Article Text

Abstract
The percentage of patients with cancer eligible for checkpoint inhibitor (CPI) therapy has increased rapidly over the past few years and approaches 45%. As a result, more cases of CPI-related nephrotoxicity, including a rare subset with vasculitis, are being reported. To elucidate the clinical presentation of CPI-associated renal vasculitis and its possible mechanisms, treatment options and prognosis, we describe cases from a comprehensive cancer center and reviewed the literature for similar cases. We retrospectively reviewed the charts of all patients with cancer from 2014 to 2020 who were diagnosed with CPI-related nephrotoxicity and underwent a kidney biopsy. We identified five cases of renal vasculitis: three patients were diagnosed with seronegative antineutrophil cytoplasm antibody (ANCA)-associated vasculitis, one case with seropositive ANCA-associated vasculitis and one case was diagnosed with IgA vasculitis. Of these cases, four patients were receiving nivolumab, and one patient was receiving tremelimumab. All patients had microscopic hematuria, four out of five patients had negative ANCA serology, one patient had concurrent lung involvement and positive ANCA serology, and all had severe acute kidney injury with creatinine >4.50 mg/dL on diagnosis. All patients were treated by discontinuing CPI and initiating corticosteroids and rituximab. Three patients received plasmapheresis; two of these required renal replacement therapy including the patient with lung involvement. All patients after rituximab had a partial or complete renal response. Two patients died within 8 months of diagnosis due to malignancy progression. None of the patients had a relapse of vasculitis. We demonstrated that CPI can be associated with different types of renal vasculitis that are predominantly ANCA negative and manifest as severe acute kidney injury. Despite the lack of strong evidence, treatment similar to treatment of primary seropositive ANCA-associated vasculitis with corticosteroids and rituximab is well tolerated with favorable renal outcomes.
- autoimmunity
- immunotherapy
- lymphocyte activation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Around 3% of patients treated with checkpoint inhibitors (CPIs) develop acute kidney injury (AKI) that is attributed to CPI treatment, where acute interstitial nephritis (AIN) is the most frequently reported kidney pathology.1 Less common pathologies include vasculitis, which, unlike other medication-associated vasculitis, can be anti-neutrophil cytoplasmic antibodies (ANCA) negative.2–6 Clinically, renal vasculitis commonly presents as severe AKI with microscopic hematuria.
There is no consensus or established recommendation for treatment of medication-associated vasculitis aside from stopping the offending medication and consideration of immunosuppression in severe cases.7 Risk of relapse after therapy is unknown. Rituximab use is associated with disease remission and improvement in kidney survival in patients with primary ANCA vasculitis as well as in patients with ANCA-negative vasculitis.8 9 In case reports, rituximab has shown a good response to CPI-related antibody-mediated adverse events, including idiopathic thrombocytopenia purpura, hemolytic anemia and encephalitis, but outcomes of its use in CPI-related renal vasculitis have not been reported. Here, we present five cases from our cancer center of CPI-associated vasculitis that we treated using the guidelines for primary ANCA vasculitis: with immunosuppressants (corticosteroids and rituximab) and discontinuation of CPI.
Materials and methods
We retrospectively reviewed the medical records of all patients who received CPI treatment, developed AKI and had a diagnostic renal biopsy at The University of Texas MD Anderson Cancer Center from 2014 to 2020.We identified the patients with diagnosis of vasculitis with kidney involvement. For each of these patients, we collected information about age, sex, cancer diagnosis, name and class of CPI received, potentially nephrotoxic medications, serum creatinine at baseline and during AKI, date of last follow-up, urine sediment, proteinuria, serological markers, renal pathology findings and tumor status.
We defined AKI using Kidney Disease: Improving Global Outcomes (KDIGO) classification guidelines (increase in serum creatinine ≥0.3 mg/dL within 48 hours or ≥50% of baseline within 7 days).10 Complete kidney function recovery was defined as post-AKI improvement in creatinine level to <0.35 mg/dL above baseline. Partial recovery was defined as creatinine level ≥0.35 mg/dL above baseline and <2 times baseline.2 Vasculitis remission was defined as a stabilization or improvement in kidney function with resolution of microscopic hematuria after discontinuation of corticosteroids.11 12 Each patient’s renal function was followed up for at least 3 months after AKI before categorizing renal recovery as remission, complete recovery or partial recovery based on stable creatinine levels and presence or absence of microscopic hematuria. Glomerular filtration rate (GFR) was estimated based on Modification of Diet in Renal Disease (MDRD) GFR formula (mL/min/1.73 m2).
We classified glomerulonephritis with necrosis and crescents on light microscopy and no or weak immunofluorescence (IF) intensity ( ≤ +2) IgA deposits that are limited to the mesangium as pauci-immune vasculitis rather than IgA nephropathy or overlapping syndrome.13 Pauci-immune glomerular lesions were classified as sclerotic, focal, crescentic or mixed class (per Berden classification).14
Clinical diagnosis of IgA vasculitis was made in patients with histopathological evidence of proliferative glomerulonephritis with predominant IgA deposits and lower extremities purpuric skin rash (as per the European League Against Rheumatism criteria).15
ANCA serology was measured by multiplex flow immunoassay (reference value is <0.4 U, negative) as recommended by the 2017 revised international consensus.16 Characteristics and outcomes of the five patients included in this study and previously reported cases of CPI-associated vasculitis with kidney involvement are listed in table 1.
Characteristics and outcomes of patients with checkpoint inhibitor-related vasculitis with kidney involvement
Case 1
A Caucasian man in his early 40s with metastatic non-small cell lung cancer and thyroiditis, as a CPI-related immune-related adverse event (IrAE), was hospitalized for evaluation of AKI prior to receiving the fifth cycle of nivolumab. Creatinine had increased from 0.97 (estimated GFR (eGFR) of 85 mL/min/1.73 m2) to 4.52 mg/dL over the 5 weeks prior to admission. He did not have any skin rash, extremity weakness, joint swelling or upper respiratory symptoms. He was normotensive and afebrile, with clear lungs on auscultation. Urinalysis (UA) with microscopy revealed hematuria (320 red blood cells per high power field (RBC/HPF)), pyuria (19 white blood cells per high power field (WBC/HPF)) and subnephrotic range proteinuria (urine microalbumin-to-creatinine ratio of 1025 mg/g). C3 and C4 levels were within normal range (143 and 28 mg/dL, respectively) and ANCA panel testing was negative (myeloperoxidase antibody (MPO antibody) and proteinase 3 (PR3) antibody, anti-PR3, were <0.2 units). Renal biopsy showed acute focal segmental necrotizing glomerulonephritis with segmental necrosis in 2 of 12 examined glomeruli. IF showed no significant staining (figure 1). Nivolumab was discontinued, and the patient received 1 mg/kg prednisone followed by a single dose of rituximab (1 g) around 4 weeks later as his creatinine rose again on tapering down the prednisone dose. Prednisone dose was increased back to 1 mg/kg then tapered down again over the following 6 weeks. He had full renal recovery at 8 weeks, with creatinine levels improving back to baseline (0.89 mg/dL, eGFR of 94 mL/min/1.73 m2) but he had persistent microscopic hematuria that we attributed to possible glomerular scarring, although a resolving glomerulonephritis could not be ruled out as the last available UA was 3 months postvasculitis diagnosis and we did not have enough follow-up since he died 4 months after vasculitis diagnosis.

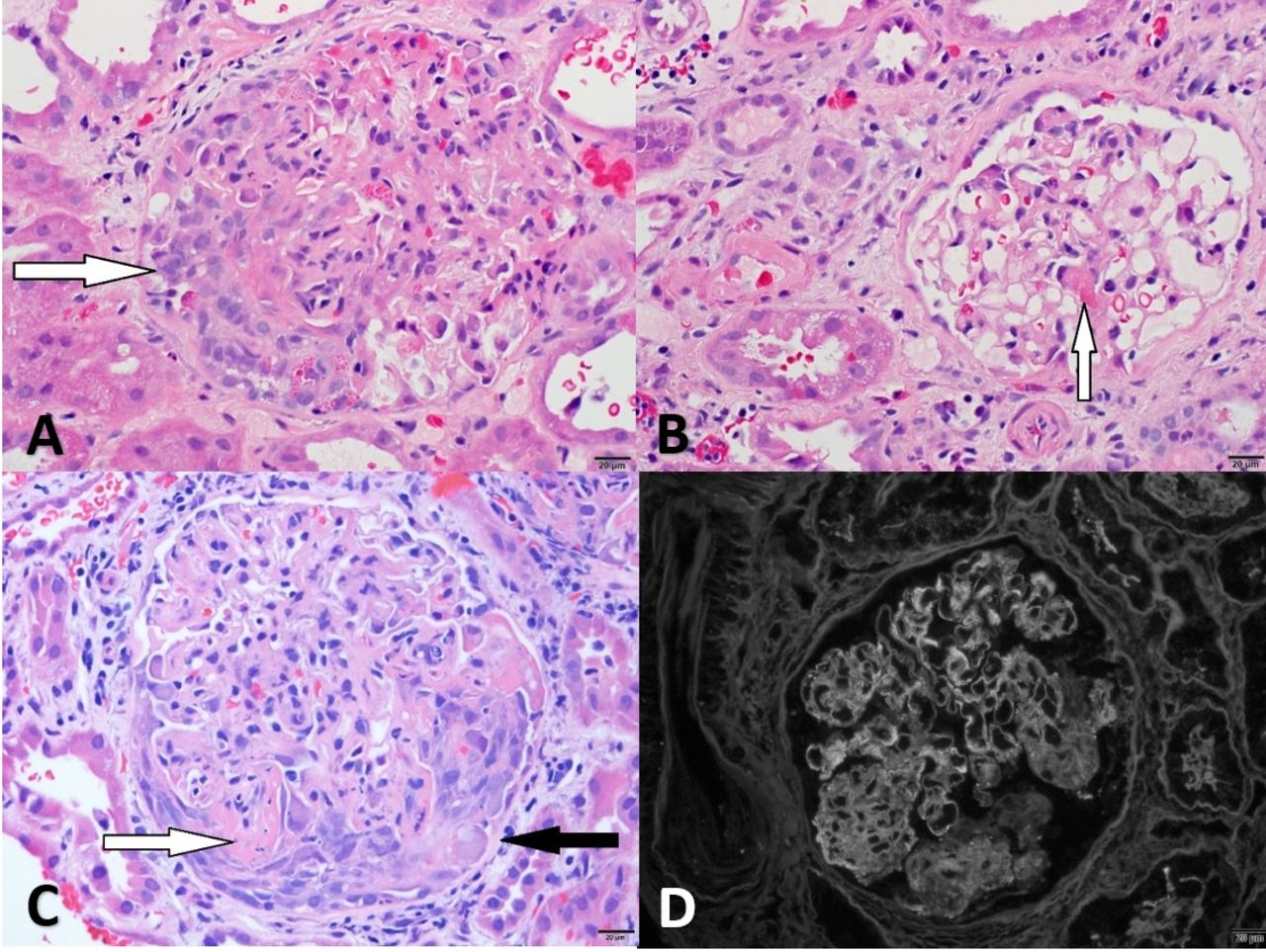
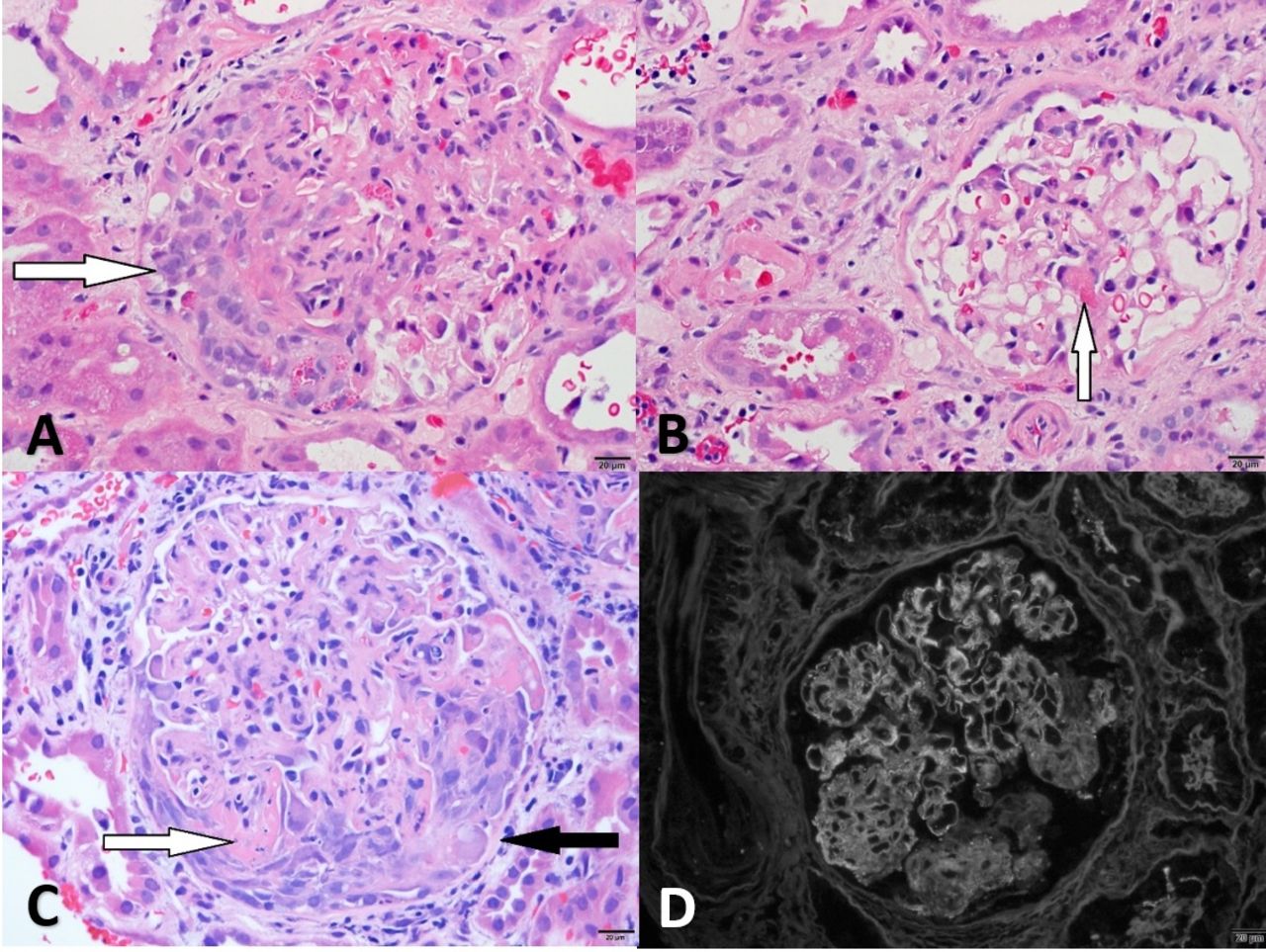
(A) Glomerulus from case 1 with segmental fibrinoid necrosis (arrow) (H&E, 40×), (B) glomerulus from case 2 with segmental necrosis (arrow) (H&E, 40×), (C) arteriole from case 3 with fibrinoid necrosis (arrow) and surrounding granuloma (H&E, 20×), (D) arteriole from case 3 (black arrow) with surrounding granuloma (white arrow) (periodic acid-Schiff, 20×).
Case 2
A Caucasian man in his mid-70s with hypertension, for which he was receiving hydralazine; stage 3 chronic kidney disease; dermatitis (IrAE) and metastatic renal cell carcinoma status postpartial nephrectomy, was hospitalized for evaluation of new-onset cough and AKI after receiving the second infusion of tremelimumab. His creatinine level had increased from 2.14 (eGFR of 30 mL/min/1.73 m2) to 4.58 mg/dL over the 2 weeks prior to admission. He did not have any skin rash, extremity weakness or joint swelling. He was normotensive and afebrile. Physical examination revealed decreased breathing sounds at lung bases on auscultation. Chest imaging did not show any significant infiltrate or cavities. UA with microscopy revealed hematuria (67 RBC/HPF), pyuria (5 WBC/HPF) and subnephrotic range proteinuria (protein-to-creatinine ratio of 1430 mg/g). Tests for antinuclear antibody (ANA) and MPO antibody were positive (1:160 and >8 units, respectively). Anti-PR3 titer and antiglomerular basement membrane (GBM) antibody were negative (<0.2 units). Renal biopsy showed acute focal segmental pauci-immune necrotizing glomerulonephritis with necrosis in 38% of glomeruli (figure 1). Hydralazine and tremelimumab were discontinued, and the patient was treated with intravenous methylprednisolone for 3 days followed by a tapered dose of prednisone, six sessions of plasmapheresis and four cycles of rituximab (0.375 g/m2 weekly). At 4 months after AKI, he had partial renal recovery (resolution of hematuria with partial improvement in creatinine to 2.87 mg/dL (eGFR of 22 mL/min/1.73 m2) with negative ANA titer. He received a maintenance dose of rituximab (500 mg) at 1 year, as his MPO titer remained positive (>8 units); however, he has had no clinical relapse over 24 months after his initial vasculitis diagnosis. He was under observation for 1 year and recently began chemotherapy for cancer progression.
Case 3
A Caucasian woman in her late 60s with hypertension, diabetes, stage 3 chronic kidney disease, IrAEs (thyroiditis, dermatitis, adrenal insufficiency) and metastatic uveal melanoma was hospitalized for evaluation of AKI after completing the third cycle of ipilimumab plus nivolumab. The patient had been feeling weak with poor appetite a few days prior to hospitalization. She had no skin rash, extremity weakness, joint swelling or upper respiratory symptoms. She was normotensive and afebrile. She had clear lung sounds on auscultation. Her creatinine level increased from 1.45 (eGFR of 36 mL/min/1.73 m2) to 4.96 mg/dL over the 3 weeks prior to admission. UA with microscopy revealed hematuria (7 RBC/HPF), pyuria (15 WBC/HPF) and subnephrotic range proteinuria (urine protein-to-creatinine ratio of 400 mg/g). C3 and C4 levels were not decreased (137 and 55 mg/dL, respectively) and ANCA panel testing (MPO antibody and anti-PR3) and anti-GBM antibody were negative (<0.2 units). Renal biopsy showed granulomatous necrotizing vasculitis with moderate patchy interstitial infiltration by plasma cells and eosinophils. Granuloma formation involving predominantly arterioles and small arteries was observed. No glomerular crescents or hypercellularity was noted. IF showed no significant staining (figure 1). CPI was discontinued and the patient was treated with 1 mg/kg of prednisone followed, over 3 weeks, by a tapered dose of prednisone. However, as prednisone dose was tapered down to 20 mg/day, the creatinine rose again (Cr 2.96>4.07 mg/dL). A single dose of rituximab (0.375 g/m2), at the end of third week of corticosteroid therapy, was given and prednisone dose was increased back to 1 mg/kg. Further doses of rituximab were held as the patient had no relapse and to avoid further immunosuppression considering she developed descending colon perforation complicated with sepsis and later reactivation of cytomegalovirus viremia. She had full renal recovery at 9 weeks (resolution of hematuria and creatinine level returned to baseline, serum creatinine of 1.40 mg/dL, eGFR of 38 mL/min/1.73 m2). She developed AKI 5 months later during a hospitalization for evaluation of sepsis. UA with microscopy showed hematuria and pyuria; however, no further renal investigation was done per the patient’s wishes, and she was transitioned to hospice care.
Case 4
A Caucasian man in his mid-60s with history of aortic valve replacement (on warfarin), urinary diverticulum with urinary tract infections and recurrent nephrolithiasis with gross hematuria who was hospitalized for left retroperitoneal and inguinal canal liposarcoma tumor resection. He had just completed the second cycles of nivolumab neoadjuvant therapy prior to hospital admission. Postoperatively, he developed methicillin-resistant Staphylococcus aureus (MRSA) bacteremia secondary to possible intra-abdominal abscess. He was treated with daptomycin for 6 weeks and had no evidence of distant metastatic infections. During the fourth week of antibiotic therapy and as the sepsis was subsiding, he developed AKI with a rise in creatinine from 1.02 (eGFR of 76 mL/min/1.73 m2) to 7.53 mg/dL over 14 days. UA with microscopy revealed hematuria (>182 RBC/HPF), pyuria (10 WBC/HPF), subnephrotic range proteinuria (urine protein-to-creatinine ratio of 1190 mg/g) and negative urine culture. ANA, antidouble-strand DNA, anti-GBM antibody and ANCA (anti-PR3 and MPO-antibody) serum tests were negative (<1:40, <12.3 IU/mL, <0.2 unit and <0.2 unit, respectively). C3 and C4 levels were within normal range (112 and 32 mg/dL, respectively). Renal biopsy was performed and showed focal (3 of 24 glomeruli) segmental necrotizing glomerulonephritis with focal global sclerosis (in 13% of glomeruli) and ATN. Minimal mesangial IgA (+1) and C3 deposits were observed under IF, but no mesangial deposits were noted by electron microscopy (EM) (figure 2). Despite Staphylococcus-associated glomerulonephritis could be considered as a differential diagnosis in this case, the timeline for the resolving MRSA infection, the normal C3 complement level, the IgA deposits present in IF that did not coincide with the active glomerular lesions and seemed to be incidentally found, and the absence of mesangial deposits under EM made us lean toward renal vasculitis diagnosis. The patient was started on renal replacement therapy (RRT) for metabolic clearance and treated with intravenous methylprednisolone for 3 days followed by a tapered dose of prednisone. In addition, patient received two sessions of plasmapheresis followed by two doses of rituximab (0.375 g/m2); the patient had a partial renal recovery and no longer requiring hemodialysis, with a creatinine level of 1.71 mg/dL (eGFR of 41 mL/min/1.73 m2) at 8 weeks of immunosuppressive therapy. His condition remained stable at 56 weeks after the vasculitis diagnosis. He continues to have microscopic hematuria with pyuria and positive urine cultures that is attributed to chronic bladder diverticulum. Of note, the patient had no UA in our medical records prior to developing AKI for which his reported microscopic hematuria could be of a chronic nature.
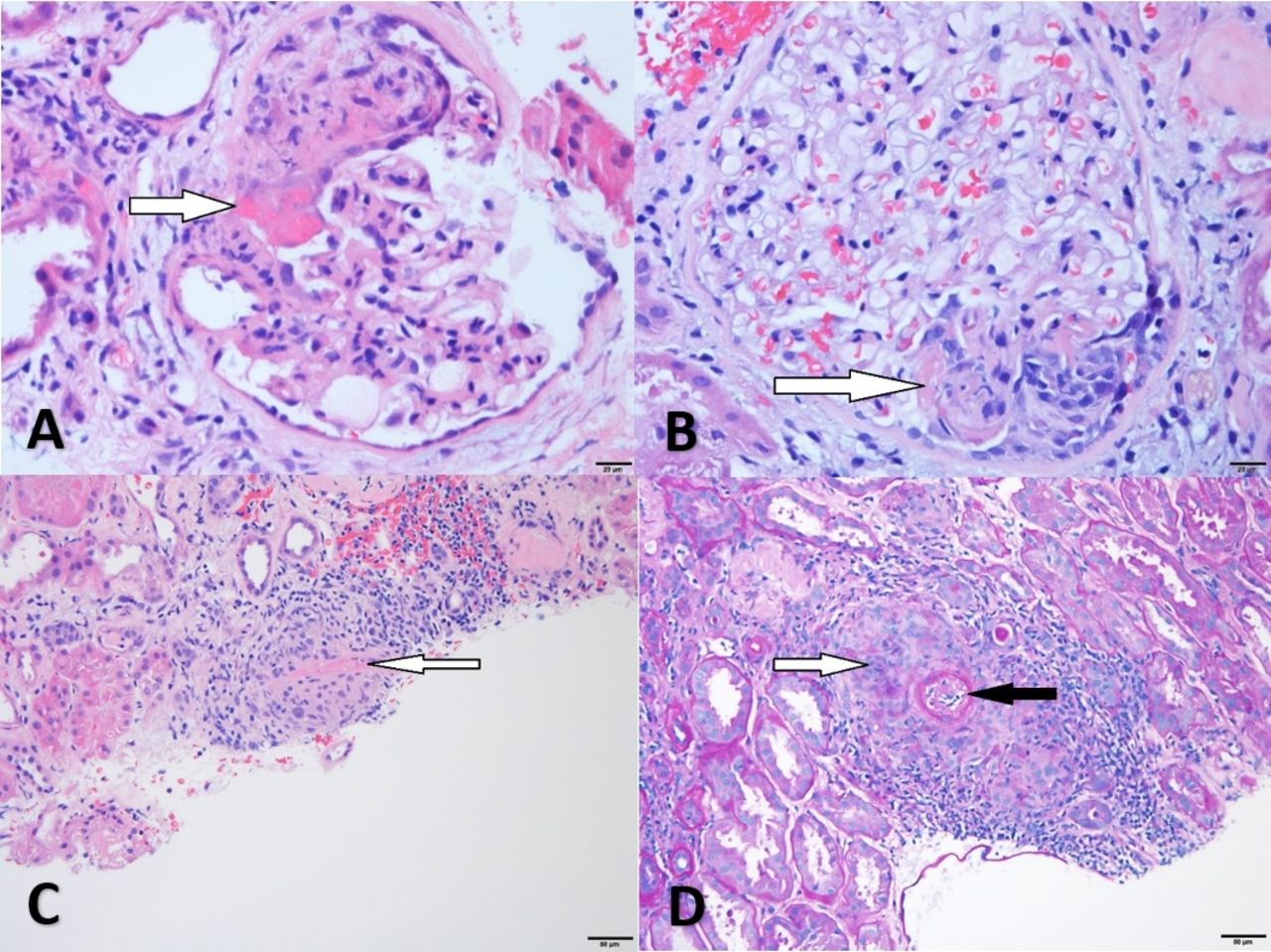
(A) Glomerulus from case 4 with cellular crescent (arrow) (H&E, 40×), (B) glomerulus from case 4 with segmental necrosis (arrow) without mesangial or endocapillary proliferation (H&E, 40×), (C) glomerulus from case 5 with segmental necrosis (white arrow) and crescent formation (black arrow) (H&E, 40×), (D) case 5, granular mesangial and capillary IgA-positive deposits (immunofluorescence, 40×).
Case 5
A Caucasian woman in her mid-50s with metastatic uveal melanoma developed diffuse purpuric skin rash on all extremities after receiving a second infusion of combination therapy with ipilimumab and nivolumab. Skin biopsy suggested leukocytoclastic vasculitis (figure 3). A blood test indicated transaminitis, and random urine sample revealed nephrotic range proteinuria (9.0 g); both conditions were attributed to CPI toxicity (IrAEs). Both nivolumab and ipilimumab were discontinued, and the patient was started on a tapering dose of prednisone (starting 1 mg/kg). Two weeks later, she presented to the emergency room for having tea-colored urine, headache and worsening fatigue. Her blood pressure was elevated (165/95 mm Hg). She had edema in the lower extremities and rales at the lung bases. Her creatinine level increased from 1.01 mg/dL (eGFR of 62 mL/min/1.73 m2) to 5.07 mg/dL over 2 weeks. UA with microscopy revealed hematuria (>182 RBC/HPF) and pyuria (117 WBC/HPF), and 24 hours urine protein collection indicated nephrotic range proteinuria (13.74 g). ANA, antidouble-strand DNA, anti-GBM antibody and ANCA (anti-PR3 and MPO antibody) serum tests were negative (<1:40, <12.3 IU/mL, <0.2 unit and <0.2 unit, respectively). C3 and C4 levels were within normal range (108 and 23 mg/dL, respectively). Renal biopsy showed diffuse necrotizing glomerulonephritis with IgA deposits. Nine of 12 examined glomeruli had global necrosis with crescents. IF microscopy showed diffuse, granular mesangial and capillary IgA deposits (+3) (figure 2). The patient was treated with 3 days of intravenous methylprednisolone (500 mg) followed by a tapered dose of prednisone over 3 months, daily plasmapheresis for 7 days and a biweekly dose rituximab (2 g). She required RRT with hemodialysis for 10 weeks and had partial renal recovery. Her creatinine level is currently stable at 2.68 mg/dL (eGFR of 19 mL/min/1.73 m2). She has minimal microscopic hematuria (2–4 RBC/HPF), and her melanoma is stable off therapy.

{kind=link}
{kind=link}
{kind=link}
Patient 5: purpuric skin rash involving both legs.
Discussion
Immune-related adverse events are common complications of CPI therapy, and 2%–5% of patients develop nephrotoxicity.1 17 CPI-associated vasculitis is considered a rare adverse event; a systematic review of the reported cases of CPI-associated vasculitis up to 2018 found only 20 confirmed cases.18 In systematic review, large vessel and central/peripheral nervous system vasculitis were the main reported types, and only one case of vasculitis with kidney involvement was included. Since then, 10 additional cases of vasculitis involving the kidney had been reported (table 1).2–6
A diagnosis of medication-associated vasculitis is based on very high titers of ANCA (anti-MPO or anti-PR3, with or without other antineutrophil protein antibodies) and a temporal relationship between its use and discontinuation with the development and recovery of vasculitis-like manifestations.7 19 Different classes of medications are linked with induction of ANCA, while only a small percentage of patients with a positive titer develop vasculitis.20 Renal involvement occurs in 20%–30% of vasculitis cases associated with antithyroid drugs and in 80%–90% of cases associated with hydralazine.19 The severity of renal involvement also varies among these medications.
Interestingly, three of our four cases of non-IgA vasculitis with kidney involvement had renal-limited manifestations and negative ANCA serology. Similar findings have been observed in patients with ANCA-negative vasculitis.21
We speculate, based on recent evidence in ANCA-negative vasculitis, that the negative ANCA serology in our reported cases might be related to a limitation of the current assay in detecting pathogenic ANCA that is reactive to a restrictive epitope.22 Further investigation is needed to evaluate this speculation.
Case 2 in our series was receiving hydralazine and a CPI when he developed lung and kidney injury with positive ANCA (anti-MPO antibody) serology. Two previously reported cases of ANCA vasculitis related to CPI therapy in the literature, however, were PR3 positive, and both had extrarenal manifestations.3 4 Both patients developed fever, arthralgia, purpuric skin rash, dyspnea and microscopic hematuria after receiving combination immunotherapy with programmed cell death ligand 1 and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) inhibitors, nivolumab with ipilimumab in one case and pembrolizumab following ipilimumab in the second case, for treatment of metastatic melanoma. The presence/absence of AKI was not reported. They were treated by discontinuation of CPI, corticosteroids and an additional immunosuppressive agent (ciclosporin and rituximab). The outcome of microscopic hematuria after treatment was not stated.
Based on the patient characteristics and renal outcomes of the few known cases, ANCA positivity did not appear to be associated with the use of a certain class of CPI or with a worse renal outcome. Our patient had been on hydralazine for a long period of time without any manifestations of vasculitis, and only after starting CPI he developed vasculitis. This is consistent with proposed mechanisms of CPI-associated acute tubulointerstitial nephritis where loss of tolerance to effector T cells primed during prior drug exposure (eg, proton pump inhibitors) occurs after CPI exposure.1 Nevertheless, more cases need to be evaluated before a conclusion can be reached.
The mechanism for renal vasculitis associated with CPI use is poorly understood. The antibody production by B lymphocytes that target neutrophils leading to endothelial injury have a major role in the pathogenesis of pauci-immune vasculitis.23 Nonetheless, there is growing evidence of T lymphocytes’ role in the pathogenesis of ANCA vasculitis,24 25 and promoting the unopposed activity of these cells, as observed with CPI use, might explain to some degree the observed association between CPI use and renal vasculitis.24–26 For example, CPIs can expand T cells (effector and follicular cells) and increase T and B cell interaction with potentially subsequent production of interferon gamma and interleukin 21—both cytokines have been previously reported to be associated with ANCA vasculitis.27–30 CD4+ regulatory T cells (Tregs) are a highly immune-suppressive subset of CD4+ T cells and their expression of regulatory transcription factor FOXP3 is critical in maintaining self-tolerance. CTLA-4 and programmed cell death protein 1 (PD-1) promote FOXP3 expression which enhances suppressive activity of Tregs. With use of CPI, Tregs lose their suppressive function and autoimmune disease can ensue. This has been demonstrated in ANCA vasculitis, which is induced by the disruption of the suppressive Tregs.31 Additionally in the case of IgA vasculitis, CPIs have also been proposed to upregulate C-X-C motif chemokine ligand (CXCL)9 and CXCL10 facilitating T cell recruitment and associated IgA vasculitis-related tissue injury.32 33
The manifestation of medication-associated vasculitis can be severe and fatal. In a retrospective study involving 10 patients diagnosed with hydralazine-associated vasculitis who were treated with glucocorticoids and cyclophosphamide (8 of 10), 2 patients died despite therapy and 3 patients had severe AKI requiring hemodialysis.19 Two of these three patients with AKI had subsequent recovery in kidney function. However, as vasculitis manifestations and severity can vary among medications and the evidence of using immunosuppressant in these patients is limited to observational studies, the current practice for treatment of CPI-associated vasculitis with kidney involvement is limited to stopping the offending agent with consideration for immunosuppression based on disease severity and the patient’s comorbidities.7
In a recent case series by Gallan et al that included four patients with a diagnosis of CPI (all with PD-1 inhibitors) induced renal vasculitis, three of the patients were treated with corticosteroids.5 Patient number 2 of the series did not have AKI but had proteinuria and hematuria, both of which resolved with therapy. Patients 3 and 4 both had severe AKI, arteritis and AIN on biopsy. No glomerular lesion was noted in patient 5, and no crescents but focal glomerulosclerosis (involving 40% of glomeruli) was described in patient 4. Patient 4 required RRT and subsequently had partial recovery, and patient 5 had complete renal recovery. In all patients there was complete or partial renal recovery with no long-term follow-up. In addition, there was no comments on tumor response. It should be pointed out that patients with CPI-related renal vasculitis often have concomitant renal pathologies, most commonly AIN, that by itself might influence the kidney recovery outcome. For example, Person et al described a case of man aged 55 years with activated protein C resistance and metastatic melanoma that was treated with two cycles of nivolumab with ipilimumab and developed IrAEs including pneumonitis, uveitis colitis and severe AKI. Renal biopsy was significant for granulomatous interstitial nephritis, vasculitis and thrombotic microangiopathy. He was treated with corticosteroids, mycophenolic acid and tumor necrosis factor-alpha blocker; however, he had no renal recovery after 6 months of AKI and remained dialysis dependent.
The patient characteristics and renal outcomes of these cases are listed in table 1.
In our reported cases, creatinine was >4.50 mg/dL on diagnosis in all patients, two patients required RRT (patients 4 and 5) and all patients had either partial of full recovery of renal function. Three patients had a remission of vasculitis as the microscopic hematuria resolved. Two patients (patients 1 and 4) had persistent microscopic hematuria despite the recovery of renal function and no vasculitis-associated symptoms. In these cases, persistent hematuria may not be related to persistent/relapsed vasculitis but rather to glomerular scarring, or resolving vasculitis as in case number 1, and being on anticoagulation with urinary bladder diverticulum, recurrent UTI and recurrent nephrolithiasis, as in case number 4.
None of the patients relapsed; however, only three patients were followed up for >6 months, and one of these patients (patient number 3) had recurrent AKI 7 months after renal vasculitis diagnosis. This patient had sepsis, and no further investigations were done. Two patients died due to presumed underlying cancer progression at 4 and 8 months after renal vasculitis diagnosis.
All five of our cases were treated with prednisone and rituximab. Plasma exchange was done for three patients who had severe AKI, required hemodialysis or had concurrent lung involvement per KDIGO recommendations.34 However, since the patients we presented have been treated, plasma exchange and glucocorticoid dosing in the treatment of antineutrophil cytoplasm antibody-associated vasculitis (PEXIVAS) study has been published and demonstrated no difference in renal or survival outcomes in patients with severe vasculitis defined as GFR <50 cc/min or pulmonary involvement.35 Nonetheless, there was no evaluation of crescentic glomerulonephritis in PEXIVAS which all our cases pathologically had. In addition, we believe that CPI-induced vasculitis is not exactly the same as de novo vasculitis since we have noted that these patients can be ANCA positive or negative and there is likely an antibody inducing the injury that we did not accounted for. As far as our decision in the use of rituximab in addition to corticosteroids in our cohort, it was based mainly on the supporting evidence from a trial of patients with primary ANCA vasculitis and rituximab’s pharmacological characteristics. Here, we summarize these supportive points. 1) The presence of crescents, decreased percentage of normal glomeruli or low eGFR at diagnosis are reported to be associated with poor renal outcome.36–38 2) The risk of relapse is not yet known in patients with CPI-associated vasculitis. 3) Corticosteroid monotherapy has a lower rate of remission and might be insufficient to induce renal recovery as observed in patient 5, who developed severe AKI despite 2 weeks of corticosteroids for a non-renal manifestation.11 4) Rituximab can shorten the course of corticosteroid therapy. 5) Patients with ANCA-negative vasculitis have a good response to rituximab therapy.8 6) As opposed to depleting T cells with cyclophosphamide, rituximab can disrupt the pathogenic B cell/CD8+ T cell axis, leading to a reduction in T cell cytokine production and subsequently less renal endothelial damage without hindering CPI antitumor activity.39 40 In addition, the malignancy risk was reported to be lower in rituximab-treated ANCA-associated vasculitis patients than in cyclophosphamide-treated patients.41 42
In our experience, rituximab was well tolerated with no reported major side effects in all patients in our series. Of note, two patients (case 3 and 4) received a reduced dose of rituximab to avoid relapse of a concurrent infection that was controlled with antibiotics and case 1 who died 4 months postdiagnosis, had evidence of cancer progression prior to receiving rituximab.
In conclusion, CPI use can be associated with vasculitis with kidney involvement causing severe kidney injury. Treating this condition in the same manner as primary ANCA vasculitis, with corticosteroids and rituximab, can be well tolerated with partial to complete renal recovery and no relapse. This analysis is limited as it is retrospective with low number of cases; therefore, prospective clinical studies are needed for establishing benefit of addition of rituximab to corticosteroids in CPI-associated renal vasculitic syndromes.
Acknowledgments
Editorial support was provided by Bryan Tutt in Scientific Publications Services, Research Medical Library at The University of Texas MD Anderson Cancer Center.
References
Footnotes
Twitter @JamieLinMD
Contributors Data acquisition was performed by AA and OM. WFG and AST performed the histological examination of the kidney biopsies and contributed in writing the pathology section of the manuscript. The manuscript was prepared by AA, OM, JSL, and edited by OM, JSL, AST, WFG, MA, US, MB, NA-W and AA. All the authors contributed to the quality control data, analysis, interpretation of data and writing and final proof of paper. All authors read and approved the final manuscript.
Funding The University of Texas MD Anderson Cancer Center is supported in part by the National Institutes of Health through Cancer Center Support Grant P30CA016672.
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval This study was approved by the institutional review board in accordance with the principles of the Declaration of Helsinki.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article