Article Text

Statistics from Altmetric.com
Introduction
The use of intact human cells to treat important diseases is attracting considerable attention. Autologous chondrocytes are under investigation for treatment of cartilage defects1; induced pluripotent stem cells are being differentiated into islet cells for treatment of diabetes,2 cardiomyocytes are being explored for therapy of damaged heart tissue3 and retinal cells are being investigated for replacement of defective retinal pigment epithelial cells in patients with macular degeneration.4 In the growing field of immunotherapy, dendritic cell vaccines are being exploited for induction of immunity against cancer-specific and virus-specific antigens,5 6 and chimeric antigen receptor (CAR) T cells are being used for targeted elimination of hematopoietic cancers.7 While the promise associated with each of these technologies is very encouraging, concerns remain over how sensitively such therapeutic cells can be manipulated following their reintroduction into patients. Thus, will it be possible for genetically modified cell activities to be upregulated or downregulated when changes in their phenotypes are required for success of the therapy? Can differentiation programs of therapeutic stem cells be redirected if the cells begin to stray from their intended destinies? Can transplanted/engineered immune cells be rejuvenated after their activities become suppressed by the tumor microenvironment or can they be terminated when their function becomes counterproductive?
Approaches to these and related problems currently rely on different solutions for each different need. Thus, termination of a cells activity has been largely approached by introduction of a suicide gene that can be activated by an inducer.8 9 Control of a cells functional status has been primarily pursued by engineering control over activation receptors by small molecule inducers.10 Inactivation of critical pathways has been recently achieved by induced ubiquination and degradation of essential proteins (eg, Proteolysis targeting chimera, PROTAC).11 However, no universal method has been developed that can mediate all of these regulatory changes with a single modification.
We describe a ‘chimeric endocytosing receptor’ (CER) that can be transfected into any cell type and then exploited to deliver any drug selectively into that cell type following its reinfusion into a patient in vivo. Construction of this CER simply requires fusion of a protein domain that constitutively endocytoses and recycles to the cell surface (eg, a glycosylphosphatidylinositol (GPI)-anchored folate receptor (FR))12 with a ligand binding domain (eg, FK506 binding protein (FKBP)13 or an scFv against fluorescein14) that exhibits high affinity for a ligand that is not naturally found in humans (figure 1A). Attachment of the desired ligand (ie, FK506 or fluorescein in the above examples) to any drug via a cleavable linker then affords a ligand–drug conjugate that will bind to the CER with high affinity and constitutively enter the transfected cell via receptor-mediated endocytosis (figure 1B). Because both the FR and the FKBP are human in origin, a fusion receptor constructed from these domains should result in minimal immunogenicity. Moreover, since no receptors for either FK506 or fluorescein exist on normal human cell surfaces, little or no drug accumulation should occur in any cell except the cell expressing the transfected receptor.15 16 Using the human FRα as the constitutively endocytosing domain fused to either a human FKBP or an antifluorescein scFv (scFv FITC) as the ligand-binding domain, we demonstrate that delivery of effective concentrations of both cytotoxic and immunosuppressive drugs can be achieved both in vitro and in vivo via the same CER following its transfection into anti-CD19 CAR T cells.

Design of chimeric endocytosing receptors and cognate ligand–drug conjugates. (A) Diagram to show the working mechanism of chimeric endocytosing receptor (CER). In the case illustrated, the fusion receptor is composed of an NH2-terminal FK506 binding protein linked via a glycine4-serine spacer to a COOH-terminal glycosylphosphatidylinositol-anchored folate receptor fragment.12 Because the fusion receptor contains the high affinity binding site for FK506, conjugation of FK506 to any drug allows the attached drug to enter CER-expressing cells by receptor-mediated endocytosis. (B) Chemical structures of FK506- or FITC-linked conjugates of any desired drug. To avoid causing immunosuppression with FK506-targeted drugs, we have carefully selected the site of attachment of FK506 to its cargo to disrupt FK506 binding to calcineurin while preserving its binding to FKBP12. Note also that the drug is linked to the targeting ligand (FK506 or FITC) via a disulfide bond that is rapidly reduced following trafficking to intracellular endosomes.39 (C) Diagram of FKBP-FR and FITC-FR fusion receptor constructs, as described in text. (D) Diagram of anti-CD19 CAR, using FKBP-FR as an example. COOH, C terminal; CD8sp, human CD8 signal peptide; FKBP-FR, FK506 binding protein folate receptor; scFv, short chain variable fragment; sp, human folate receptor alpha signal peptide; T2A, self-cleaving peptide; VH, variable heavy chain; VL, variable light chain.
Results
Design, expression and characterization of the FKBP-FR and FITC-FR fusion receptors
In order to develop a ‘CER’ that would allow targeted delivery of any drug into an engineered cell, we hypothesized that the engineered passageway should exhibit the following properties. It must (1) be expressed on the cell surface of the desired therapeutic cell, (2) contain a high affinity binding site for a ligand not recognized by any other cell type, (3) endocytose constitutively into the therapeutic cell, (4) recycle rapidly back to cell surface for additional rounds of endocytosis, (5) be assembled from largely human components and (6) exhibit good stability in vivo. With such a receptor expressed exclusively in the therapeutic cell, any desired drug should be deliverable into that cell by linking the drug to a ligand that recognizes the engineered receptor.
In the studies below, two engineered CERs are described that employ either an antifluorescein scFv (FITC) or FKBP12 as the high affinity ligand binding domain of the fusion receptor (figure 1). Although other binding domains could have been selected, the above two were chosen because they met the criteria described above and exhibited high affinity for their specific ligands (ie, 50 fM14 and 0.2 nM,13 respectively). Moreover, to assure that both ligand binding domains would endocytose constitutively into their engineered cells, each ligand binding domain was tethered via a flexible peptide linker to the GPI-anchored domain of human FRα. Because this domain constitutively enters cells by receptor-mediated endocytosis and then rapidly recycles back to the cell surface,12 it satisfies the internalization and recycling requirements mentioned above. Further, to promote proper trafficking of de novo synthesized fusion receptors to the cell surface, the N-terminal signal peptide from FRα was attached to the NH2-terminus of the fusion receptor (figure 1C). Finally, in situations where coexpression of a second engineered protein was desired (eg, a CAR), the gene for the CER was ligated via an oligonucleotide encoding the T2A self-cleaving peptide17 to the 3’ end of the gene of the coexpressed protein (figure 1D), that is, ensuring proper expression and independent trafficking of both engineered proteins.18 The entire construct was then placed under control of the PGK (phosphoglycerate kinase) promoter of the lentiviral vector.
To determine whether the above CERs might exhibit their intended properties following transfection into target cells in vitro, we expressed both fusion constructs in different cell lines and evaluated their properties. First, the level of expression of the GPI-anchored fusion proteins was assayed in Jurkat T cells by incubation of the transfected cells with fluorescent dye conjugates of their respective cognate ligands (figure 2A). The number of bound fluorescent conjugates was then measured by flow cytometry. As shown in figure 2B, incubation of the FKBP-FR transfected cells with FK506-rhodamine caused a shift of nearly two orders of magnitude in the mean fluorescence intensity of the transfected Jurkat cells (red line). That this cell-associated fluorescence was bound to the GPI-anchored FKBP-FR could be demonstrated by treating the cells with a GPI-directed phosphoinositide phospholipase C (PI-PLC) and showing that the shift in fluorescence was abrogated (figure 2A, blue line). Importantly, as seen in the adjacent panel of figure 2A, a similar outcome was obtained when the FITC-FR containing cells were incubated with FITC-AlexaFluor 647.

Analysis of ligand–drug conjugate binding to FKBP-FR and FITC-FR fusion receptors. (A) Chemical structures of ligand–drug conjugates (FK506-rhodamine (left) and FITC-AlexFluor647 (right)) used to analyze conjugate binding to CER-expressing cells in vitro. (B) Flow cytometric analysis of FK506-rhodamine (left) and FITC-AlexFluor647 (right) binding to Jurkat cells transfected with FKBP-FR and FITC-FR fusion receptors, respectively. Note that phosphoinositide-specific phospholipase C (PI-PLC) treatment (blue line) cleaves the chimeric endocytosing receptor from the cell surface and thereby abolishes conjugate binding. (C) Relative expression of the two fusion receptors on Jurkat cells compared with expression FRα on KB cells, as quantitated by counting of EC20-99mTc binding (black bar: FRα in KB cells, gray bar: FKBP-FR in Jurkat cells, hatched bar: FITC-FR in Jurkat cells). Bar graph represents mean±SD, n=3. (D) Western blot of FITC-FR in Jurkat cells, FKBP-FR in Jurkat cells and FRα in KB cells detected with anti-FRα monoclonal antibody. FKBP-FR, FK506 binding protein folate receptor; FRα, folate receptor alpha.
Next, to obtain a more quantitative measure of the number of CERs expressed in the transfected Jurkat T cells, we exploited the fact that the fusion receptor also contained a functional binding site for folic acid, that is, allowing the number of FKBP-FR and FITC-FR fusion receptors to be determined by measuring the binding of folate-99mTc to the transfected cells (figure 2C). As shown in figure 2C, the FKBP-FR transfected Jurkat cells bound ~48% as much folate-targeted radio-imaging agent as FR-expressing KB cells, while the FITC-FR transfected cells bound ~60% as much as KB cells. Assuming that KB cells express ~3 million FR/cell,19 the above data suggest that the transfected Jurkat T cells express between 1.4 and 1.8 million CERs/cell. Because naturally occurring T cells are more difficult to transduce, the number of CERs that can be expressed on peripheral blood T cells may be significantly lower. Immunoblots of the transfected cells further demonstrated that the expressed fusion receptors have the anticipated molecular weights of ~50 and ~60 kDa for FKBP-FR and FITC-FR, respectively (figure 2D).
The affinity of each CER for its cognate ligand was then determined by quantitating FK506-rhodamine or FITC-AlexaFluor 647 binding to the same two transfected Jurkat T cell lines as a function of each conjugate’s concentration (figure 3A). Analysis of the derived binding isotherms yielded Kd values of 4 nM and 8 nM, respectively. Based on prior experience with other ligand-targeted drug conjugates, these affinities should be optimal for drug delivery, since they are sufficiently high to allow receptor saturation at very low systemic concentrations of conjugate in vivo, but not so high that conjugates cannot dissociate from their receptors following endocytosis into their target cells.
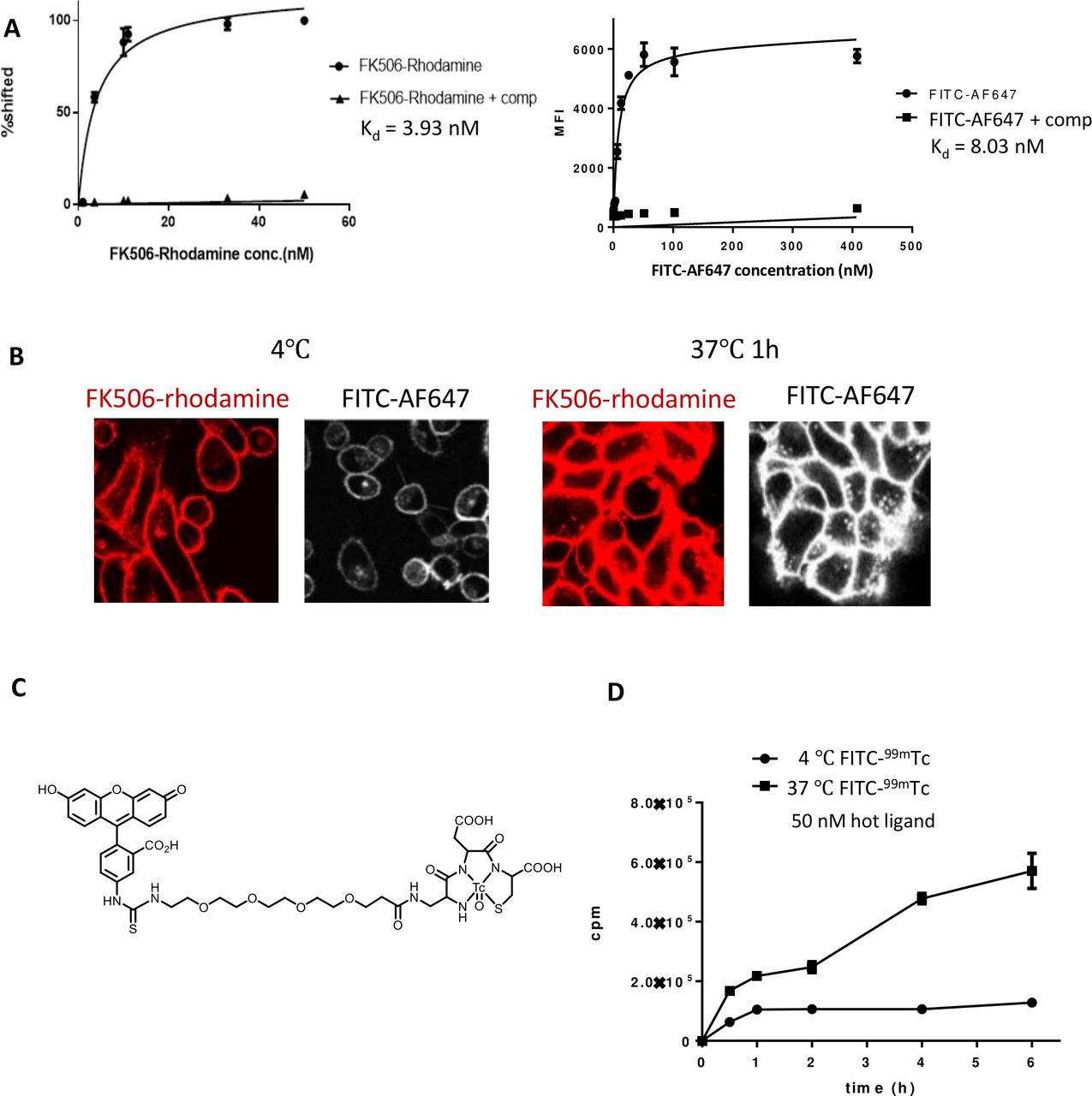
Quantitation of cell surface binding and internalization of ligand–drug conjugates by chimeric endocytosing receptor expressing HEK293 cells. (A) Evaluation of binding of FK506-rhodamine and FITC-AlexFluor 647 to FKBP-FR (left) and FITC-FR (right) expressing HEK293 cells in the presence and absence of 100-fold excess competitor (ie, glucosamine conjugates of the respective ligands). MFI: mean fluorescence intensity. (B) Internalization of FKBP-FR and FITC-FR fusion receptors in HEK293 cells at 4℃ and 37℃ detected by confocal imaging of the fluorescence of their ligand–dye conjugates, that is, FK506-rhodamine and FITC-AF647, respectively. (C) Chemical structure of FITC-99mTc conjugate used for radiolabeling of cells expressing FITC-FR fusion receptor. (D) Binding and accumulation of FITC-99mTc in FITC-FR expressing HEK293 cells. FITC-FR expressing cells were incubated with 50 nM FITC-99mTc at either 4℃ or 37℃ for the times indicated, and then radioactivity was determined by gamma counting. Graphs represent mean±SD, n=3. FITC-FR, FK506 binding protein folate receptor.
To document the generality of CER expression, HEK293 cells were similarly transfected with the FKBP-FR or FITC-FR lentiviral vectors and analyzed for internalization of the fluorescent conjugates by confocal microscopy. As shown in figure 3B, all of the fluorescence remained on the cell surface at 4°C (ie, at a temperature where endocytosis is inhibited), whereas much of the fluorescence became intracellular when cells were incubated at 37°C (ie, a temperature where endocytosis is permitted12). These data demonstrate that the FKBP-FR fusion receptor can internalize with its fluorescent cargo by receptor-mediated endocytosis at 37°C. Since similar results were obtained when FITC-FR transfected HEK293 cells were incubated with FITC-AlexaFluor 647 (right panels of figure 3B), we conclude that both CERs internalize their bound cargoes at 37°C, regardless of the structures of the attached drugs.
Next, to quantitate the rates of internalization of the ligand-drug conjugates at 37°C, we conjugated each ligand to a 99mTc-chelating agent and examined its uptake as a function of time by stripping the cells and gamma counting. As shown for the FITC-99mTc-chelate conjugate in figure 3C, binding of the radioactive conjugate to the FITC-FR HEK293 cells became saturated within an hour at 4°C, after which uptake leveled off due to the blockade of internalization (figure 3D). In contrast, uptake of the radioconjugate at 37°C continued linearly and indefinitely due to constitutive recycling of the fusion receptor back to the cell surface. Based on the difference between the curves at 4°C and 37°C, the rate of intracellular delivery of the 99mTc conjugate at 37°C can be estimated to be ~1000 molecules/cell/min.
Finally, the stability of both fusion receptors was examined by comparing their conjugate binding affinities over a period of months. Analyses (data not shown) demonstrate that no change in binding affinity occurred, suggesting that expression of functional fusion receptors could be maintained for prolonged periods of time.
Targeted delivery of therapeutic quantities of drugs to human T cells
With the ability of the CER to deliver a variety of cargoes to different CER-containing cell lines established, the question next arose whether sufficient quantities of therapeutic drug could be delivered to change cell properties. Because a major need of many CAR T cell therapies has been to terminate the cell’s activity when its behavior became life threatening,20 we undertook to determine whether we could kill CER-expressing T cells with passageway-targeted cytotoxic conjugate. As shown in figure 4, treatment of FITC-FR transfected fresh human T cells with a maytansine-FITC conjugate (ie, DM4, a microtubule inhibitor,21 conjugated to FITC via a disulfide linker22; figure 4A) resulted in killing of the T cells with an IC50 of 4.2 nM. Similar but less potent results were also obtained when an FK506-tethered DM1 conjugate was incubated with FKBP-FR (online supplemental figure S1). Moreover, addition of excess FITC-glucosamine to block maytansine-FITC binding to the anti-FITC CER reduced the conjugate’s toxicity ~10-fold, suggesting that most of the conjugate’s uptake was mediated by the CER. Indeed, this conclusion was strengthened by data in online supplemental figure S1 showing that the toxicity of the FK506-DM1 conjugate was similarly suppressed by addition of excess FK506-glucosamine. Taken together, these results demonstrated that suppression of an overactive T cell can be achieved by killing the T cell with a CER-targeted cytotoxic drug.
Supplemental material
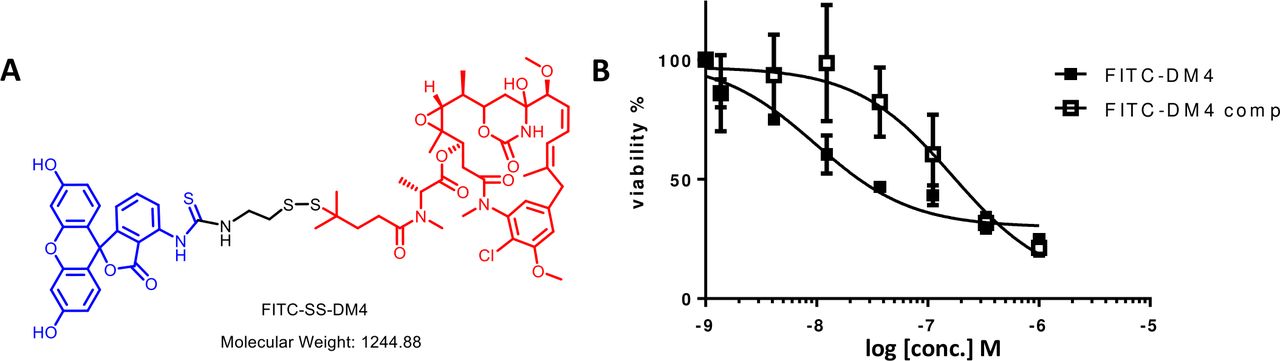
Effect of a FITC-cytotoxic drug conjugate on the viability FITC-FR transfected human T cells. (A) Structure of FITC-maytansine conjugate (FITC-DM4) constructed with a protected self-immolative disulfide releasable linker. Blue: FITC; Black: linker; Red: DM4. This self-immolative linker is designed to remain intact during transit through the vasculature to the chimeric endocytosing receptor expressing cell, but release an unmodified maytansine following endocytosis into a reducing compartment within the targeted cell.22 (B) Freshly isolated human T cells were transfected with FITC-FR and then incubated for 2 housr at 37°C with different concentrations of FITC-DM4 in the absence or presence of 100-fold excess competitor (FITC-glucosamine). After washing to remove drug conjugates, incubation was continued at 37°C for 72 hours, and cell viability was determined using an LDH (lactate dehydrogenase) release assay. Graphs represent mean±SD, n=3. FITC-FR, K506 binding protein folate receptor
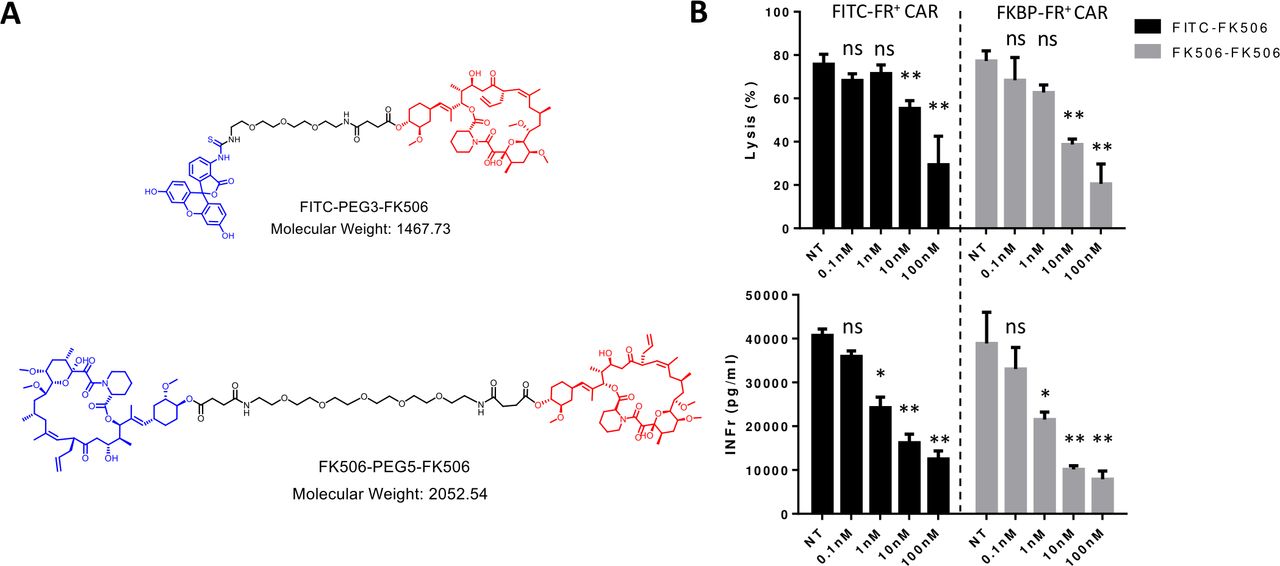
Next, to test whether the CER might be analogously used to reduce a T cell’s activity without killing it, we exploited the same two CERs to deliver FK506, a non-cytotoxic immunosuppressant of T cell activity.23 As shown in figure 5A, delivery of FK506 to FITC-FR expressing CAR T cells was achieved by constructing a conjugate of FITC linked to FK506 (upper structure), whereas delivery of FK506 to FKBP-FR expressing CAR T cells was attained by synthesizing FK506 linked to FK506 (lower structure). Regardless of the fusion receptor used, increasing doses of targeted drug facilitated enhanced suppression of CAR T cell activity (figure 5B). Moreover, the immunosuppressive effect of targeted FK506 is significantly reduced when the CERs are blocked by competition with excess ligand (ie, FK506-glucosamine or FITC-glucosamine, online supplemental figure S2). Because no decrease in cell viability was observed during these treatments, we conclude that elevating the concentration of either targeted FK506 conjugate can suppress both CAR T cell-mediated lysis of CD19+ Raji cells (figure 5B, top panels) and CAR T cell-derived secretion of interferon (IFN) gamma (figure 5B, lower panels) without loss of viability. This capability should prove generally useful, since control of CAR T cell activity via activation of a suicide gene (ie, the alternative approach to controlling a cytokine storm) will often terminate the immunotherapy.

Suppression of anti-CD19 CAR T cell activity following chimeric endocytosing receptor-mediated delivery of non-lethal immunosuppressant conjugates. (A) Chemical structures of FITC-PEG3-FK506 and FK506-PEG5-FK506 ligand immunosuppressant conjugates. Blue: targeting ligands FITC (upper panel) or FK506 (lower panel); Black: linker; Red: FK506. (B) Inhibition of FITC-FR or FKBP-FR expressing anti-CD19 CAR T cell lysis of Raji cells (top) and IFNγ release (bottom) on incubation for 2 hours at 37°C with FITC-FK506 or FK506-FK506, respectively. Following incubation with ligand–drug conjugates, cells were washed to remove drugs and incubation was continued for 12 hours before analyses. Bar graphs represent mean±SD, n=3, * denotes a p-value < 0.05, ** < 0.01, ns = not significant. NT, no treatment containing only vehicle control. CAR, chimeric antigen receptor; FKBP-FR, FK506 binding protein folate receptor; IFNγ, interferon-γ.
Control of CAR T cell numbers and/or activities in vivo using a CER
Next, to evaluate the ability of the CER to enable manipulation of CAR T cell functions in vivo, we prepared CER-expressing CAR T cells by transfecting fresh human T cells with an anti-CD19 CAR linked via a self-cleaving T2A peptide to our FITC-FR CER (see the Methods section). After injecting the engineered CAR T cells into NSG mice bearing CD19-transfected MDA-MB-231 human tumor xenografts, we first explored whether a FITC-targeted 99mTc radioconjugate (figure 3C) might accumulate selectively in the CAR T cells in vivo. To obtain this information, 14 days after tail vein injection of the CAR T cells, mice were reinjected with FITC-99mTc and then sacrificed 4 hours later in preparation for biodistribution analysis. As shown in figure 6A, the FITC-99mTc conjugate was found to bind to CER-expressing CAR T cells in vitro with a Kd ~2.8 nM. Moreover, following intravenous injection, the conjugate was seen to accumulate primarily in tumor and liver, with significantly less retention in all other tissues (panels B and C, solid bars). That the vast majority of this tissue retention was CAR T cell-mediated is demonstrated by the observations that (1) FITC-99mTc uptake is largely prevented by co-administration of unlabeled conjugate (see hatched bars) and (2) FITC-99mTc uptake is essentially absent in all tissues except liver in mice not receiving engineered CAR T cells (open bars). Taken together, these data demonstrate that FITC-linked conjugates accumulate almost exclusively in FITC-FR fusion receptor-expressing CAR T cells where the only known FITC receptor exists. The fact that some accumulation of FITC-99mTc remains in livers of mice not receiving CAR T cells is probably due to non-specific uptake of the conjugate by scavenger receptors in the liver.24
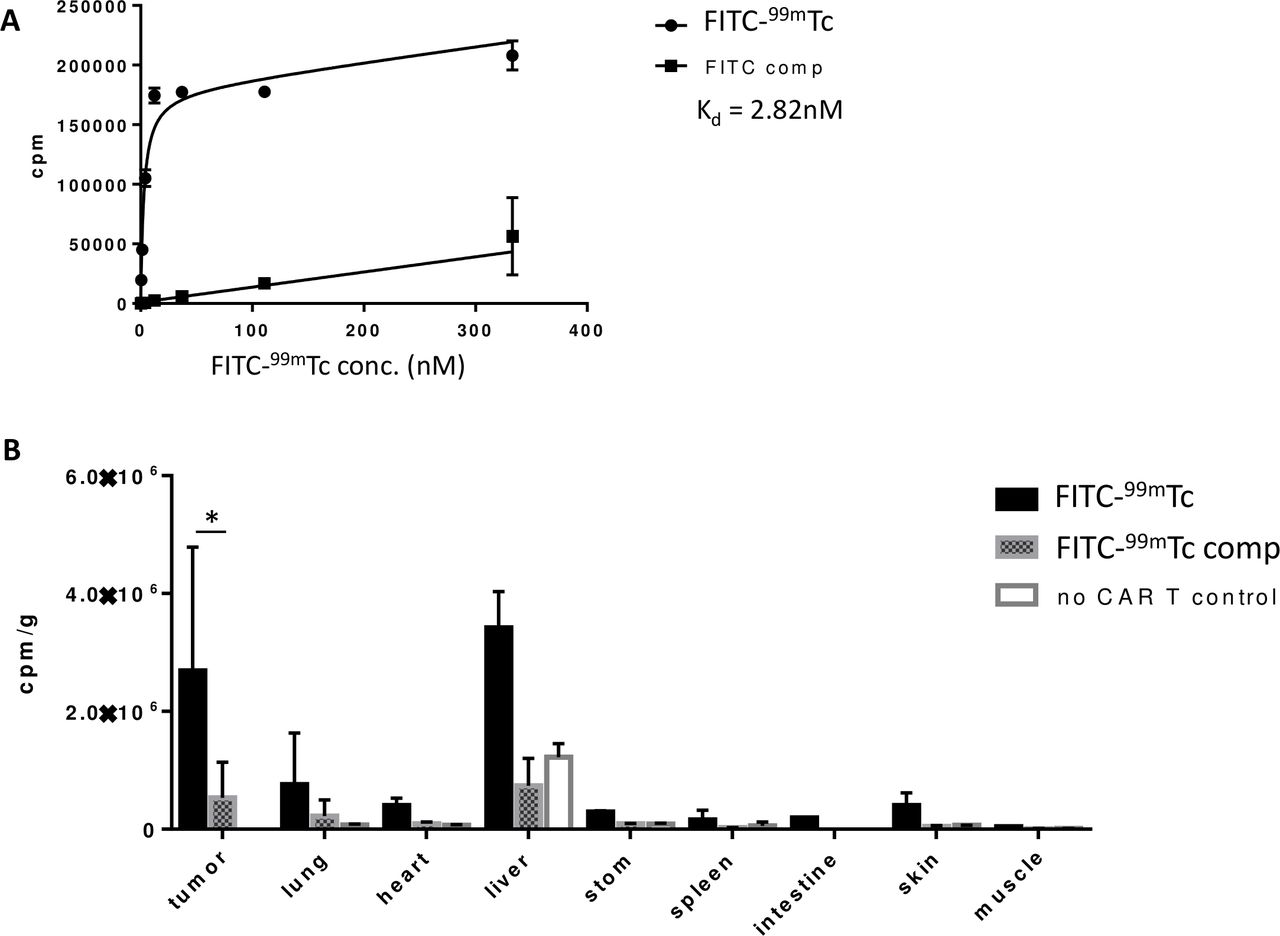
Confirmation of in vivo specificity of FITC as a targeting ligand for drug delivery through FITC-FR chimeric endocytosing receptor. (A) Evaluation of binding affinity of FITC-99mTc for FITC-FR fusion receptor in the presence and absence of 100-fold excess competitor (FITC-glucosamine). (B) Biodistribution of FITC-99mTc 4 hours post-tail vein injection. Black bars: FITC-99mTc; Hatched bars: FITC-99mTc plus competition with 100-fold excess cold conjugate; Open bars: no CAR T cells injected; Bar graph represents mean±SD, n=3, * denotes a p-value < 0.05. CAR, chimeric antigen receptor; FITC-FR, FK506 binding protein folate receptor.
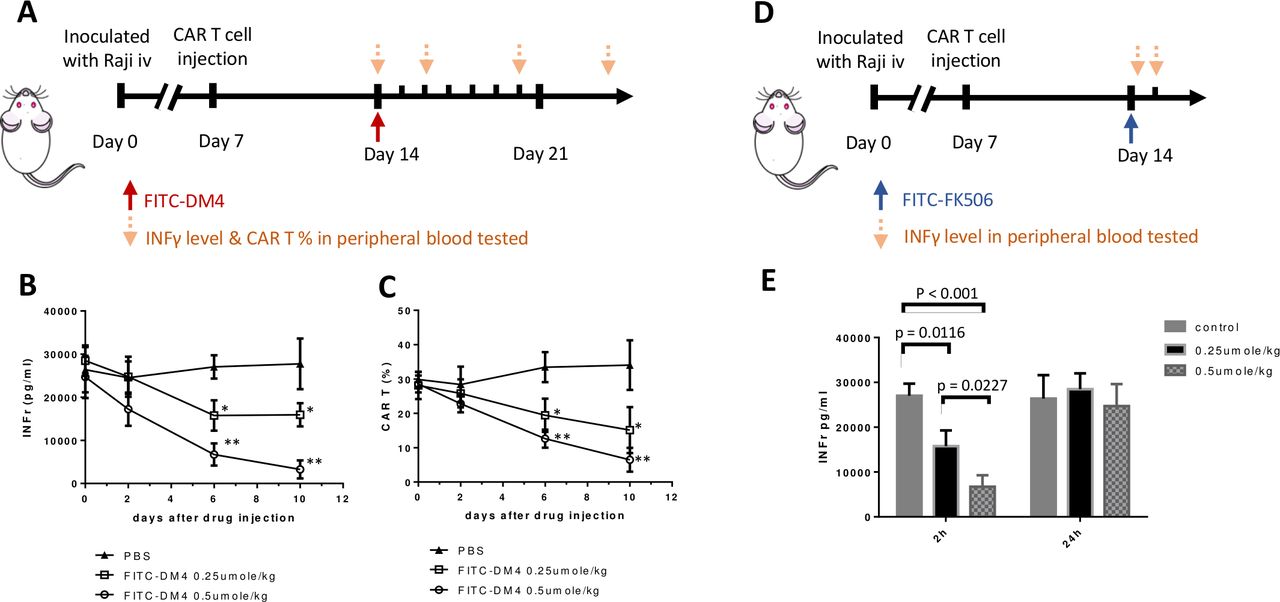
Finally, to assess the ability of this CER to facilitate control of CAR T cell activity in vivo, NSG (NOD-scid IL2Rgnull) mice were inoculated intravenously with CD19-expressing Raji cells to mimic a disseminated hematopoietic cancer, and the malignant cells were allowed to proliferate until their numbers exceeded ~8% of the total white cell count and their body weights decreased by ~10%. Anti-CD19 FITC-FR CAR T cells were then injected and CAR T cell-derived (ie, human) IFNγ levels were permitted to rise to >25 000 pg/mL to mimic a cytokine release syndrome (CRS).25 The mice were then injected with a single dose of FITC-DM4 to determine whether CAR-mediated uptake of the cytotoxic drug would reduce CAR T cell numbers and decrease associated IFNγ levels without causing systemic toxicity. As seen in figure 7B, human IFNγ (ie, CAR T cell-derived IFNγ) began to decline immediately after FITC-DM4 injection and continued to drop until the experiment was terminated. Not surprisingly, CAR T cell numbers also declined with similar kinetics, suggesting that the diminution of IFNγ likely arose from killing of the human CAR T cells. More importantly, although non-targeted DM4 was observed to increase serum aspartate transaminase concentrations (ie, a marker of liver damage), FITC-DM4 induced no elevation in aspartate transaminase above control mice (online supplemental figure S3). Because only 0.25 µmoles/kg FITC-DM4 was sufficient to reduce cytokine expression and since the CAR receptors do not saturate until ~0.8–1.0 µmol/kg,26 occupancy of all receptors was not required to achieve a significant biological effect.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Suppression of a CAR T-mediated cytokine release syndrome (CRS) via use of the chimeric endocytosing receptor to deliver either a cytotoxic or immunosuppressive payload. (A) NSG mice were intravenously injected with 2×106 Raji cells on day 0 and then treated on day 7 with 107 anti-CD19 CAR T cells containing the FITC-FR fusion receptor. Following emergence of CRS symptoms (significantly elevated plasma IFNγ), mice were injected on day 14 with a single dose of FITC-DM4 (0.25 μmol/kg or 0.5 μmol/kg) and monitored for changes in IFNγ levels (B) and CAR T numbers (C) at the indicated days (down arrows). (D) Alternatively, mice treated as above on days 0 and 7 were injected on day 14 with a single dose of FITC-FK506 (0.25 μmol/kg or 0.5 μmol/kg) and monitored for changes in IFNγ levels both 2 hours and 24 hours after treatment (E). n=5 mice per group. All data represent mean±SE, * denotes a p-value < 0.05, ** < 0.01. CAR, chimeric antigen receptor; FITC-FR, FK506 binding protein folate receptor; IFNγ, interferon-γ.
To test the ability of a non-cytotoxic FITC conjugate of FK506 to suppress CAR T cell activities without terminating the CAR T cell therapy, we next treated a similar cohort of NSG tumor-bearing mice with an FITC conjugate of FK506; that is, a non-lethal suppressor of CAR T cell activity (figure 7D). As shown in figure 7E, IFNγ levels decreased dramatically within 2 hours of FITC-FK506 administration, but increased to pretreatment levels by 24 hours postadministration. These data demonstrate that a transient inhibition of CAR T cell activity can be achieved through use of a FITC-targeted non-toxic inhibitor of T cell activity, allowing the user to decide the duration and magnitude of CAR T cell suppression via control of the timing and concentration of FITC-FK506 administered.
Discussion
Although adoptive cell therapies (ACTs) provide attractive options for treatment of many diseases, their translation into the clinic has been limited by an inability to control their activities following reinfusion into patients. Thus, overactivated CAR T cells can generate lethal concentrations of cytokines,27 while exhausted CAR T cells can adapt to comfortably coexist with cancer cells.28 Genetically modified ACTs can transform into malignant cells if inserted genes alter control of oncogenes29 30 and transplanted stem cells can differentiate into unwanted cell types when microenvironmental signals alter their intended differentiation.31 32 Because drugs that might otherwise regulate ACTs have no specificity for ACT cell types, only drugs that have little or no systemic toxicity can be exploited to manipulate the behaviors of re-infused ACTs. While the recent use of dasatinib to suppress a CRS appears to constitute a safe method to control the excessive release of toxic cytokines33 34 no obvious remedy exists to differentiate, rejuvenate, proliferate or otherwise modulate the properties of ACTs in vivo. To address this problem, we have engineered a ‘CER’ that will allow delivery of any drug selectively into an ACT, thereby avoiding the potential toxicity associated with uptake of the same drug by healthy cells throughout the body. Thus, when ACT activity must be terminated, the re-infused cells plus all of their progeny can be killed by delivery of a cytotoxic drug through the ACT-specific CER. Similarly, when a cell therapy becomes exhausted, overactivated, underdifferentiated or wrongly differentiated, its properties can be analogously manipulated by targeting of the appropriate modulators specifically to the CER-expressing ACT; that is, regardless of the effect of the same modulator on other cells in the body. Because patient safety constitutes a major criterion on which FDA authorization depends, an ability to control multiple ACT properties by use of a single fusion protein could prove critical to ACT approval. While activation of suicide genes has already been explored for termination of ACTs,9 when less terminal manipulations of cell behavior are desired, the use of a CER constitutes an excellent option.
In our design of a CER, we desired a receptor that would: (1) express at a high level in any ACT cell type, (2) traffic to the exoplasmic surface of this cell type, (3) recycle rapidly through nonlysosomal compartments back to the cell surface, (4) be composed of primarily of human components to minimize antigenicity, (5) contain a binding site for a low molecular weight targeting ligand that would have no other binding site in the body and (6) exhibit very high affinity for this targeting ligand. Use of human FRα for the recycling domain assured criteria 1–4 (12), while use of either FKBP or human scFv against fluorescein largely satisfied criteria 4–6. Most other high affinity protein–ligand interactions required either use of a non-human component (eg, the streptavidin–biotin interaction) or would have been compromised by additional binding sites existing on non-ACT cells (eg, a cannabinoid receptor). Although concern might have still arisen over a possible interaction between an FK506-drug conjugate and an intracellular FKBP (ie, all FKBPs that bind FK506 are intracellular), this apprehension has been allayed by both designing our ligand–drug conjugates to be membrane impermeable, thereby preventing their interaction with cells lacking a surface-exposed FKBP. While either fusion protein could turn out to be immunogenic, following humanization of the anti-FITC scFv, the only non-human peptides that could be presented in an HLA-restricted manner would reside at the junction between the two human domains, thus minimizing the probability of immunogenicity.
Although most envisioned applications of this CER strategy occur in the area of ACT regulation, it did not escape our attention that the same receptor might be exploited for imaging, detection and quantitation of ACTs. Thus, when the number, location, pharmacokinetics or pharmacodynamics of a transplanted cell type must be monitored, delivery of a radiolabeled payload with selective affinity for the ACT should allow an accurate method for their quantitation. Thus, in cases where a transplanted CAR T, islet, or stem cell might appear to lose efficacy, a decrease in cell number might be distinguished from a decline in cell activity by administering a radiolabeled conjugate of FITC or FK506 and determining the number of ACT cells remaining in the tissue. With the remedies for a decrease in ACT number differing from the remedies for a decline in ACT activity, information on the cause of any diminution in activity can be useful.
Materials and methods
Cell lines and human T cells
RPMI 1640 (Gibco) containing 10% heat-inactivated fetal bovine serum and 1% penicillin–streptomycin was used for culture of Raji and Jurkat cell lines. DMEM (Gibco) containing 10% heat-inactivated fetal bovine serum and 1% penicillin–streptomycin was employed for culture of MDA-MB-231 and MDA-MB-231 CD19+ cells. Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll density gradient centrifugation (GE Healthcare Lifesciences) of whole human blood obtained from healthy volunteers. Pure CD3+ T cells were enriched from PBMCs using an EasySep Human T Cell Isolation Kit (STEM CELL technologies) and then cultured in TexMACS medium (Miltenyi Biotech) containing 1% penicillin and streptomycin sulfate plus 2% human serum (Valley Biomedical) in the presence of human interleukin 2 (IL-2) (100 IU/mL, Miltenyi Biotech). Human T cells were counted every 2–3 days and maintained at 0.5×106 cells/mL. All cells were cultured in 5% CO2 at 37°C and were regularly tested for contamination of mycoplasma.
Preparation and use of lentiviral vectors encoding anti-CD19 CAR T2A FKBP-FR and anti-CD19 CAR T2A FITC-FR
For coexpression of a classical anti-CD19 CAR35 together with our CER fusion protein in the same human T cell, a lentiviral vector was constructed containing genes for the above two proteins linked via a T2A self-cleaving sequence.18 For preparation of the gene for the FKBP-containing a CER (FKBP-FR), a construct of the following sequences in frame from 5’ to 3’ end was prepared: residues 1–24 of human (FRα, NM_016724.2), the entire sequence for human FKBP12 (NM_000801.5), the sequence of a flexible peptide linker (GGGGS)3, and the sequence for the remainder of human FRα (residues 25–257) (see figure 1C). For coexpression of this gene in the same vector as the anti-CD19 CAR, the sequence for the anti-CD19 CAR was ligated in frame to the sequence for the T2A self-cleaving peptide,17 which in turn was ligated in frame to the sequence for FKBP-FR (figure 1D). The resulting construct was then inserted into a pHR lentiviral expression vector by restriction enzyme digestion with MluI and NotI, and then human T cells were transfected with the lentiviral vector using standard methods.36 Briefly, purified human T cells were first activated using Dynabeads coupled to anti-CD3/CD28 antibodies (Life Technologies) for 12–24 hours in the presence of human IL-2 (100 IU/mL) and then infected with the aforementioned lentivirus. After 3–5 days of transfection,37 the T cells were harvested and analyzed by flow cytometry to establish that ~70% of the CD3 +T cells were expressing a CER. The antifluorescein scFv containing CER (FITC-FR) was prepared similarly, except the sequence for the anti-FITC scFv14 was used instead of the sequence for FKBP.
Synthesis, purification and analytical characterization of FK506 and FITC derivatives
See text in Supporting Information.
CER binding and internalization assays
For analysis of FK506-rhodamine binding to CER-expressing cells, FKBP-FR transfected Jurkat cells or non-transfected cells (5×105) were preincubated in the presence or absence of FK506-glucosamine (100-fold excess to block all available fusion receptors) for 30 min at 4℃, and then incubated with the indicated concentration of FK506-rhodamine again for 30 min at 4 ℃. Cells were washed three times, then resuspended in 2% FBS PBS and submitted for analysis by flow cytometry. The binding affinity assays for FITC-AlexFluor 647 were determined similarly, except 100-fold excess FITC-glucosamine was employed as the competitor. For analysis of the effect of PI-PLC treatment, cells were removed from the monolayer with scrapers and washed in PBS, then suspended at 5×106 cells/mL in 0.025 M Tris-HCl, 0.25 M sucrose, 0.01 M glucose, 1 x protease inhibitor cocktail (Roche), pH 7.5. 100 µL of the cell suspension were then incubated with 5 units of PI-PLC (ThermoFisher, #P6466) at 37 ℃ for 1 hour, after which the reaction was stopped by cooling to 4℃. Cells were then washed and analyzed for FK506-rhodamine or FITC-AlexFluor 647 binding by incubation with the above conjugates at 4℃ following by washing in PBS and analysis by flow cytometry. Results were evaluated using FlowJo software. GraphPad Prism V.7 software was used to analyze binding affinity.
For evaluation of ligand–drug conjugate internalization, 1×104 FKBP-FR expressing HEK293 cells in 0.5 mL of growth media were seeded in covered glass chambers (ThermoFisher, #155383) and incubated overnight at 37 ℃ to achieve full adherence. Fresh medium containing 50 nM FK506-rhodamine was then added, and cells were further incubated either at 4℃ or 37℃ for the times indicated. Cells were then washed with warm cell medium and imaged using a confocal laser scanning microscope. The internalization assay for the FITC-FR fusion receptor was performed similarly, except FITC-AlexFluor 647 was used as the ligand–drug conjugate.
Quantification of CER expression using FA-99mTc
A folate complex of 99mTc (EC20-99mTc) was prepared as previously described.38 KB, FKBP-FR Jurkat or FITC-FR Jurkat cells were then grown in a 24-well plates and incubated with 50 nM EC20-99mTc at 4℃ for 30 min. After incubation, the cells were washed and lysed in 10% SDS solution, and the radiation level of each sample was determined using a gamma counter.
Measurement of internalized and membrane-bound FITC-99mTc
FITC-99mTc was synthesized and chelated with technetium similar to EC20-99mTc (see Supporting Information). To quantitate the amount of surface-bound and internalized FITC-99mTc following incubation with HEK293 cells expressing FITC-FR, cells were seeded at ~30% confluence and cultured for ~2 days in 24-well plates and then incubate with FITC-99mTc for the desired times at either four or 37°C. Cells were then washed with PBS and lysed in 0.5 mL 10% SDS solution. The radiation levels of each sample were then determined with a gamma counter.
Characterization of FITC-FR and FKBP-FR fusion receptor molecular weights by Western blotting
Expression of FITC-FR and FKBP-FR in HEK293 cells was evaluated by lysing the cells in SDS-PAGE solution and centrifuging the solution at 16 000xg for 10 min at 4°C to remove any residual debris. Supernatants were then resolved by electrophoresis on 4%–20% SDS-PAGE gels and electrophoretically transferred to nitrocellulose filters. Blots were probed with anti-FRα or antiactin antibody, followed by staining with horseradish peroxidase-conjugated secondary antibody. Stained blots were then visualized using the enhanced chemiluminescence method.
Analysis of cell killing using FITC-maytansine (DM4) ligand–drug conjugate
Human T cells transfected with FITC-FR were incubated for 2 hours at 37°C with different concentrations of an FITC-targeted conjugate of the potent cytotoxin, maytansine DM4 in either the absence or presence of 100-fold excess of FITC-glucosamine (competition). Cells were then washed twice, supplemented with fresh medium, and cultured for another 72 hours. CellTiter-GloTM reagent (Promega, #G7570) was then added to each well at a 1:1 vol/vol ratio and luminescent readings were obtained to determine cell viability using a plate reader according to the manufacture’s protocol.
Inhibition of human T cell lysis of cancer cells using an FK506 conjugate of either FITC or FK506
Anti-CD19 CAR T cells coexpressing either FITC-FR or FKBP-FR were cocultured with CD19-expressing Raji cells at an effector : target cell ratio of 5:1. FITC-FK506 or FK506-FK506 was then added to the coculture and the incubation was continued for an additional 2 hours in the absence or presence of competing ligands (FITC-glucosamine or FK506-glucosamine). Cells were then washed, supplemented with fresh medium and the incubation was continued for an additional 12 hours. Culture plates were then centrifuged at 350 × g for 10 min, and supernatants were assayed for both cancer cell death by evaluating released lactate dehydrogenase activity using CytoONE (Promega) and IFNγ levels using human IFNγ ELISA kit (BioLegend).
Evaluation of the biodistribution of CER positive CAR T cells with FITC-99mTc conjugate
To quantify the biodistribution of CER expressing anti-CD19 CAR T cells in vivo, a CD19-expressing murine solid tumor model was desired. For this purpose, MDA-MB-231 human breast cancer cells were transfected with lentivirus encoding CD19 (NM_001178098.1), and 106 MDA-MB-231 CD19+ cells were implanted subcutaneously in immunodeficient NSG mice (Jackson Laboratory). After allowing the tumors to grow to ~100 mm3, ~107 anti-CD19 FITC-FR CAR T cells were intravenously injected and permitted to redistribute and proliferate naturally for 2 weeks. Two hundred µCi of FITC-99mTc dissolved in 100 µL PBS was then intravenously injected and 4 hours later all major organs were collected and counted by gamma counting. Tumor volumes were estimated using a caliper and the equation V= ½(L x W2), where L is the longest axis of the tumor and W is the axis perpendicular to L.
Evaluation of FITC-DM4 and FITC-FK506 on FITC-FR CAR T cell activity in a Raji tumor model
Immunodeficient NSG mice (Jackson Laboratory) were implanted intravenously with 2×106 Raji cells (a CD19 positive lymphoma cell line). Seven days later, mice were injected intravenously with 107 FITC-FR anti-CD19 CAR T cells and bodyweights were monitored every other day. Seven days after CAR T cell injection, FITC-DM4 or FITC-FK506 was administered intravenously and peripheral blood IFNγ levels were assayed at the times indicated using an IFNγ ELISA kit (BioLegend). Peripheral blood human CAR T cell numbers were similarly determined in peripheral blood samples by c-myc-APC staining, followed by by flow cytometry in the presence of counting beads.
Acknowledgments
The authors would like to acknowledge the Purdue Center for Cancer Research, the Purdue Institute for Drug Discovery, the Purdue Flow Cytometry Core and the Chemical Genomic Facility for their services. This work was funded by a gift from the Hurvis Family Foundation.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors BZ contributed to the design of study, acquisition of in vitro and in vivo data, analysis/interpretation of data, and writing of the manuscript. PSL conceived of the concept and contributed to the design of study, analysis/interpretation of data, writing of the manuscript, and supervision of the project. QL conducted the toxicity study. JVN, XL and MS designed and synthesized related molecules. All authors edited and approved the final manuscript.
Funding This work was funded by a gift from the Hurvis Family Foundation.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All animal care and use were followed by NIH guidelines, and all experimental protocols were approved by the Purdue Animal Care and Use Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as online supplemental information.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.