%, 23.0%, and 12.9% of T cells expressing both PD-1 and TIM-3, respectively (figure 4A,D). We did not observe a significant difference in the percentage of T cells expressing the lymphocyte-activation gene-3 (LAG-3) immune checkpoint (figure 4E); however, we also see a significant decrease in T-cell immunoreceptor with IG and ITIM domains (TIGIT+) T cells when panning is used, as compared with PreREP and FTD+ beads, with an average of 50.8%, 67.9%, and 62.1% of T cells expressing TIGIT, respectively (figure 4F,G). Taken together, our results demonstrate a decrease in coexpression of inhibitory immune checkpoints when TILs are expanded by panning, which may indicate a decreased relative state of exhaustion.

Tumor-infiltrating lymphocytes (TILs) generated by panning have reduced coexpression of immune checkpoints. (A) Representative T-cell immunoglobulin and mucin-domaincontaining-3 (TIM-3) versus programmed cell death-1 (PD-1) staining of autologous TIL products. (B) Frequency of PD-1+ T cells in the final TIL products. (C) Frequency of TIM-3+ T cells in the final TIL products. (D) Frequency of PD-1+/TIM-3+ double-positive T cells in the final TIL products. (E) Frequency of lymphocyte-activation gene-3 (LAG-3+) T cells in the final TIL products. (F) Representative T-cell immunoreceptor with IG and ITIM domain (TIGIT) staining of autologous TIL products. (G) Frequency of TIGIT+ T cells in the final TIL products. FTD, fresh tumor digest; NS, not significant; PreREP, Pre-Rapid Expansion Protocol.
Bead-based expansion generates TIL products with increased clonal diversity
TILs are known to respond to a variety of tumor-associated antigens or tumor-specific neoantigens created by somatic mutations within protein coding genes which are transcribed, translated, degraded and ultimately presented as mutated peptide fragments on MHCs. TILs recognize these peptide fragments/MHCs through their unique T-cell receptors (TCRs). The most diverse region of the TCR is the complementarity-determining region 3 (CDR3) on the beta chain and it is statistically improbable that two different T-cell clones share the same TCRB CDR3 sequence; therefore, this region can be used to identify unique T-cell clones. We preformed TCRB CDR3 sequencing on autologous TILs expanded by panning, PreREP, and FTD+ beads from three donors. Donor 68 was selected because it was the outlier in which TIL yields were nearly identical using all three methods (22.4×107, 24.7×107, and 22.8×107, respectively) and donors 57 and 62 were selected because TILs from these samples fit the trends in yield, phenotype and function which we are reporting. We found that TILs expanded using pan-CD3 stimulation via anti-CD3/CD28 beads (panning and FTD+ beads) results in a TIL product with a greater absolute number of T-cell clones (figure 5A). An average of 13,822, 4279, and 11 381 clones were detected in the TIL products expanded by panning, PreREP and FTD+ beads, respectively, based on productive TCRB CDR3 DNA sequences, which result in a unique amino acid sequence (ie, unique but synonymous DNA sequences were considered identical clones). The top 20 most abundant clones detected in the TIL products expanded by the three methods accounted for an average of 16.5%, 54.8%, and 17.2% of the total TCR repertoires, respectively (figure 5B). In agreement with these trends, the productive Simpson clonality was greatest in the TIL products expanded by PreREP (figure 5C). Finally, we show that although the overall clonal diversity within the TIL products differed, the most abundant clones expanded by each method tend to be identical (figure 5D). This is exemplified by TILs expanded from donor 68 in which gross TIL yields were nearly identical and the frequency of the most abundant clones were also highly comparable.
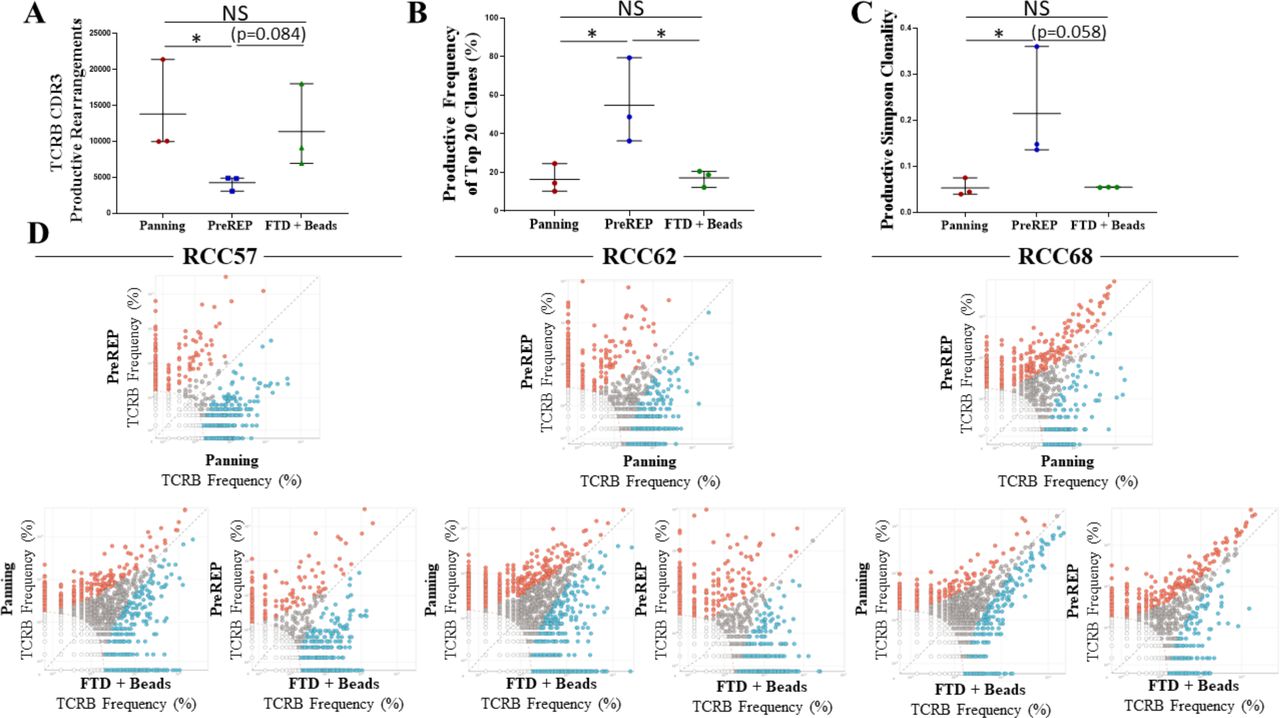
Bead-based tumor-infiltrating lymphocyte (TIL) expansion generates TIL products with increased clonal diversity; however, dominant clones are identical and expanded by all methods. (A) Absolute number of productive TCRB CDR3 rearrangements detected in the TIL products expanded by each method. (B) The combined productive frequency of the top 20 clones expanded by each method. (C) The productive Simpson clonality metric for the TIL products expanded by each method. (D) Comparison of clonal frequency for each specimen and method: clear dots represent clones which were too infrequent to be analyzed (each dot may represent multiple clones), gray dots represent clones which were not significantly enriched due to expansion method, blue dots represent clones which were significantly enriched by the method on the x-axis, and red dots represent clones which were significantly enriched by the method on the y-axis. CDR3, complementarity-determining region 3; FTD, fresh tumor digest; NS, not significant; PreREP, Pre-Rapid Expansion Protocol; RCC, renal cell carcinoma; TCR, T-cell receptor.
TILs generated by panning are tumor reactive
A potential concern when expanding TILs using immediate pan-CD3 stimulation via anti-CD3/CD28 beads is that the increased clonal diversity of the TILs may be the result of expanding bystander T cells which are not specific for tumor antigens. This could create a dilute TIL product relative to the PreREP protocol. However, on the contrary the increased clonal diversity could be attributed to tumor-reactive T-cell clones which are eliminated as a result of the suppressive signaling that occurs when leaving adherent cells in the TIL culture. Surprisingly, there was not a significant difference in the proportion of TILs which degranulated (CD107a+) after co-culture with autologous tumor cells as a result of the expansion method, although the average percentage was greatest using the PreREP protocol with a mean of 2.2%, 3.8%, and 1.9% of TILs expanded by panning, PreREP, and FTD+ beads degranulating in response to autologous tumor cells, respectively (figure 6A,B). Of note, there were as many as 3.2% of TILs degranulating in the unstimulated condition after PreREP expansion and up to 1.8% using the FTD+ beads protocols. This observation may indicate tumor cells and/or antigen are still in the culture at the 14-day mark using these protocols. Furthermore, over 80% of TILs expanded by all methods expressed granzyme B (presumably within the CD107a+ granules) with no significant differences in GZMB expression (online supplemental figure 13). Both a CD8+ and CD8- response was observed; the CD8- response is a result of CD4+, CD56+, and CD4-/CD56- TIL with no discernable trend in CD8- responses as a result of expansion method (online supplemental figure 14A,B). There was also no significant difference in the percentage of TILs which produced IFN-ɣ in response to autologous tumor cells with an average of 1.5%, 2.0%, and 1.0% of TILs producing this effector molecule after expansion by panning, PreREP, and FTD+ beads, respectively. A similar phenomenon of IFN-γ production in the unstimulated populations, which were not subjected to panning, was noted (figure 6C,D). Interestingly, in response to autologous tumor cells, TILs generated by all protocols were rarely polyfunctional (ie, simultaneously degranulated and produced IFN-ɣ) (figure 6E). As shown, only ~16.5% (0.89/5.39) of the TILs which responded in co-culture with autologous tumor cells produced IFN-γ and degranulated, even though of the cells capable of degranulating and/or producing IFN-γ (OKT3 positive control)~40.8% (17.3/42.4) were polyfunctional. This could be a result of bystander TILs, which are only activated in the positive control, having increased levels of polyfunctionality or it may be that the tumor cells are suppressing a polyfunctional response by an unknown mechanism. These results parallel the report by Markel and colleagues who showed that non-pooled PreREP RCC TIL cultures displayed either lytic activity or IFN-ɣ secretion, but not both.30 The average percentage of tumor-reactive T cells generated—defined as CD107a+ and/or IFN-γ+ when co-cultured with autologous tumor cells—was greatest using the PreREP protocol (figure 6F). An average of 3.4%, 5.3%, and 2.8% of TIL were tumor reactive in the final TIL products expanded by panning, PreREP and FTD+ beads, respectively. However, when we multiplied the percentage of tumor-reactive TIL (figure 6F) by the absolute number of TILs generated in each condition from each sample (figure 3A), we observed an average of 8.0×106, 2.7×106, and 4.6×106 total tumor-reactive TILs generated by panning, PreREP, and FTD+ beads, respectively (figure 6G). These results indicate that although a greater percentage of bystander TIL is potentially being generated when anti-CD3/CD28 beads are added to the initial TIL culture, the combination of ACD and bead-based TIL stimulation generates an increased total number of RCC-reactive TIL in a 14-day time frame.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Tumor-infiltrating lymphocytes (TILs) generated by panning are tumor reactive. (A) Degranulation (CD107a+) of autologous TIL products left unstimulated, OKT3 stimulated or in response to the autologous tumor cell line (auto-TCL). (B) Frequency of degranulation of the final TIL products. (C) Interferon gamma (IFN-γ) expression by autologous TILs in the indicated conditions. (D) Frequency of IFN-γ expression within the final TIL products. (E) Costaining IFN-γ versus CD107a of panning TIL in response to autologous tumor cells. (F) Percentage of CD107a+ and/or IFN-γ+ TIL (tumor-reactive TIL) in the final TIL products. (G) Tumor-reactive TIL yield—calculated by multiplying the percentage of tumor-reactive TIL by the total TIL yield for each condition and sample. FTD, fresh tumor digest; PreREP, Pre-Rapid Expansion Protocol.
Discussion
Consistent generation of TIL cultures from RCC has been historically problematic which has hindered the development of this potentially curative cell therapy for patients with kidney cancer.15 Here we demonstrate that a novel protocol for TIL expansion allows for consistent production of tumor-reactive TIL from ccRCC. Critically, the panning approach advances the field of T-cell therapy for RCC by demonstrating that the phenotype of the resulting TIL product is superior for adoptive transfer relative to autologous TILs produced by other protocols. Mechanistically, we hypothesize these phenotypic differences are a result of expanding TILs in the absence of immunosuppressive tumor cells. An activated T cell within a whole FTD secretes inflammatory molecules (eg, IFN-γ) and as a result adherent tumor cells in culture actively upregulate PD-L1 potentially suppressing TIL activation. Simultaneously, secretion of Gal-9 by tumor cells may link TIM-3 to the supramolecular activation cluster.24 The significance of Gal-9/TIM-3 interaction alone has been somewhat controversial; however, this interaction has been shown to lead to the phosphorylation of tyrosine 265 on the cytoplasmic tail of TIM-3 and most evidence points to this interaction playing a role in suppressing type 1 T-cell responses.24 37–40 Other ligands of TIM-3 which have been shown to modulate function that can be restored with TIM-3 blockade are phosphatidylserine (PS), carcinoembryonic antigen cell adhesion molecule 1, and high-mobility group box 1, and these ligands have binding clefts separate from Gal-9s on the TIM-3 receptor.39 41 Therefore, if the activated T cell does manage to degranulate and lyse an adherent tumor cell during TIL expansion, the flipping of PS to the outermembrane of tumor cell-derived apoptotic bodies may deliver this second immunosuppressive signal via the Gal-9 primed TIM-3 receptor. We believe this combination of activating and inhibitory signals is responsible for inducing a regulatory and exhausted phenotype which can be minimized by ACD prior to TIL expansion.
The proposed panning method also likely reduces TIL exposure to the CD103 ligand, E-cadherin, which is expressed by adherent epithelial cells. We hypothesize that decreased ligand exposure is the mechanisms which leads to a TIL product with fewer CD103+ tissue-resident memory T cells. Although TRM cells have been implicated in antitumor responses,42 we believe they are not ideal for adoptive transfer because durable systemic responses will likely require tumor-reactive T cells with a memory phenotype which allows them to recirculate. Therefore, the phenotypic differences that result from expanding RCC TILs after ACD are optimal for adoptive transfer.
We show that the proposed panning method produces a greater absolute number of tumor-reactive T cells as determined by in vitro co-culture with autologous tumor cells. However, we believe it is likely that both the per cent and absolute number of tumor-reactive T-cells are being underestimated using this method. Indeed, the NGS data (ie, >98% allele frequency for a given mutation) suggests that short-term in vitro propagation of the adherent tumor cells leads to clonal populations. However, it does not represent the more heterogeneous tumor cell populations that exist in vivo. Therefore, there may be tumor-reactive TILs expanded by all methods that are not appreciated in these assays because a clonal tumor cell population was used as the target.
Finally, it has been suggested that there is an inverse correlation between in vitro culture time and the efficacy of the TIL product; however, it has also been demonstrated that there is a direct correlation between the absolute number of TILs infused and response rates. Balancing these two opposing curves is an overarching challenge in the field of TIL therapy. We are proposing the panning protocol as a single-phase rapid expansion protocol to generate a TIL product in an unprecedented 14-day time frame. The absolute yields reported in this manuscript are a result of starting with only a fraction of the tumor digests which were generated using discard tissue after surgical pathology processed the samples. In the clinical trial setting, a larger portion of tissue would likely be obtained for TIL generation and the entire sample could be used to generate a TIL product via panning which would drastically scale up TIL yields in the same time frame. TIL counts of 1010 to 1011 are the usual target yields prior to cell infusion using the two-phase PreREP/REP protocol; however, cell manufacturing can take up to 6 to 8 weeks to achieve these counts using this method. A 2-week TIL product would be of great interest to patients dealing with the mental and physical stresses associated with waiting for TIL production as well as for clinicians monitoring disease progression during this same time frame; therefore, we propose further evaluation of this single-phase protocol to expand TILs with a firm 14-day timeline from surgery to infusion regardless of TIL yield.
This report demonstrates the consistent and rapid generation of tumor-reactive TILs from RCC is possible. The stigma from the 1990s that TIL therapy is not feasible for RCC should be disregarded and renewed efforts at translating this promising therapy into the clinics for patients with advanced ccRCC is warranted.
Acknowledgments
We would like to thank all members of the Godwin research program for their advice and support. We acknowledge the University of Kansas Cancer Center’s BRCF and staff, in particular Alex Webster, who is sponsored, in part, by the National Institutes of Health (NIH)/National Cancer Institute (NCI) Cancer Center Support Grant P30 CA168524. We acknowledge the Flow Cytometry Core Laboratory, which is sponsored, in part, by the NIH/National Institute of General Medical Sciences COBRE grant P30 GM103326 and the NIH/NCI Cancer Center grant P30 CA168524. The IL-2 used in this study was obtained through the NCI Biological Resources Branch Preclinical Biologics Repository under HHSN 75N91019D00024. The muse cell counter used in this study was acquired with an equipment grant from an NIH Clinical and Translational Science Award grant (UL1 TR002366).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
ND and AKG contributed equally.
Contributors MWB, ND, HBP, and AKG designed experiments. MWB, ML, SH, and ZP were involved in data acquisition and analysis. AKG directed the collection of human materials and the gathering of clinical information, MWB, HA, ND, and AKG wrote the paper, and all authors reviewed the manuscript.
Funding This work has been supported in part by the NIGMS COBRE grant P20 GM130423 and the Kansas Bioscience Authority (KBA) Eminent Scholar grant to AKG.
Competing interests AKG is the Chancellors Distinguished Chair in Biomedical Sciences Endowed Professor at the KUMC.
Patient consent for publication Not required.
Ethics approval All patients who provided deidentified tumor tissue samples for this study did so under written informed consent and KUMC approved the collection protocol (HSC no: 5929).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. The data sets acquired during the current study are available from the corresponding author on reasonable request.