Article Text

Abstract
Background Immunosuppressive therapy or T-cell depletion in transplant patients can cause uncontrolled growth of Epstein-Barr virus (EBV)-infected B cells resulting in post-transplant lymphoproliferative disease (PTLD). Current treatment options do not distinguish between healthy and malignant B cells and are thereby often limited by severe side effects in the already immunocompromised patients. To specifically target EBV-infected B cells, we developed a novel peptide-selective chimeric antigen receptor (CAR) based on the monoclonal antibody TÜ165 which recognizes an Epstein-Barr nuclear antigen (EBNA)−3C-derived peptide in HLA-B*35 context in a T-cell receptor (TCR)-like manner. In order to attract additional immune cells to proximity of PTLD cells, based on the TÜ165 CAR, we moreover generated T cells redirected for universal cytokine-mediated killing (TRUCKs), which induce interleukin (IL)-12 release on target contact.
Methods TÜ165-based CAR-T cells (CAR-Ts) and TRUCKs with inducible IL-12 expression in an all-in-one construct were generated. Functionality of the engineered cells was assessed in co-cultures with EBNA-3C-peptide-loaded, HLA-B*35-expressing K562 cells and EBV-infected B cells as PTLD model. IL-12, secreted by TRUCKs on target contact, was further tested for its chemoattractive and activating potential towards monocytes and natural killer (NK) cells.
Results After co-cultivation with EBV target cells, TÜ165 CAR-Ts and TRUCKs showed an increased activation marker expression (CD137, CD25) and release of proinflammatory cytokines (interferon-γ and tumor necrosis factor-α). Moreover, TÜ165 CAR-Ts and TRUCKs released apoptosis-inducing mediators (granzyme B and perforin) and were capable to specifically lyse EBV-positive target cells. Live cell imaging revealed a specific attraction of TÜ165 CAR-Ts around EBNA-3C-peptide-loaded target cells. Of note, TÜ165 TRUCKs with inducible IL-12 showed highly improved effector functions and additionally led to recruitment of monocyte and NK cell lines.
Conclusions Our results demonstrate that TÜ165 CAR-Ts recognize EBV peptide/HLA complexes in a TCR-like manner and thereby allow for recognizing an intracellular EBV target. TÜ165 TRUCKs equipped with inducible IL-12 expression responded even more effectively and released IL-12 recruited additional immune cells which are generally missing in proximity of lymphoproliferation in immunocompromised PTLD patients. This suggests a new and promising strategy to specifically target EBV-infected cells while sparing and mobilizing healthy immune cells and thereby enable control of EBV-associated lymphoproliferation.
- CD8-positive t-lymphocytes
- cell engineering
- immunotherapy
- receptors
- chimeric antigen
- transplantation immunology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- CD8-positive t-lymphocytes
- cell engineering
- immunotherapy
- receptors
- chimeric antigen
- transplantation immunology
Background
Epstein-Barr virus (EBV) infection or reactivation is a frequent complication of hematopoietic stem cell and solid organ transplantation. While EBV-infected B cells are under the control of EBV-specific T cells in immunocompetent individuals, delayed immune reconstitution and immunosuppressive treatment after transplantation can lead to the uncontrolled proliferation of malignantly transformed EBV-infected B cells, resulting in post-transplant lymphoproliferative disease (PTLD). The incidence of PTLD ranges from 1% to 20%, depending on the transplanted organ, the patient’s EBV serostatus, and the intensity of immunosuppression.1 PTLD has a 3-year overall survival rate of only 49% to 80% and significant side effects, emphasizing the need for novel treatment approaches.2 3
Current treatment options, such as reduction of immunosuppression, chemotherapy and B-cell depletion, exhibit varying response rates and are often limited by severe side effects.4–6 Recently, it was shown that adoptively transferred EBV-specific T cells expanded or directly isolated from the blood of EBV-seropositive donors can restore the EBV-specific T-cell immunity and persist for several years.6–9
An immunotherapeutic approach gaining increasing importance in the last years is to genetically redirect T cells by introducing chimeric antigen receptors (CARs) that combine antigen-binding properties with the activating signals of cellular receptors (eg, CD3ζ chain).10 Most often, an antibody-based single-chain variable fragment (scFv) is used as the antigen recognition domain, which mediates binding to extracellular antigens.11 Current limitations to CAR-T cell (CAR-T) applications, such as an immunosuppressive tumor microenvironment and heterogeneous antigen expression, prompted the development of T cells redirected for universal cytokine-mediated killing (TRUCKs), which further endow T cells with the ability to deliver a transgenic effector molecule that accumulates in the target tissue.12 TRUCKs engineered to secrete a proinflammatory cytokine, such as interleukin (IL)-12 or IL-18, on tumor cell engagement exhibit improved effector functions; they act by recruiting and activating innate immune cells (eg, natural killer (NK) cells and macrophages), and by re-programming the immunosuppressive tumor microenvironment. When an inducible expression cassette is used, the cytokine is only released after CAR activation, which reduces systemic toxicity by locally restricting the presence of the cytokine.13 Impressively, TRUCKs were shown to eliminate heterogeneous solid tumor lesions and antigen-loss variants that would otherwise remain invisible to conventional, antigen-dependent CAR-Ts.14
The major challenge in CAR design is to choose an appropriate target antigen only expressed on tumor and not on healthy cells to prevent on-target off-tumor toxicity.15 In order to specifically address malignant EBV-positive PTLD cells and sparing healthy cells, so-called T-cell receptor (TCR)-like monoclonal antibodies (mAbs) can be utilized to direct CARs against an intracellular EBV antigen. TCR-like mAbs specifically bind to an epitope consisting of a defined peptide in complex with an HLA molecule and their usage as therapeutic agents is increasingly being investigated, since tumor or virus-infected cells could be targeted with a high specificity.16–22 Using the binding specificity of TCR-like mAbs for the generation of CARs, researchers could circumvent the need for frequent antibody administration and enable a persisting in vivo response to tumor-associated peptides in the HLA context.19 23–25
In the present study, we generated a CAR based on the scFv of the TCR-like mAb TÜ165. TÜ165 is one of the first TCR-like mAbs with human specificity and exclusively binds to an EBV-infected, HLA-B*35-positive B-cell line but not to cell lines lacking HLA-B*35 or non-EBV-infected HLA-B*35-positive cells.26 It was shown that TÜ165 recognizes LPPHDITPY when bound to HLA-B*35:01, a peptide derived from the Epstein-Barr nuclear antigen (EBNA)−3C, which is expressed in EBV latency type III associated with PTLD.27 The peptide is predicted to bind to the majority of HLA-B*35 subtypes (www.cbs.dtu.dk/services/NetMHCpan; July 7, 202028) and, depending on the geographic region, 11.3% to 37.9% of the population are positive for HLA-B*35 alleles (www.allellefrequencies.net; July 7, 202029). The antibody’s unique TCR-like binding specificity allows the CAR to address an intracellular EBV target antigen, thus sparing healthy B cells in the immunocompromised PTLD patients.
By additionally introducing an inducible IL-12 (iIL-12) expression cassette into the TÜ165 CAR construct, we developed iIL-12-secreting TÜ165 TRUCKs with improved effector functions that recruit and activate bystander immune cells, which otherwise occur rarely in close proximity to lymphoproliferative cells in PTLD patients. Our results demonstrate that TÜ165 CAR-Ts and TÜ165 CAR-based TRUCKs that are equipped with inducible IL-12 expression are able to specifically and effectively respond to an intracellular EBV target and may thereby enable control of EBV-associated lymphoproliferation in immunocompromised patients.
Methods
Human sample materials
All experiments were performed with residual blood samples from routine platelet collection. Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation with Lymphosep (c.c.pro).
Cell lines and peptide loading
Cell lines used and their cultivation medium are listed in online supplemental table S1. K562 were transduced with a lentiviral vector to express HLA-A*35:01 (K562-B*35). B-lymphoblastoid cell lines (B-LCLs) were generated by infecting PBMCs with cell culture supernatant of B95-8 (#ACC-100, DSMZ) and adding 1 µg/mL ciclosporin A (Sigma).
Supplemental material
Peptide loading was performed by incubating cells in serum-free RPMI 1640 with 10 µg/mL EBNA-3C-derived peptide LPPHDITPY (EZ Biolab) at 37°C and 5% CO2. In TÜ165 mAb and CAR binding studies, the aforementioned peptide and CMV_pp65 IPSINVHHY, EBV_EBNA-3A VPATQPQY, EBV_BALF2 YPLREVATL and EBV_EBNA1 HPVGEADYFEY peptides (all EZ Biolab) were added at the specified concentrations and incubated as mentioned.
Construction of TÜ165 CAR, iIL-12-secreting TÜ165 CAR and iEGFP-expressing TÜ165 CAR vectors
The TÜ165 CAR was designed by synthesizing the scFv of TÜ165 (Thermo Fisher Scientific) and cloning it into the previously described ROR1-CAR-epHIV7 vectors.30 To generate the iIL-12-secreting TÜ165 CAR and inducible enhanced green fluorescent protein (iEGFP)-expressing TÜ165 CAR constructs, we cloned the complete CAR backbone, including the co-expressed truncated epidermal growth factor receptor (EGFRt), into the newly developed pRRL.PPT.NFATenh.synTATA.IL12.PGK.newMCS.GD2CAR.PRE and pRRL.PPT.NFATenh.synTATA.EGFP.PGK.newMCS.GD2CAR.PRE.31 Further details as well as generation and titration of lentiviral vectors are provided in the online supplemental methods.
Supplemental material
Generation of TÜ165 CAR-Ts and TÜ165 TRUCKs
Untouched CD8+ T cells were isolated (Miltenyi Biotec) from PBMCs from HLA-B*35-positive donors and activated with anti-CD3/CD28 beads (Thermo Fisher Scientific) at a ratio of 1:1 in TexMACS (Miltenyi Biotec) with 3% human serum (c.c.pro; CTL medium) supplemented with 12.5 ng/mL IL-7 and IL-15 (PeproTech). After 1 day, T cells were transduced with lentiviral particles by spinoculation using an multiplicity of infection (MOI) of 3 and addition of 5 µg/mL Polybrene Infection/Transfection Reagent (Merck). Untransduced T cells were treated equally and served as controls. The anti-CD3/CD28 beads were removed on the following day. On day 7 or 9, EGFRt+ T cells were enriched as described30 or via fluorescence-activated cell sorting (FACS). After a total expansion time of 9 to 17 days, functionality of the transduced T cells was assessed by co-culturing them with target cells. For this, 5×104 target cells and effector cells according to the specified effector-to-target (E:T) ratios were seeded in 200 µL CTL medium. Since they do not express IL-12, iEGFP-expressing TÜ165 CAR-Ts (TÜ165 EGFP-TRUCKs) were expected to show the same functionality as TÜ165 CAR-Ts and were both used to compare functionality with iIL-12-secreting TÜ165 CAR-Ts (TÜ165 TRUCKs).
CAR reporter assay
A previously described reporter cell line based on the Jurkat T cell line JE6-132 33 was transduced with lentiviral particles by spinoculation using an MOI of 1 and addition of 5 µg/mL Polybrene Infection/Transfection Reagent. After two days, target cell recognition of these reporter cells was assessed by co-culturing them with unloaded K562, K562-B*35 or peptide-loaded K562-B*35 as target cells. For this, 1x105 reporter and target cells, respectively, were seeded in an E:T ratio of 1:1 in 200 µL RPMI 1640, 10% fetal bovine sereum (FBS; Sigma-Aldrich) and 2 mM L-glutamine. JE6-1 cell activation was assessed after 24 hours by measurment of EGFP and enhanced cyan fluorescent protein (ECFP) by flow cytometry.
Flow cytometry
Antibodies used are listed in online supplemental table S2. TÜ165 was purified from the supernatant of a TÜ165 hybridoma cell line. Intracellular staining was performed by using the IntraPrep Permeabilizaton Reagent (Beckman Coulter). Samples were analyzed on a BD FACSCanto Flow Cytometer (Becton Dickinson).
Specific activation marker and intracellular cytokine upregulation in response to EBNA-3C-peptide-loaded K562-B*35 (K562-B*35/pEBV) as target cells was calculated by subtracting the frequency or mean fluorescence intensity (MFI) of effector T cells co-cultured with unloaded K562-B*35 from the respective frequency or MFI of those co-cultured with K562-B*35/pEBV (Equation 1).
 (1)
(1)
Multiplex cytokine analyis
Cytokine concentrations in the supernatant were determined using a customized LEDGENDplex Multi-Analyte Flow Assay (BioLegend), which allowed for the detection of human IL-2, IL-12 (p70), granzyme B, interferon (IFN)-γ, perforin and tumor necrosis factor (TNF)-α. Samples were analyzed with LEGENDplex V.8.0 software (BioLegend). In experiments with K562-B*35 target cells, specific cytokine release was calculated by subtracting the amount of secreted cytokine of effector T cells cultured with K562-B*35 from the respective amount in those cultured with K562-B*35/pEBV (Equation 1).
Determination of cytotoxicity by LDH assay and flow cytometry
The release of lactate dehydrogenase (LDH) into the cell culture supernatant was assessed by using the Cytotoxicity Detection Kit (Roche). Cells lysed by adding Triton X-100 (Merck) to a final concentration of 1% to all control wells served as maximum controls. Absorbance was assessed at a wavelength of 490 nm with a reference wavelength of 690 nm on a Synergy 2 Multi-Mode Microplate Reader (Biotek). LDH release was calculated using Equation 2:
 (2)
(2)
B-LCL killing was assessed by flow cytometry and by gating target cells as CD3-CD8- cells. Killing was calculated with Equation 3:
 (3)
(3)
Live cell imaging
To visualize interactions, untransduced T cells labeled with 0.25 µM carboxyfluorescein succinimidyl ester (Thermo Fisher Scientific), TÜ165 CAR-Ts stained using 2 µM CellTrace Violet (Thermo Fisher Scientific), and unstained K562-B*35/pEBV were mixed at a ratio of 1:1:2 in CTL medium and monitored every 30 s for 2 hours with an Olympus IX81 microscope. Analysis was performed using 20X short-distance objective lenses and Olympus ScanR analysis software.
Migration and activation of innate immune cells
The influence of TÜ165 TRUCKs on innate immune cells was assessed by measuring their chemoattractant effect on monocytes (Mono-Mac-6 (MM6) cells) and NK cells (NK-92 cells). The migration of those cells towards prediluted (1:3) 48 hours culture supernatants of the engineered T cells with and without target cells or prediluted (1:3) IL-12 standard in the indicated concentrations was investigated in a Boyden chamber as previously described.31
The effect of TÜ165 TRUCKs on the killing capacity of NK-92 cells was measured using 0.4 µm transwell inserts (Greiner Bio-One) that allowed the exchange of soluble mediators but not cell migration. T cells were co-cultured with K562-B*35/pEBV in the transwell above a co-culture of NK-92 cells with CellTrace Violet-stained SPI-801. After 48 hours, SPI-801 killing by NK-92 cells was measured by flow cytometry and gating on dying cells SPI-801 based on the forward and side scatter properties of CellTrace Violet-positive cells.
The influence of IL-12 released by TÜ165 TRUCKs on IFN-γ expression by NK-92 was also assessed. NK-92 cells were co-cultured with SPI-801 cells in 48 hours culture supernatants of the engineered T cells with and without target cells. The supernatants were pooled from two donors, respectively, and prediluted 1:2.5 in RPMI 1640 with 10% FBS and 2 mM L-glutamine. After 4 hours, total RNA was isolated using the RNeasy Mini Kit (Qiagen) and complementary DNA (cDNA) was reverse-described with the High-capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Expression of IFN-γ was quantified by inventoried mixes (Thermo Fisher Scientific) whereby Glyceraldehyde-3-phosphate dehydrogenase served as internal control.
Statistical analysis
Statistical analysis was performed with GraphPad Prism V.5.02 using the one-tailed Mann-Whitney U test. Values are shown as mean with SEM. ***p≤0.001; **p≤0.01; *p≤0.05; ns>0.05.
Results
TÜ165 specifically binds to EBNA-3C-derived peptide/HLA-B*35 complexes
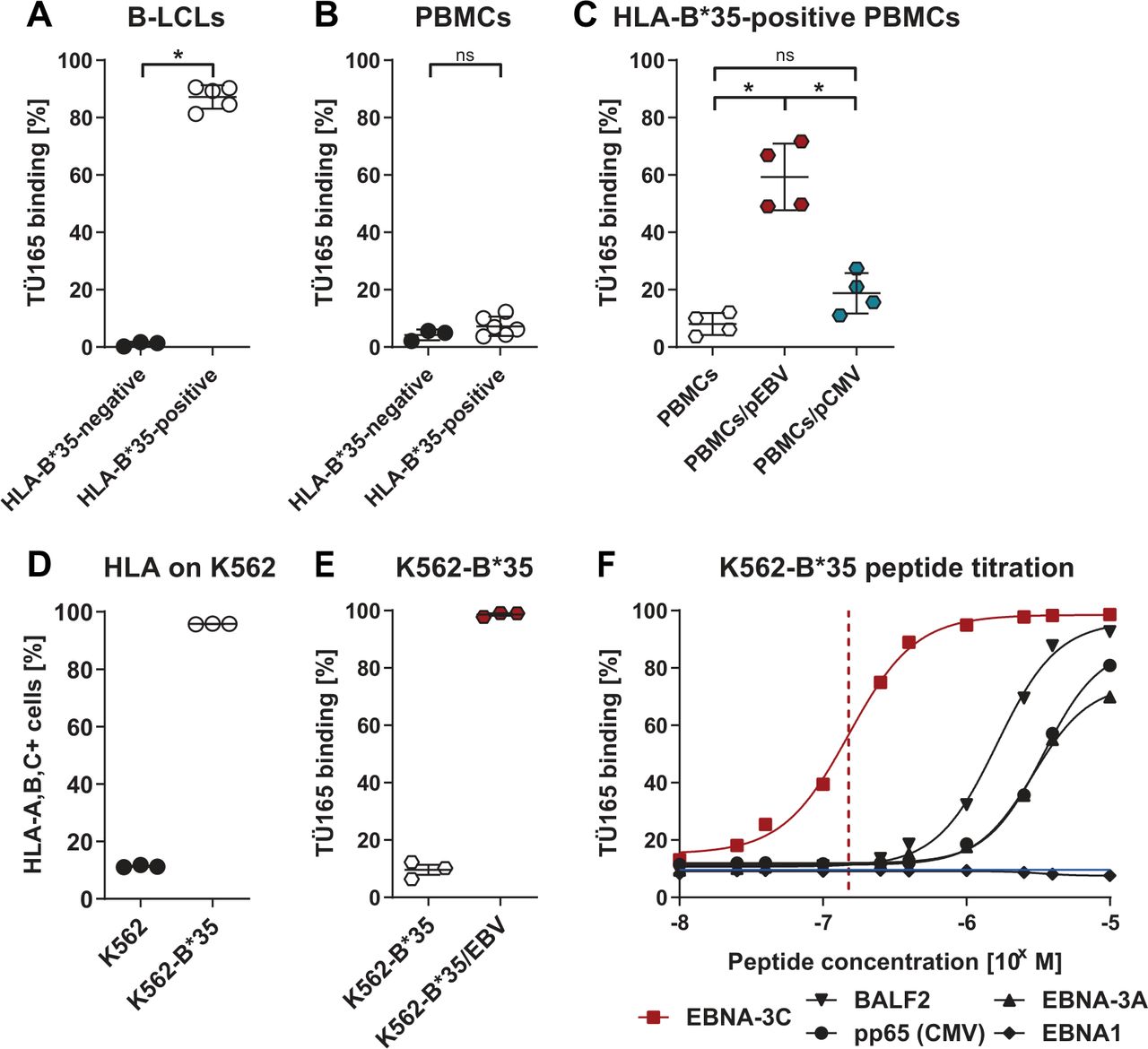
B-LCLs expressing EBV latency type III proteins similar to malignant EBV-PTLD cells6 were generated by infecting PBMCs with EBV. In the binding specificity assay, EBV-positive B-LCLs and PBMCs from the same donor were stained with TÜ165 culture supernatant, which recognized B-LCLs from HLA-B*35-positive donors at a frequency of 87%, but did not recognize HLA-B*35-negative B-LCLs and non-EBV-infected PBMCs (figure 1A,B). HLA-B*35:01-typed donors were used for these binding studies; however, all other HLA-B*35 alleles are also predicted to present the EBNA-3C-derived peptide LPPHDITPY enabling TÜ165 binding, which could be shown for HLA-B*35:03-positive B-LCLs (online supplemental figure S1). Binding of TÜ165 to PBMCs was enabled by exogenously loading HLA-B*35-positive PBMCs with the EBNA-3C-derived peptide (referred to as PBMCs/pEBV); this indicates that presentation of the EBNA-3C peptide in the HLA context is mandatory for antibody binding (figure 1C). Conversely, loading with an HLA-B*35-restricted control peptide derived from CMV_pp65 (IPSINVHHY; referred to as PBMCs/pCMV) resulted in significantly lower antibody binding.
Supplemental material

TÜ165 specifically binds to EBNA-3C-derived peptide/HLA-B*35 complexes. TÜ165 binding to (A) B-LCLs and (B) PBMCs from HLA-B*35-negative and HLA-B*35-positive donors. (C) TÜ165 binding to PBMCs from HLA-B*35-positive donors loaded with 10 µg/mL of EBNA-3C peptide LPPHDITPY or CMV pp65 peptide IPSINVHHY, respectively. (D to F) Studies of K562 cells lentivirally transduced to express HLA-B*35:01 (K562-B*35). (D) Confirmation of HLA class I expression. (E) TÜ165 binding to K562-B*35 loaded with 10 µg/mL EBNA-3C-derived peptide or (F) the indicated concentrations of peptides derived from EBV proteins EBNA-3C, BALF2 (YPLREVATL), EBNA-3A (VPATQPQY), and EBNA1 (HPVGEADYFEY) and CMV protein pp65 (IPSINVHHY). The red dashed line indicates the half-maximal binding of TÜ165 to K562-B*35/pEBV. The blue line indicates background binding of TÜ165 to unloaded K562-B*35. Data are shown as mean of 3 donors with a sigmoidal regression curve. (A to E) Data are shown as mean±SEM on scatter plots where each point represents one donor (n≥3). Statistical analysis was performed using the one-tailed Mann-Whitney U test. *p≤0.05. B-LCLs, B-lymphoblastoid cell lines; EBNA, Epstein-Barr nuclear antigen; ns, not significant; PBMCs, peripheral blood mononuclear cells; PBMCs/pCMV, pp65-peptide-loaded PBMCs; PBMCs/pEBV, EBNA-3C-peptide-loaded PBMCs.
In an additional model, K562 cells endogenously lacking HLA expression were lentivirally transduced to express HLA-B*35:01 (referred to as K562-B*35), resulting in HLA expression by 96% of cells (figure 1D). Binding assays showed that TÜ165 weakly recognized unloaded K562-B*35, whereas exogenous loading of K562-B*35 with the EBNA-3C-derived peptide LPPHDITPY (referred to as K562-B*35/pEBV) resulted in TÜ165 binding to 99% of the cells (figure 1E). Binding of TÜ165 to different peptide/HLA-B*35 complexes was peptide concentration-dependent (figure 1F). TÜ165 recognized K562-B*35/pEBV in a highly specific manner, with half-maximal binding at a low peptide loading concentration of 1.5×10–7 M, whereas K562-B*35 loaded with peptides derived from EBV_BALF2, EBV_EBNA1 and EBV_EBNA-3A or CMV_pp65 were bound at low levels of 9% to 12% when used at the same loading concentration.
TÜ165 CAR-Ts and TRUCKs with inducible IL-12 generated using a clinical manufacturing and expansion protocol have a favorable phenotype
A second-generation TÜ165 CAR was generated based on the TÜ165 scFv (figure 2A). TÜ165 TRUCKs were developed by inserting a nuclear factor of activated T-cells (NFAT)-driven inducible IL-12 expression module into the same vector (CAR iIL-12), as was previously described for a different CAR.31 A TÜ165 CAR with an iEGFP expression module was used as control (CAR iEGFP). As the extracellular spacer between the scFv and the transmembrane domain can strongly influence CAR functionality, we tested two different CAR constructs with a short (12 AA) and a long (228 AA) spacer, respectively, to determine the optimal distance between effector-cell and target-cell epitope, as was previously described for CARs with other specificities.30 34 35 T cells transduced with the TÜ165 CAR long-spacer construct showed lower functionality (no increase in CD137 and markedly lower cytotoxicity towards target cells) than the corresponding short-spacer construct with TÜ165 CAR-Ts (online supplemental figures S2A,B). For this reason, all constructs used for further experiments contained the short spacer.
Supplemental material
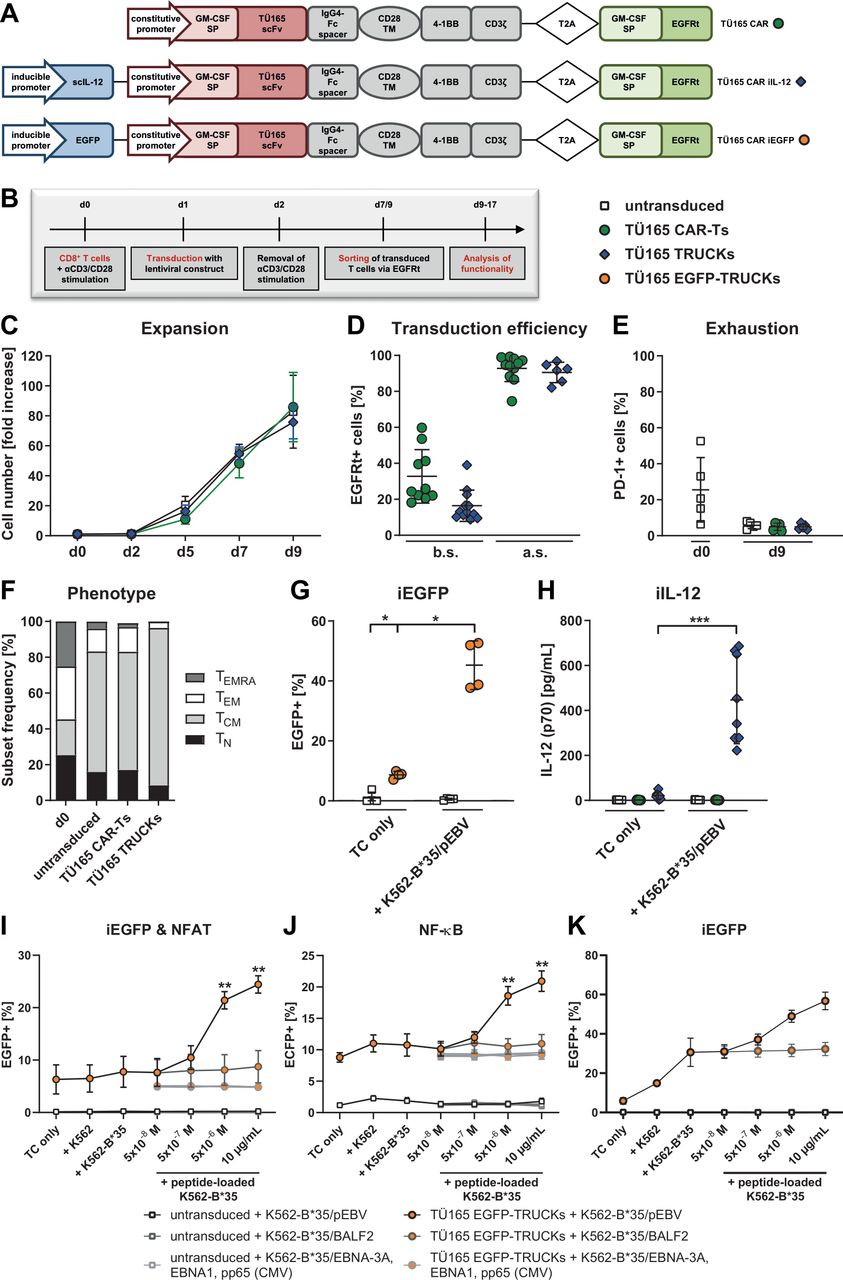
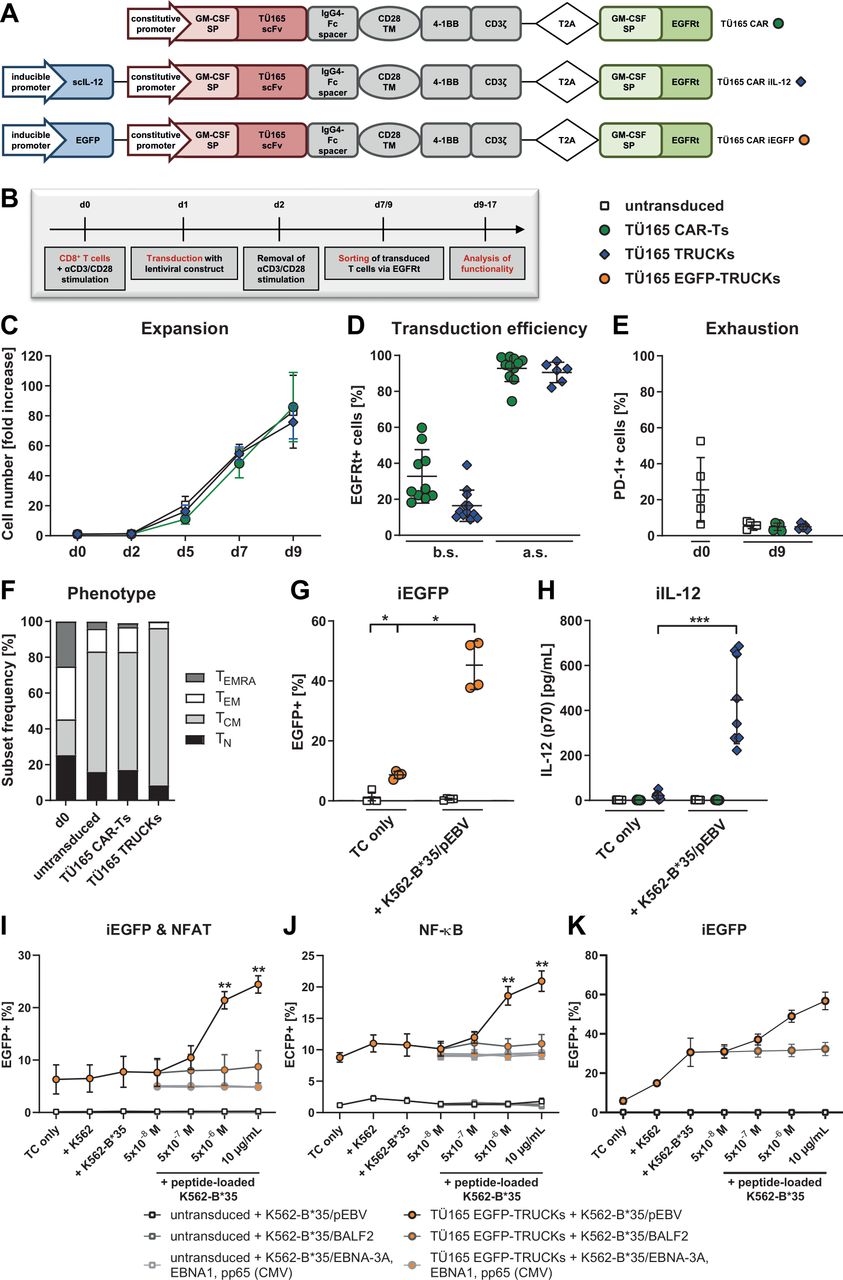
TÜ165 CAR-Ts and TRUCKs generated using a clinical manufacturing protocol have a favorable phenotype. (A) The TÜ165 CAR construct contained a constitutive promoter for the expression of granulocyte-macrophage colony-stimulating factor (GM-CSF) signal peptide (SP), a single-chain variable fragment (scFv) of TÜ165, a 12 AA short spacer based on IgG4-Fc, a CD28 transmembrane domain (TM), the cytoplasmic signaling domains of 4-1BB and CD3ζ, and a GM-CSF signal peptide with a truncated epidermal growth factor receptor (EGFRt) sequence connected by a T2A ribosomal skip element. TÜ165 CAR constructs with iIL-12 and iEGFP additionally contained an inducible promotor element with six NFAT response elements and a synthetic promoter (NFATsyn) in addition to human IL-12 or EGFP, respectively. (B) Experimental layout for CD8+ T-cell transduction with lentiviral constructs, sorting and expansion. (C) T cell expansion, as assessed by cell counting: The fold increase in cells was calculated by dividing the cell numbers on a given day by the respective cell numbers on day 0 (d0). Data are shown as mean±SEM of 4 donors. (D) Transduction efficiency, as measured by staining T cells with biotin-anti-EGFRt and PE-streptavidin antibody on d9 before sorting (b.s.) and after sorting (a.s.). (E) PD-1 expression during expansion served as an indicator of exhaustion. (F) T cells were phenotyped on d0 and d9 and characterized as either naïve (TN, CD45RA+CD62L+), central memory (TCM, CD45RA-CD62L+), effector memory (TEM, CD45RA-CD62L-) or terminally differentiated effector memory cells re-expressing CD45RA (TEMRA, CD45RA+CD62L-). Data are shown as mean of 5 donors. (G) Induction of EGFP expression and (H) IL-12 release by T cells cultured alone (TC only) or with the specified target cells for 48 hours. (I, J, K) A reporter JE6-1 Jurkat T cell line (I, J) or primary T cells (K) were transduced with the TÜ165 CAR iEGFP construct to generate TÜ165 EGFP-TRUCKs. They were co-cultured with the indicated unloaded target cells or K562-B*35 loaded with the specified concentrations of peptides derived from EBV proteins EBNA-3C, BALF2 (YPLREVATL), EBNA-3A (VPATQPQY), and EBNA1 (HPVGEADYFEY) and CMV protein pp65 (IPSINVHHY) at an E:T ratio of 1:1. After 24 hours (I, J) or 48 hours (K), induction of the iEGFP cassette (I, K) and the NFAT EGFP reporter (I) as well as the NF-κB ECFP reporter (J) was assessed by flow cytometry. Data are shown as mean±SEM of 2–5 donors. (D, E, G, H) Data are shown as mean±SEM on scatter plots where each point represents one donor (n≥4). Statistical analysis was performed using the one-tailed Mann-Whitney U test. *p≤0.05, ***p≤0.001. CAR-T, chimeric antigen receptor-T cells; iEGFP, inducible enhanced green fluorescent protein; iIL-12, inducible interleukin-12; NFAT, nuclear factor of activated T-cells; TRUCK, T cells redirected for universal cytokine-mediated killing.
CD8+ T cells were lentivirally transduced and expanded under conditions similar to those used for clinical manufacturing (figure 2B).36 As with the untransduced T cells, we performed 76-fold to 86-fold expansion of TÜ165 CAR-Ts and iIL-12-secreting TÜ165 CAR-based TRUCKs (referred to as TÜ165 TRUCKs) in 9 days (figure 2C). The transduction efficiency was 33% for the CAR without and 16% for the CAR construct with iIL-12, which was enriched to 93% and 91%, respectively, by using the co-expressed marker EGFRt for purification (figure 2D). Both untransduced and transduced T cells showed no sign of exhaustion after 9 days of expansion as PD-1 expression, an indicator of exhaustion and activation, decreased to 5% (figure 2E). LAG-3 and TIM-3 were shown to be upregulated following anti-CD3/CD28 stimulation but also decreased again until day 16 (data not shown). The T-cell phenotypes largely shifted to an increased proportion of central memory (TCM; 67% to 88%) and lower frequencies of naïve (TN; 8% to 17%), effector memory (TEM; 3% to 14%) and effector memory RA (TEMRA; 0 to 4%) T cells after expansion (figure 2F).
To confirm CAR-induced cytokine release in TÜ165 TRUCKs, T cells harboring the TÜ165 CAR iEGFP control (referred to as TÜ165 EGFP-TRUCKs) were co-cultured with K562-B*35/pEBV as target cells for 48 hours before EGFP expression measurement. EGFP expression was significantly upregulated (45%) in TÜ165 EGFP-TRUCKs cultured with the target cells, but remained low (8.7%) in those cultured without them (figure 2G). IL-12 assays showed that iIL-12-equipped TÜ165 TRUCKs cultured with the same target cells released much higher amounts of IL-12 (447 pg/mL) than those cultured without them (21.1 pg/mL), and that TÜ165 CAR-Ts without the iIL-12 cassette did not express IL-12 (figure 2H). These findings demonstrate that the inducible cassette was activated in TÜ165 TRUCKs after target-cell engagement.
Specificity of the TÜ165-based CAR was determined by using a previously described reporter assay and the Jurkat T cell line JE6-1.32 33 TÜ165 CAR iEGFP-transduced Jurkat cells did not respond to co-cultivation with K562, K562-B*35 or K562-B*35 loaded with peptides derived from EBV_BALF2, EBV_EBNA1 and EBV_EBNA-3A or CMV_pp65, whereas both, the reporter for NFAT as well as the indicator for nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), were induced upon recognition of K562-B*35/pEBV in a concentration-dependent manner (figure 2I,J). The same peptide specificity could be observed for the induction of the inducible EGFP cassette in primary EGFP TRUCKs (figure 2K). Background activation of these cells following co-cultivation with K562 and K562-B*35 can, most likely, be attributed to an endogenous TCR-mediated, allogeneic response towards K562, since the JE6-1 cell line harboring the TÜ165 CAR but lacking any TCR did not respond towards unloaded K562.
TÜ165 CAR-T-cell response to HLA/EBV peptide recognition, characterized by activation marker and cytokine upregulation, is improved by inducible IL-12
Target recognition by engineered T cells was assessed by measuring activation marker and cytokine expression in response to co-culture with K562-B*35/pEBV versus K562-B*35. Untransduced CD8+ T cells did not show CD137 expression (<2%) in response to either target-cell type, whereas TÜ165 CAR-Ts showed increased CD137 expression (4.7%) in response to the EBV peptide-loaded target cells (figure 3A). TÜ165 TRUCKs showed markedly increased CD137 expression (17.7%) in response to K562-B*35/pEBV. Specific CD137 upregulation was defined as the difference between expression levels in T cells cultured with K562-B*35/pEBV and unloaded K562-B*35 (individual data sets are shown in online supplemental figures S3A–G). CAR-Ts and TRUCKs showed significant specific CD137 upregulation (3% and 6%, respectively), whereas untransduced T cells did not (−0.5%) (figure 3B). The specific upregulation of activation markers was further confirmed by detecting the MFI of CD25 on T cells. Compared with levels in untransduced T cells, CD25 MFIs in both TÜ165 CAR-Ts and TRUCKs increased significantly in response to the EBV peptide (MFI of 960 and 5300, respectively), and TÜ165 TRUCKs showed a significantly stronger response than TÜ165 CAR-Ts (figure 3C).
Supplemental material

TÜ165 CAR-T-cell activation and cytokine response to HLA/EBV peptide recognition is improved by inducible IL-12. Engineered T cells were co-cultured with EBNA-3C peptide-loaded K562-B*35 (K562-B*35/pEBV) at an effector-to-target (E:T) ratio of 1:1 for 48 hours. (A) CD137 expression after co-culture of the specified CD8+ T cells with unloaded (K562-B*35) or loaded (K562-B*35/pEBV) target cells (representative plots). (B) Specific upregulation of CD137 (%), (C) upregulation of CD25 mean fluorescence intensity (MFI), (E) TNF-α upregulation (%), and (G) IFN-γ upregulation (%) on CD8+ T cells as well as specific release of (D) TNF-α, (F) IFN-γ and (H) IL-2 (pg/ml) into cell culture supernatants were calculated by subtracting the respective read-outs after co-culture with unloaded K562-B*35 from those after co-culture with K562-B*35/pEBV. (B to H) Data are shown as mean±SEM on scatter plots where each point represents one donor (n≥3). Statistical analysis was performed using the one-tailed Mann-Whitney U test. *p≤0.05, **p≤0.01, ***p≤0.001. CAR-T, chimeric antigen receptor-T cells; EBV, Epstein-Barr virus; IFN-γ, interferon-γ; IL, interleukin; TNF, tumor necrosis factor; ns, not significant; TRUCK, T cells redirected for universal cytokine-mediated killing.
The release of TNF-α into the cell culture supernatant increased significantly (by 1.4 pg/mL) in TÜ165 TRUCKs co-cultured with target cells, whereas untransduced T cells did not secrete more TNF-α on contact with EBV-loaded versus unloaded K562-B*35 (figure 3D). This finding was confirmed by intracellular staining of TNF-α, which revealed a slight upregulation in TÜ165 CAR-Ts in response to the EBV peptide (9.5%) and a significant upregulation in TÜ165 TRUCKs (17%) (figure 3E). IFN-γ was released similarly: TÜ165 TRUCKs secreted significantly more (3300 pg/mL) IFN-γ as target response than both, untransduced T cells (2.2 pg/mL) and TÜ165 CAR-Ts (4.5 pg/mL), and intracellular staining confirmed this (figure 3F,G). TÜ165 TRUCKs also showed a proliferative response to the EBV target: IL-2 secretion by TÜ165 TRUCKs was upregulated by 9.4 pg/mL, whereas no measurable increase was detected in untransduced T cells or TÜ165 CAR-Ts (figure 3H).
TÜ165 CAR-T response to EBV-infected cells, characterized by activation marker and cytokine upregulation, is improved by inducible IL-12
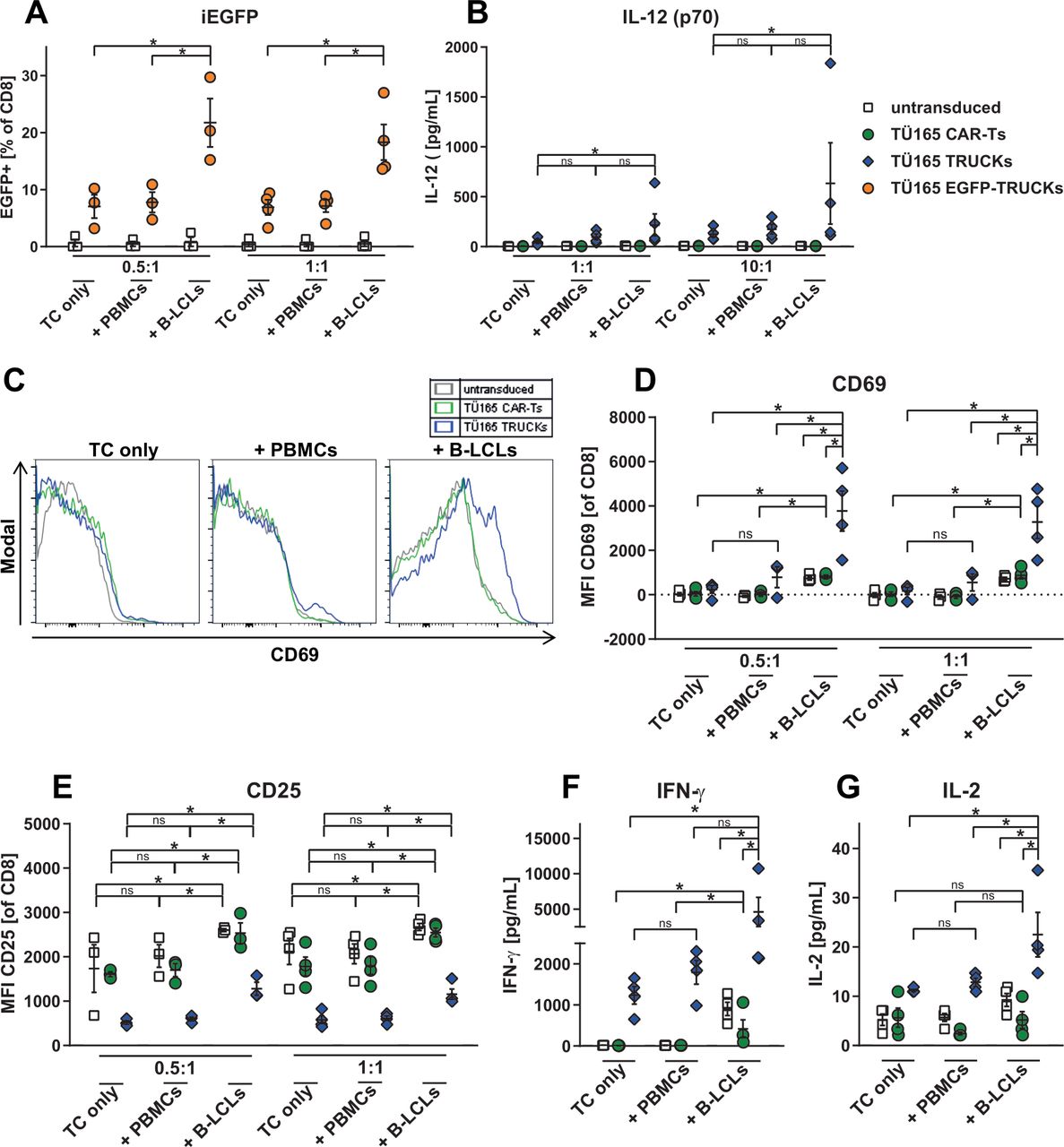
Further, mimicking an EBV lymphoproliferation model, we used autologous EBV-infected B-LCLs as target cells and the respective uninfected PBMCs as controls. The results confirmed activation of the inducible expression cassette in TÜ165 TRUCKs on B-LCL engagement: EGFP expression was upregulated (18% to 22%) in TÜ165 EGFP-TRUCKs co-cultured with the EBV-infected B-LCLs at different E:T ratios, but remained at background levels (7% to 8%) when the TÜ165 EGFP-TRUCKs were cultured alone or with uninfected PBMCs (figure 4A). TÜ165 TRUCKs equipped with the inducible IL-12 cassette responded to B-LCLs by releasing higher amounts of IL-12 (45 to 216 pg/mL), but showed no significant increase in response to the uninfected target cells (figure 4B). As proof of target recognition and activation response, we measured CD69 expression on engineered T cells as MFI. After co-culture with B-LCLs at different E:T ratios, MFI CD69 values for TÜ165 CAR-Ts increased (840 to 870) compared with those for TÜ165 CAR-Ts cultured alone or with PBMCs (<42) (figure 4C,D). TÜ165 TRUCKs showed the same increase pattern, but the effect was even higher. CD25 MFI analysis revealed a reduced pre-activation of TÜ165 TRUCKs after expansion, but confirmed upregulation of activation marker expression on TÜ165 CAR-Ts and TÜ165 TRUCKs co-cultured with B-LCLs, but not on those co-cultured with PBMCs (figure 4E). However, CD25 upregulation most likely can be attributed to a TCR-mediated response of EBV-specific memory T cells as also untransduced T cells reacted equally and only EBV-seropositive donors were utilized. As specific response to B-LCLs, TRUCKs released IFN-γ: IFN-γ levels were threefold higher in TÜ165 TRUCKs co-cultured with B-LCLs than in those co-cultured with PBMCs or cultured alone (figure 4F). TÜ165 CAR-Ts showed a similar response to a smaller extent, equally to untransduced T cells. Moreover, IL-2 release in TÜ165 TRUCKs cultured with B-LCLs was enhanced; TÜ165 CAR-Ts showed only slight and non-significant increases in IL-2 (figure 4G).

TÜ165 CAR-T activation and cytokine response to EBV-infected cells is improved by inducible IL-12. Engineered T cells were cultured for 48 hours alone (TC only), with autologous EBV-infected B-LCLs (+BLCLs) or with uninfected PBMCs (+PBMCs), respectively. (A) Expression levels of EGFP and (B) release of IL-12 were determined in T cells cultured alone and with the specified target cells at the indicated E:T ratios for proof of induction of the NFAT-driven inducible expression cassette. (C, D) CD69 median fluorescence intensity (MFI) values for CD8+ T cells cultured alone and with PBMCs or B-LCLs, respectively, at an E:T ratio of 0.5:1 in representative histograms (C) and in scatter plots at E:T ratios of 0.5:1 and 1:1 (D). (E) CD25 MFI values for the aforementioned cells and E:T ratios. (F) IFN-γ and (G) IL-2 concentrations (pg/mL) were measured in cell culture supernatants at an E:T ratio of 10:1 by Legendplex analysis. (A, B, D–G) Data are shown as mean±SEM on scatter plots where each point represents one donor (n≥3). Statistical analysis was performed using the one-tailed Mann-Whitney U test. *p≤0.05. B-LCLs, B-lymphoblastoid cell lines; CAR-T, chimeric antigen receptor-T cells; iEGFP, inducible enhanced green fluorescent protein; IFN-γ,interferon-γ; IL, interleukin; ns, not significant; NFAT, nuclear factor of activated T-cells; PBMCs, peripheral blood mononuclear cells; TRUCK, T cells redirected for universal cytokine-mediated killing.
TÜ165 CAR-Ts kill EBV-positive target cells and release effector molecules
Cytotoxicity of TÜ165 CAR-Ts and TRUCKs towards target cells was determined by measuring the release of LDH into cell culture supernatants. When co-cultured with unloaded K562-B*35, untransduced T cells and TÜ165 CAR-Ts exhibited similarly low cytotoxicities of 11% and 12% to 13%, respectively (figure 5A). Whereas untransduced T cells showed the same level of cytotoxicity towards K562-B*35/pEBV, TÜ165 CAR-Ts and TÜ165 TRUCKs showed significantly higher cytotoxicity (20% and 25%, respectively) after target-cell recognition. The amount of released effector molecules (perforin and granzyme B) in the supernatant confirmed these results since target-specific secretion of both mediators was significantly higher in both TÜ165 CAR-Ts and TÜ165 TRUCKs compared with levels in untransduced T cells (figure 5B,C; individual data sets in online supplemental figures S3H,I). TÜ165 TRUCKs secreted more perforin and significantly more granzyme B than TÜ165 CAR-Ts, indicating that the induced IL-12 resulted in improved effector T-cell function.

TÜ165 CAR-Ts kill EBV-positive target cells and release effector molecules. (A to C) Results for engineered T cells co-cultured with K562-B*35/pEBV for 48 hours at an effector-to-target (E:T) ratio of 1:1. (A) The release of lactate dehydrogenase (LDH) into the cell culture supernatant was measured as an indicator of the killing of target cells (unloaded K562-B*35 or K562-B*35/pEBV) by T cells. LDH levels are expressed as a percentage of the maximum lysis level obtained using controls lysed with 1% Triton X-100. (B) Specific release of perforin and (C) specific release of granzyme B; specific release into the co-culture supernatant was calculated by subtracting the value measured after co-culture with K562-B*35 (unloaded) from the value after co-culture with K562-B*35/pEBV. (D to F) Engineered T cells were co-cultured with autologous B-LCLs for 48 hours at (D,E) the indicated E:T ratios in an representative experiment or (F) an E:T ratio of 1:1. (D) B-LCLs were gated as CD3-CD8- cells. (E) B-LCL killing was calculated by dividing the frequency after 48 hours by the respective frequency on day 0 and subtracting it from 100%. (F) Release of granzyme B into the cell culture supernatant of the indicated T cells cultured alone (TC only) or in co-cultures with B-LCLs (+B LCLs). (G) Carboxyfluorescein succinimidyl ester (CFSE)-labeled untransduced T cells (green), CellTrace Violet-stained TÜ165 CAR-Ts (blue), and unlabeled K562-B*35/pEBV were set at a ratio of 1:1:2 and monitored under the microscope (Olympus IX81) at the indicated time points. Tracking experiments were performed using 20X short-distance objective lenses, and the results were analyzed using Olympus ScanR analysis software. Cell sizes of effector and target cells were estimated using a digital field of view of 433×330 µm and a resolution of 1344×1024 pixels instead of analog scale bars. (A to C, F) Data are shown as mean±SEM on scatter plots where each point represents one donor (n≥4). Statistical analysis was performed using the one-tailed Mann-Whitney U test. *p≤0.05, **p≤0.01, ***p≤0.001. B-LCLs, B-lymphoblastoid cell lines; CAR-T, chimeric antigen receptor-T cells; EBV, Epstein-Barr virus; ns, not significant; TRUCK, T cells redirected for universal cytokine-mediated killing.
Using the EBV lymphoproliferation model, we assessed the number of B-LCLs after 48 hours of co-culture with TÜ165 TRUCKs by FACS. An initial experiment showed killing of B-LCLs by TÜ165 TRUCKs, whereas B-LCLs expanded when co-cultured with untransduced T cells (figure 5D,E). This was confirmed by an increased release of granzyme B in co-cultures of TÜ165 TRUCKs with autologous B-LCLs (figure 5F).
Live cell imaging revealed specific target-cell contact of TÜ165 CAR-Ts. For this experiment, TÜ165 CAR-Ts and untransduced T cells were added in equal amounts and co-cultured with K562-B*35/pEBV. After 45 min of culture, transduced TÜ165 CAR-Ts became active and elongated on contact and were recruited by the target cells, whereas untransduced T cells remained mainly inactive and round (figure 5G, online supplemental figure S4). After 2 hours of culture, large clusters of TÜ165 CAR-Ts and target cells were detected.
Supplementary video
TÜ165 TRUCKs facilitate the recruitment of MM6 and NK-92 cells and improve effector functions of NK-92 cells
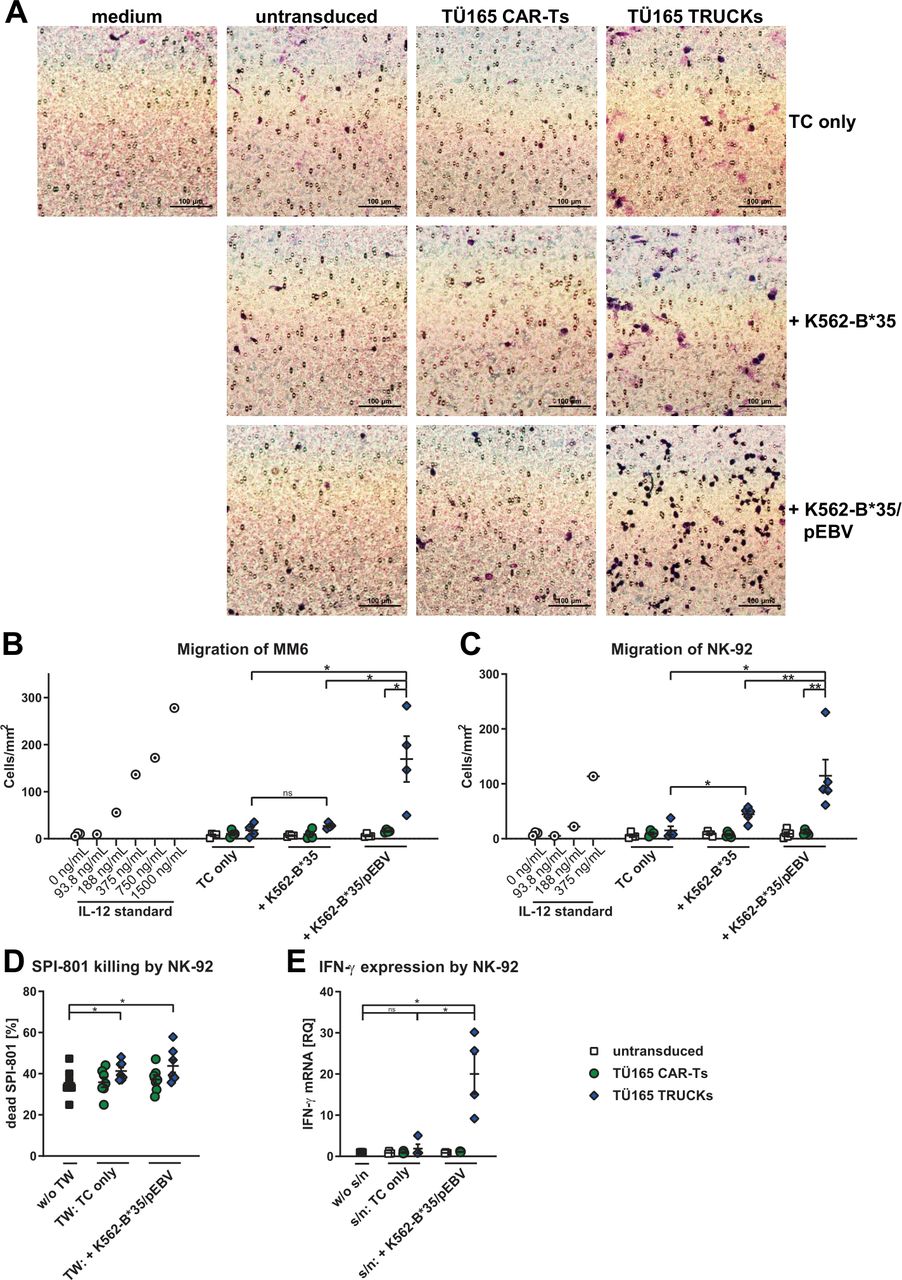
To determine if the IL-12 released by TÜ165 TRUCKs after target-cell contact specifically recruits and activates macrophages and NK cells as described for other TRUCKs,13 we tested supernatants of engineered T cells co-cultured with K562-B*35/pEBV for 48 hours for their chemoattractive potential towards monocytes (Mono-Mac-6 (MM6) cells) and NK cells (NK-92 cells) by using a Boyden chamber. MM6 cells migrated through a membrane with a pore size of 8 µm at a density of 170 cells/cm2 when attracted by supernatants of TÜ165 TRUCKs co-cultured with K562-B*35/pEBV compared with a density of only 16 to 18 cells/cm2 when attracted by supernatants of TÜ165 TRUCKs cultured alone or with unloaded K562-B*35 (figure 6B). Similarly, NK-92 cells migrated towards supernatants of TÜ165 TRUCKs co-cultured with peptide-loaded K562-B*35 at a higher density (figure 6A,C). Supernatants of untransduced T cells and TÜ165 CAR-Ts did not attract MM6 or NK-92 cells.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
TÜ165 TRUCKs recruit MM6 and NK-92 cells and improve effector functions of NK-92 cells. (A to C) Chemoattractive potential of IL-12 released by TÜ165 TRUCKs on target recognition in terms of the migration of (A,C) NK cells (NK-92 cells) and (B) monocytes (MM6 cells). Supernatant of engineered T cells cultured alone (TC only) or together with unloaded K562-B*35 or K562-B*35/pEBV, respectively, for 48 hours were diluted 1:3, placed in a Boyden chamber, covered with an 8 µm polycarbonate membrane and incubated for another 4 hours. Medium served as the control supernatant. The membranes were Giemsa stained. (A) Representative pictures of Giemsa-stained NK-92 cells (violet) on the bottom of the membrane. (B, C) The number of cells that migrated through the membrane was determined. Different concentrations of IL-12 served as control supernatants. (D) Engineered T cells were cultured alone (TC only) or with K562-B*35/pEBV at an E:T ratio of 1:1 in a transwell (TW) insert with a pore size of 0.4 µm above a co-culture of NK-92 cells with CellTrace Violet-stained SPI-801 cells at an E:T ratio of 1:1. NK-92 cells co-cultured with SPI-801 cells with no cells in the transwell (w/o TW) served as controls. Dead SPI-801 cells were detected based on forward and side scatter properties of CellTrace Violet+ cells. (E) Supernatants (s/n) of engineered T cells cultured alone or together with K562-B*35/pEBV for 48 hours were diluted 1:2.5 and utilized as culture medium for a co-culture of NK-92 cells with SPI-801 at an E:T ratio of 1:1. A co-culture in RPMI 1640 with 10% FBS and 2 mM L-glutamine (w/o s/n) served es control. After 4 hours, total RNA was isolated, reverse-described and quantified by quantitative PCR whereby Glyceraldehyde-3-phosphate dehydrogenase served as internal control for calculation of RQ values. (B to E) Data are shown on scatter plots where each point represents one donor (n≥4; IL-12 standards n=1). Statistical analysis was performed using the one-tailed Mann-Whitney U test. *p≤0.05, **p≤0.01. CAR-T, chimeric antigen receptor-T cells; IFN-γ, interferon-γ; MM6, Mono-Mac-6; mRNA,messenger RNA; NK, natural killer; ns, not significant; TRUCK, T cells redirected for universal cytokine-mediated killing.
To measure the effect of the released IL-12 on NK-cell activity, we cultured engineered T cells in a transwell allowing cytokine exchange but not cell-cell contact with an NK-92 co-culture with HLA-class I-negative SPI-801 cells. The target-induced release of IL-12 by TÜ165 TRUCKs resulted in an increased killing capacity of NK-92 cells towards SPI-801 cells: killing increased from 35% to 41% in transwell-conditions without induced IL-12 to 44% in transwell conditions with target-activated TRUCKs (figure 6D). Further, the effect of IL-12 containing supernatants of engineered T cells co-cultured with K562-B*35/pEBV was measured in co-cultures of NK-92 and SPI-801. Quantitative PCR revealed a significantly increased expression of IFN-γ after 4 hours when the cells were cultivated in prediluted supernatants of TÜ165 TRUCKs co-cultured with K562-B*35/pEBV (RQ of 20) compared with cultivation in the control supernatants (RQ of 1 to 2) (figure 6E). This indicates that the released target-induced IL-12 by TÜ165 TRUCKs was biologically active. TÜ165 CAR-Ts had no impact on the functionality of NK-92 cells.
Discussion
Currently, the two dominant strategies for engineering T cells are to introduce either a recombinant TCR or a CAR into T cells. TCR-like CAR-Ts combine the advantages of TCR- and CAR-engineered T cells. Major concerns when generating TCR-engineered T cells (TCR-Ts) are the development of unexpected specificities due to mispairing of the introduced TCR with the endogenous TCR and low expression of the introduced TCR; however, some strategies were recently developed to circumvent these limitations.37 The use of CAR-Ts, on the other hand, prevents dimer formation with the endogenous TCR. Some studies already showed impressive anti-tumor responses, leading to the recent approval of two CD19-targeting CAR-T therapies for the treatment of different B-cell malignancies.38 Since the depletion of all B cells includes EBV-infected lymphoma cells, CD19 CAR-T transfer for the treatment of PTLD is conceivable. However, as is the case for current PTLD treatment options like chemotherapy and antibody-mediated B-cell depletion, this strategy would be limited by the patient’s poor condition and the high level of immunosuppression needed to prevent organ rejection.4–6 Thus, an approach targeting only EBV-infected B cells and sparing healthy B cells to treat PTLD without diminishing the already low numbers of immune cells in these patients would be very desirable. Adoptively transferred EBV-specific T cells address this problem, but their use is often limited by time constraints or by the lack of an appropriate donor.6 Building on this strategy in the present study, we utilized the TCR-like mAb TÜ165, which binds to a PTLD-associated, EBNA-3C-derived peptide presented in the HLA-B*35 context. TCR-like antibodies have shown promise as therapeutic agents designed for the targeting of tumor and virus-infected cells.16–22 Otherwise, for the HLA-B*35/LPPHDITPY epitope no naturally occurring TCRs could be found (online supplemental figure S5). The absence of T cells with natural specificity against the epitope can be explained by the fact that the epitope is not immunodominant, respective T-cell frequencies are too low to induce a detectable response or that specific T cells have been deleted via tolerance mechanisms due to self-reactivity.
Supplemental material
In the present study, TÜ165 bound to EBV-positive B-LCLs from HLA-B*35-positive donors expressing similar EBV proteins as PTLD cells.6 B-LCLs were generated by using the laboratory EBV strain B95.8, but also naturally occurring strains C666-1 (GenBank no. AB828190.1), HKNPC1 (GenBank no. JQ009376.2) and M81 (GenBank no. KF373730.1) encode the LPPHDITPY peptide indicating the recognized sequence is conserved in different EBV strains. Only the laboratory strain AG867 (GenBank no. DQ279927.1) has an amino acid exchange at the first position of the peptide (leucine → phenylalanine; L → F; FPPHDITPY), to which the TÜ165 antibody and CAR binding would need to be assessed in further studies. Besides B-LCLs, the mAb TÜ165 bound to EBNA-3C peptide-loaded PBMCs and equally loaded HLA-B*35-transduced K562s (K562-B*35/pEBV). This confirms the reported capability of TÜ165 to bind to EBNA-3C peptide/HLA-B*35:01 epitopes.26 27 The peptide was predicted to be presented by a majority of HLA-B*35 subtypes (www.cbs.dtu.dk/services/NetMHCpan; 07.07.202028) and HLA-B*35 is a prevalent allele (www.allellefrequencies.net; 07.07.202029). HLA-B*35:03-positive B-LCLs could be stained with the mAb TÜ165 with the same or slightly lower frequency confirming the predicted peptide binding and indicating TÜ165 is also able to recognize the EBNA-3C peptide in context of other HLA-B*35 subtypes than the reported HLA-B*35:01. It would be interesting to assess the binding capability of the TÜ165 antibody and CAR to the EBV peptide in complex with different HLA-B*35 subtypes in further studies. TÜ165 binding was moreover peptide concentration-dependent. EBNA-3C peptide-loaded K562-B*35 were recognized at low loading concentrations, indicating a high binding specificity, whereas other EBV/CMV peptides were not. Importantly, unloaded K562-B*35 and PBMCs can be expected to present a variety of peptides derived from self-proteins on their HLA-B*35 molecules,39 but TÜ165 did not bind to them. Consequently, we conclude that TÜ165 binding is restricted to the EBNA-3C-derived peptide when presented by HLA-B*35 and that binding to other peptides will occur only at unphysiologically high peptide loading concentrations. Overall, the observed differential recognition of B-LCLs and PBMCs as well as the recognition of EBNA-3C peptide-loaded versus unloaded K562-B*35 confirms the suitability of TÜ165 to specifically distinguish between EBV-associated target and healthy cells.
TÜ165-based CAR-Ts and TRUCKs showed a favorable phenotype with low PD-1 expression, indicating that the cells were not exhausted despite being expanded around 100-fold in the first 9 days using an expansion protocol similar to the one used for clinical manufacturing. Only following activation with CD3/CD28 beads, expression of PD-1, TIM-3 and LAG-3 was transiently increased on T cells (data not shown). However, since these markers are also activation markers, only long-term expression would demonstrate an exhausted state.40 Here, after cultivation for 16 days, expression of all markers markedly decreased again (data not shown) and cells were shown to be functional. Therefore, we deduced that T-cell dysfunction might not be a major problem in TÜ165 CAR-T and TRUCK generation. Previous studies have shown that the phenotype of adoptively transferred T cells predicts their long-term persistence and engraftment in vivo.41 42 In this context, less differentiated TCM cells, which formed the main population after expansion in the present study, showed superior functionality in vivo: they exhibited long-term persistence and occupied memory T-cell niches.
When TCR-like CAR-Ts were first introduced as a new strategy, various problems had to be addressed. For example, one study using CARs based on high-affinity TCR-like antibodies recognizing Wilms tumor suppressor gene 1 (WT1) peptide/HLA-A02 complexes showed a loss of specificity and elimination of HLA-A*02-positive, peptide-negative cells, whereas the low-affinity TCR-like CAR-Ts maintained potent cytotoxicity and specificity.43 Maus et al 44 later confirmed that lowering the affinity of a TCR-like CAR to the TCR affinity range ensures epitope specificity and effective cytotoxicity of the CAR. This is most likely due to the ability of the CAR-Ts to sequentially engage peptide/HLA complexes, as is reportedly necessary for proper T-cell activation. In the present study, the TÜ165 CAR was shown to be activated highly specifically only on recognition of K562-B*35/pEBV in a concentration-dependent manner, whereas unloaded K562, K562-B*35 or K562-B*35 loaded with other peptides did not activate the CAR. Only when transduced into primary T cells, a background activation of TÜ165 CAR-Ts and TRUCKs following stimulation with unloaded K562 and K562-B*35 was detected. A major drawback of this model is allogenicity. Although K562 cells only express low levels of HLA (mainly HLA-C),45 untransduced T cells mildly recognize the cell line, which is shown by a very slight upregulation of CD137 and CD25 and release of TNF-α and IFN-γ in co-cultures with unloaded K562-B*35 (online supplemental figure S3). TÜ165 CAR-Ts exhibit a response towards the unloaded control cells to a similar extent showing that this response most likely is mediated by the TCR and not unspecifically by the introduced CAR towards empty HLA-B*35. TÜ165 TRUCKs, however, showed an even enhanced response to unloaded K562-B*35, which might be due to the fact that allogeneic recognition of HLA on the surface of K562 cells by the TCR induces NFAT expression and a subsequent induction of the IL-12 cassette. This hypothesis could be confirmed by testing the TÜ165 EGFP-TRUCK in JE6-1 Jurkat cells as effector cells. By the use of these cells lacking endogenous TCRs, no background response towards either K562 or K562-B*35 cells was observed. The expression of EGFP and ECFP is only induced upon loading with the EBNA-3C-derived peptide. These findings showed that the recognition of the epitope by the scFv of the CAR is highly specific.
When using primary T cells as effector cells, substracting the activation and cytokine data obtained for co-cultures with unloaded K562-B*35 from the ones in co-cultures with K562-B*35/pEBV could indicate the LPPHDITPY-specific component of the TÜ165 CAR-T-cell recognition. Here, TÜ165 CAR-Ts showed functionality by upregulating the expression of activation markers (CD137 and CD25) and by slightly increasing intracellular IFN-γ production in specific response to K562-B*35/pEBV but not to unloaded K562-B*35 controls. Moreover, they exhibited cytotoxicity by upregulating perforin and granzyme B when co-cultured with EBV peptide-loaded target cells and formed large clusters around them. When cultured with autologous B-LCLs in a model of lymphoproliferation, they increased the expression of activation markers (CD69 and CD25) and released IFN-γ in a range similar to that of untransduced T cells. Of note, expansion and off-target cytotoxicity of TÜ165 CAR-Ts towards unloaded K562 cells and uninfected PBMCs was equal to that of untransduced T cells. This indicates that they did not recognize HLA-B*35 without EBNA-3C peptide or when complexed with other naturally occurring peptides. The maintenance of specificity in TÜ165 CAR-Ts can be explained by the fact that the CAR is based on an immunoglobulin M (IgM) antibody, which normally has 10 antigen-binding sites. When reduced to a single antigen-binding site in the TÜ165 CAR, a lower avidity and weaker binding might be the consequence. This, in turn, allows the aforementioned sequential peptide/HLA engagement and yields functional CAR-Ts. In general, effector functions of the TCR-like TÜ165 CAR were rather mild when compared with other CARs, such as the well-described CD19 CAR. However, CAR activation is known to be highly dependent on the expression levels of target antigen46 explaining that the target of the TÜ165 CAR, a peptide presented in HLA context, induces less CAR activation as compared with other highly-expressed surface molecules such as CD19. Considering their weak target binding and mild functionality, it would need to be evaluated, if TÜ165 CAR-Ts are able to effectively control EBV-associated lymphoproliferation for a potential future clinical use. For this, detailed in vivo testing is now required. Regarding possible models to study TÜ165 CAR-T function, immunocompromised mice previously engrafted with human B-LCLs are a model of EBV-associated PTLD that was recently used to test an EBV-specific, TCR-like mAb for the treatment of PTLD.20 Further, generation and testing of other TCR-like CARs and TRUCKs recognizing EBV epitopes in context of PTLD would be desirable to extend the therapy to HLA-B*35-negative patients. As source for the scFv, TCR-like mAbs were described that recognize EBV-derived peptides in context of HLA-A*02:01,20 47 and more can be generated using conventional hybridoma and phage display technology.16 48
Furthermore, we generated TÜ165 TRUCKs which induced IL-12 secretion only on recognition of the TÜ165 target. We encoded constitutive CAR expression and inducible cytokine expression in a novel ‘all-in-one’ vector to further facilitate the manufacturing of these cells for clinical applications and to reduce the risk of insertional mutagenesis when introduced into other cells as previously described.31 To evaluate functionality of the inducible cassette, TRUCKs with inducible EGFP expression were used. When transduced into JE6-1 reporter cells, EGFP expression was only induced after specific target recognition. It was not induced in 100% of cells; however we only measured it after 48 hours and kinetics of CAR activation and signaling are not fully understood yet.49 There is evidence that EGFP expression might not be completely upregulated in some cells at that time point or even already be downregulated again. Furthermore, Zimmermann et al showed that T cells transduced with a GD2-specific CAR and the same inducible EGFP cassette did upregulate EGFP expression on 60% of the cells after specific target recognition.31 The slightly reduced EGFP induction of 45% in TÜ165 EGFP-TRUCKs can, most likely, again be explained by the aforementioned more rare expression of the target antigen. IL-12 has been reported to induce T helper 1 differentiation of CD4+ T cells and to enhance cellular immunity by increasing IFN-γ release, augmenting granzyme and perforin production, and improving T and NK-cell proliferation.50 In the present study, we used an inducible delivery route of IL-12 by TÜ165 TRUCKs on target recognition. This resulted in highly improved CAR function in specific response to EBV peptide as well as to malignant EBV-infected cells. Also the abovementioned allogeneicity towards unloaded K562-B*35 was enhanced, because, most likely, signaling by the endogenous TCR lead to an induction of IL-12. For a potential future clinical application, this would imply that TRUCKs should not be utilized in the allogeneic setting as they would probably cause an even more severe graft-versus-host disease. However, this should not be a problem in the autologous use and, moreover, attempts to generate universal CAR-Ts with disrupted TCR expression are currently under investigation.51 Compared with CAR-Ts, TÜ165 TRUCKs showed augmented upregulation of CD25 expression, TNF-α and IFN-γ release, and IL-2 secretion. This is in line with previous findings that other IL-12-engineered T cells show substantially increased IFN-γ and TNF-α release, which is mandatory for the accumulation of activated macrophages in the tumor lesion and enables the elimination of antigen-loss cancer cells.14 Both IFN-γ and TNF-α promote resistance to tumor-derived immunosuppressive factors often released in EBV-positive Hodgkin’s disease.52 TRUCKs with other specificities have not shown enhanced cytotoxicity14 or superior CD107a or granzyme B expression53 compared with the respective CAR-engineered or TCR-engineered T cells. Here, interestingly, TÜ165 TRUCKs released slightly more perforin and significantly more granzyme B on recognition of the EBV-associated target than TÜ165 CAR-Ts. Moreover, TÜ165 TRUCKs tended to be more cytotoxic and exhibited cytotoxicity and granzyme B release towards autologous B-LCLs. Thus, besides their usage in the treatment of PTLD, we here provide a proof of principle how to improve the functionality of weakly binding CAR-Ts with mild effector function. As described above, TCR-like CAR-Ts were shown to maintain epitope specificity and effective cytotoxicity by lowering the affinity of a TCR-like CAR to the TCR affinity range.44 Furthermore, for clinical use, one major concern is tonic CAR-T signaling leading to exhaustion.54 Hence, using low affinity CARs in combination with an inducible cytokine might provide a safer and more enduring T cell product with the same or even increased functionality, which would need to be examined in further CAR studies.
The rationale for developing TRUCKs with inducible IL-12 was not only to improve functionality but also, and even more importantly, to modulate the tumor microenvironment. In a study with TCR-Ts against EBV-associated Hodgkin’s disease, the released IL-12 mediated resistance to the anti-proliferative and -cytotoxic effects of TGF-β that normally limit T-cell functionality against these tumors.52 Moreover, IL-12 results in the recruitment of NK cells and macrophages to the tumor lesion, thereby promoting antitumor effects.55 In the present study, TÜ165 TRUCKs releasing IL-12 on target engagement recruited both monocyte and NK-cell lines and enhanced target-cell killing as well as IFN-γ expression of NK-92 cells. PTLD patients display a heterogeneous immune deficiency associated with their immunosuppressive treatment regimen.56 However, the importance of NK cells in EBV-associated malignancies was demonstrated since primary immunodeficiencies in NK cells predispose patients to EBV-driven pathologies57 and a distinct subpopulation of tonsillar NK cells could restrict B-cell transformation by EBV.58 Thus, the use of IL-12-secreting TÜ165 TRUCKs to attract and activate NK cells, which are not normally located in close proximity to PTLD cells, is a new approach to treating immunocompromised patients that not only spares healthy B cells, but also attracts as many other innate immune cells as possible to induce a broad anti-EBV response. An immunocompetent mouse model is required to study attraction and involvement of endogenous immune cells by the engineered T cells in vivo. IL-12-induced toxicities are of particular interest and should be evaluated in future studies. Most of the available studies of inducible IL-12-secreting TRUCKs report that safe adoptive transfer of those cells into mice is possible, but one group observed severe toxicities associated with increased systemic levels of TNF-α and IFN-γ due to the non-specific release of IL-12.59 Although the amounts of IL-12 secreted by unstimulated TRUCKs in vitro in the present study were lower than those in the aforementioned study, evaluation of possible toxicities should definitely be a major focus of prospective preclinical in vivo testing of TÜ165 TRUCKs.
Conclusions
In conclusion, the results of this study demonstrated the suitability of CAR-Ts based on the peptide-selective and TCR-like mAb TÜ165 to specifically recognize and react to an intracellular EBV target associated with PTLD. Equipping the TÜ165 CAR with inducible IL-12 expression to generate TÜ165 TRUCKs improved effector functions and, importantly, led to the recruitment and activation of surrounding immune cells, which normally are not found in close proximity to lymphoproliferative cells. Thus, we suggest this method of specifically targeting EBV-infected cells while sparing and mobilizing the healthy immune cells in already immunocompromised patients as a new and promising approach to PTLD treatment, thereby enabling the control of EBV-associated lymphoproliferation.
Acknowledgments
We would like to thank Andreas Ziegler (Ziegler Biosolutions, Waldshut-Tiengen, Germany) for his helpful suggestions concerning the use of TÜ165 and for providing the TÜ165-producing hybridoma cell line. We also thank Aline Häcker and Winfried Wels, Georg-Speyer-Haus, Institute for Tumor Biology and Experimental Therapy, Frankfurt, Germany, for their help in designing the experimental layout of the transwell assays. We further thank Peter Steinberger, Medical University of Vienna, Institute of Immunology, Vienna, Austria, for providing the reporter JE6-1 Jurkat T cell line and the respective protocols. Thanks also to Marina Kramer, Sarina Lukis and Elvira Schulde for technical support and to Dr Matthias Ballmeier (Core Facility for Cell Sorting, MHH, Germany) for FACS-based cell sorting of transduced T cells.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
AS, MH and BE-V contributed equally.
Contributors Conceptualization: ACD, JL, HA, MH, AS, BM-K and BE-V. Methodology: ACD, BU-Z, KZ, TN, DS, JL, SK, CK and ST-Z. Software: ACD. Validation: ACD and BE-V. Formal analysis: ACD and BE-V. Investigation: ACD and BE-V. Resources: BU-Z, RB, AS and BE-V. Data curation: ACD and BE-V. Writing — original draft preparation: ACD. Writing — review and editing: All authors. Visualization: ACD. Supervision: BM-K and BE-V. Project administration: BE-V. Funding acquisition: RB, MH, AS and BE-V.
Funding This research was funded by “From CARs to TRUCKs” grants (Krebshilfe/German Cancer Aid-Priority Program in Translational Oncology, to BE-V, MH, HA, AS), the Comprehensive Cancer Center Hannover (Claudia van Schilling-Center), Member of the CCC Lower Saxony (ZN3481 CCC), the German Federal Ministry of Education and Research (reference numbers: 01EO0802 IFB-Tx and ZN3327 CORE), and, in part, by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation; project ID 158989968, SFB 900/B11 to BE-V and 324392634, TRR 221 to MH).
Disclaimer The funders played no role in designing the study, in collecting, analyzing or interpreting the data, in writing the manuscript, or in the decision to publish the results.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All experiments were performed with residual blood samples from routine platelet collection. According to standard donation requirements, the respective donors had no signs of acute infection and no previous history of blood transfusion. Written informed consent was obtained from all donors as approved by the Ethics Committee of Hannover Medical School (2519–2014, 3639–2017).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. The data sets and protocols used and/or analysed during the current study are available from the corresponding author (eiz-vesper.britta@mh-hannover.de) on reasonable request.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.