Article Text

Abstract
Background Pancreatic ductal adenocarcinoma (PDAC) is notoriously resistant to treatment including checkpoint-blockade immunotherapy. We hypothesized that a bimodal treatment approach consisting of dendritic cell (DC) vaccination to prime tumor-specific T cells, and a strategy to reprogram the desmoplastic tumor microenvironment (TME) would be needed to break tolerance to these pancreatic cancers. As a proof-of-concept, we investigated the efficacy of combined DC vaccination with CD40-agonistic antibodies in a poorly immunogenic murine model of PDAC. Based on the rationale that mesothelioma and pancreatic cancer share a number of tumor associated antigens, the DCs were loaded with either pancreatic or mesothelioma tumor lysates.
Methods Immune-competent mice with subcutaneously or orthotopically growing KrasG12D/+;Trp53R172H/+;Pdx-1-Cre (KPC) PDAC tumors were vaccinated with syngeneic bone marrow-derived DCs loaded with either pancreatic cancer (KPC) or mesothelioma (AE17) lysate and consequently treated with FGK45 (CD40 agonist). Tumor progression was monitored and immune responses in TME and lymphoid organs were analyzed using multicolor flow cytometry and NanoString analyzes.
Results Mesothelioma-lysate loaded DCs generated cross-reactive tumor-antigen-specific T-cell responses to pancreatic cancer and induced delayed tumor outgrowth when provided as prophylactic vaccine. In established disease, combination with stimulating CD40 antibody was necessary to improve survival, while anti-CD40 alone was ineffective. Extensive analysis of the TME showed that anti-CD40 monotherapy did improve CD8 +T cell infiltration, but these essential effector cells displayed hallmarks of exhaustion, including PD-1, TIM-3 and NKG2A. Combination therapy induced a strong change in tumor transcriptome and mitigated the expression of inhibitory markers on CD8 +T cells.
Conclusion These results demonstrate the potency of DC therapy in combination with CD40-stimulation for the treatment of pancreatic cancer and provide directions for near future clinical trials.
- immunotherapy
- dendritic cells
- T-lymphocytes
- vaccination
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Pancreatic adenocarcinoma is currently the fourth-leading cause of cancer-related death in the USA and the third in Europe.1 2 The incidence is rising and it is expected that pancreatic cancer will be the second-leading cause of cancer-related death by 2030.3 The current prognosis of a newly diagnosed pancreatic cancer patient is poor with a 5-year survival of 8.5%.1 To date, surgical resection is the mainstay of curative treatment. However, this is usually not an option due to local vascular invasion or metastasis at diagnosis. Only 10%–20% of all pancreatic cancer patients are eligible for surgical resection and relapse rates are high.4 5 Adjuvant chemotherapy following surgical resection improves median overall survival, but even with new chemotherapy regimens cure is exceedingly rare.6 Therefore, new treatment modalities are desperately needed in order to achieve durable disease control in pancreatic cancer patients.
Although immunotherapy yields striking results in numerous malignancies, clinical responses in pancreatic cancer have been disappointing.7–9 Reasons for this poor clinical response are likely multifactorial. Pancreatic cancer has been considered an immunologically ‘cold’ tumor with rare infiltration of cytotoxic T cells, explaining the low response rates to immune checkpoint antibodies.10–12 A highly immunosuppressive tumor microenvironment (TME) consisting of a plethora of cells including myeloid-derived suppressor cells (MDSCs), tumor associated macrophages (TAMs) and regulatory T cells (Tregs) in conjunction with a characteristic dense desmoplastic stroma has been reported to be responsible for the observed T-cell exclusion and dysfunction in established tumors.13 Several therapeutic agents targeting the pancreatic TME have shown promising results.14 15 Seminal studies have investigated the potency of CD40-agonistic antibodies in modulating the TME and desmoplastic stroma of pancreatic cancers, thereby allowing T-cell infiltration and antitumor efficacy.16 This was later shown to be dependent on stromalysis by TAM-precursors which, following upregulation of matrix metallo-proteases, degrade fibrosis and support the influx and antitumor efficacy of T cells.17 18 Although some clinical responses to CD40-agonistic antibodies have been reported, durable responses are limited. A lack of successfully presented and high-quality tumor antigens has also been proposed to be involved in the lack of immune reactivity to pancreatic cancers.19 20 Akin to this, observational patient studies have shown rare long-term postresection pancreatic cancer survivors to have increased levels of tumor-reactive T cells in their peripheral blood and tumors.20 21 Dendritic cells (DCs) are the most potent T-cell activators of the immune system, and DC vaccination can successfully induce immune responses and clinical responses in various less-immunogenic malignancies when loaded with the appropriate tumor antigens.22 Ideally, these antigens should be derived from the patient’s own tumor. However, at this point in time, implementation of these personalized vaccines poses a logistical hurdle. An allogeneic ‘off-the-shelf’ strategy for tumor lysate could circumvent this issue and standardize treatment across patients. We have previously shown that treating mesothelioma patients with autologous DCs loaded with a allogeneic tumor lysate is feasible and induces immune responses and tumor regressions in a subset of patients.22 As several tumor-associated antigens (TAAs), such as cancer-testis antigens and tumor differentiation antigens, are shared across different tumor types, this vaccine could be effective in other tumors as well, including pancreatic cancer, which coexpresses several TAAs with mesothelioma tumors (eg, mesothelin, WT-1, MUC1).
Here, we investigated the efficacy of DC vaccination in a representative murine model of human pancreatic ductal adenocarcinoma (PDAC). We show that vaccination with mesothelioma lysate-loaded DCs yields tumor-specific immune responses against pancreatic cancer and decreases tumor progression. In established tumors, significant prolonged survival was only achieved when DC vaccination was combined with an agonistic CD40 antibody. Extensive analysis of the TME showed that whereas CD40-agonistic antibodies as monotherapy improved intratumoral T-cell infiltration, these cells displayed hallmarks of exhaustion. In the combination treatment, an improved T-cell phenotype lacking the high expression of various inhibitory receptors was observed. Therefore, CD40-agonistic antibody treatment may sensitize pancreatic tumors to tumor-specific immune responses induced by DC vaccination. These translational studies pave the way for future clinical trials investigating DC vaccination in occult disease or as part of combination immunotherapy in inoperable pancreatic cancer patients, some of which have already been initiated (REACtiVe Trial).
Results
DC vaccination with mesothelioma lysate induces T-cell immunity and efficacy against pancreatic cancer
We hypothesized that vaccination with DCs loaded with mesothelioma TAAs can generate a cross-reactive immune response against pancreatic cancer. We, therefore, evaluated whether pancreatic cancer (KPC3) lysate loaded-DCs or mesothelioma (AE17) lysate loaded-DCs induced protective immunity in mice challenged with KPC3 (figure 1A). Comparison of RNA-seq transcriptome profiles of KPC3 and AE17, based on a predefined list of validated TAAs, revealed that 63% of the TAAs were expressed by both AE17 and KPC3 (figure 1B, online supplementary table S1).23 This supports the notion of shared antigens between the two cancer types. For a more unbiased approach, we also investigated the overlap in transcriptome profiles of KPC3, AE17 and two unrelated cell lines (B16F10, MC38) (online supplementary figure S1). Shared transcripts could be found in all four tumor cell lines. Exposure of DCs to tumor lysates and CpG led to rapid upregulation of activation markers (eg, CD40, CD80/86) (online supplementary figure S2). Importantly, prophylactic vaccination of mice with DCs loaded with pancreatic or mesothelioma lysate was equally effective in delaying tumor growth and both had significant smaller tumor volumes compared with untreated mice at day 20 (figure 1C,D).
Supplemental material
Supplemental material
Supplemental material
Supplemental material

Mesothelioma lysate-DC vaccination is able to delay pancreatic tumor growth and induce strong T-cell immunity. (A) Dendritic cell (DC) vaccination study setup. (B) Expression of immunogenic tumor antigens as described by Cheever et al 23 in the tumor cell line KPC3 and AE17. Percentages indicate amount of overlapping and non-overlapping genes. (C) Tumor volumes (with SEM) measured over time of untreated and treated mice. (D) Tumor size at the time of sacrifice (day 20 after tumor injection). (E) Circulating CD4+ and CD8+T cell frequencies at day −3, 4 and 20. (F) Percentage of CD69+, Ki67+ and CD44+CD62L− subsets of CD4+ and CD8+ circulating T cells 4 days after DC vaccination. (G) CD3+, CD4+, CD8+ and CD4+CD25+FoxP3+TILs as a percentage of alive CD45 +cells at day 20 after tumor injection. (H) Expression of CD44 and Ki67 on CD4+ and CD8+T cells at day 20 in blood, spleen and tumor. n=8 per group. Significance was determined using the non-parametric Mann-Whitney U test. Data presented as the mean±SEM. *P<0.05,**P<0.01,***P<0.001.
To elucidate the mechanisms underlying DC therapy efficacy, we analyzed immune parameters in peripheral blood, spleen and tumors in both pancreatic cancer and mesothelioma lysate-loaded DC therapy treated mice. In vaccinated mice, increased frequencies of circulating CD3+, CD4+ and CD8+T cells could be detected as early as 4 days after DC treatment (day −3 before tumor inoculation). These immune responses were durable and persisted over time until day of sacrifice (figure 1E). A more in-depth phenotypic analysis demonstrated that vaccinated mice had higher frequencies of activated (CD69+), proliferating (Ki-67+) and effector memory (CD44 +CD62L-) CD4+ and CD8+T cells in the peripheral blood compared with untreated mice. This did not differ between mesothelioma-lysate and pancreatic cancer-lysate DC-treated mice (figure 1F). In contrast to changes in T-cell frequencies, the expression of CD69+, Ki-67+ and CD44+CD62L- on circulating T cells of vaccinated mice waned over time (online supplementary figure S3). Higher frequencies of intratumoral CD4+ and CD8+T cells were noted, paralleling the delayed tumor growth observed after vaccination (figure 1G). CD8+ tumor infiltrating lymphocytes (TILs) of treated mice more often expressed the memory marker CD44 and the proliferation marker Ki-67, which was not observed in the spleen and peripheral blood at the time of sacrifice (figure 1H). This was also not observed for CD4 +TILs. Importantly, the frequencies of regulatory CD4 +CD25+FoxP3+TILs remained comparable between treated and untreated mice (figure 1G). Therefore, DC vaccination is able to induce the infiltration of PDAC tumors with activated, proliferating CD4+ and CD8+T cells without concomitant Treg-induction.
Supplemental material
DC vaccination depends on tumor antigens and tumor-specific T cells
We then assessed whether T-cell responses induced by mesothelioma lysate-loaded DCs were reactive to tumor antigens present on pancreas carcinoma cells. Mice treated with mesothelioma lysate-loaded DCs had significantly smaller tumors compared with untreated mice or those treated with non-loaded DCs, suggesting that the delay in tumor outgrowth was due to a TAA-reactive immune response (figure 2A). Indeed, CD8 +T cells isolated from vaccinated mice responded in vitro specifically to autologous pancreatic cancer lysate-loaded DCs, while T cells from untreated mice or those from non-loaded DC vaccinated mice did not (figure 2B). On stimulation, higher frequencies of CD8 +T cells from mesothelioma lysate-loaded DC treated mice expressed CD107a (being a marker of cytotoxic degranulation), Granzyme B, interferon-γ (IFNγ) and tumor necrosis factor α (TNFα) compared with CD8 +T cells from untreated mice or mice treated with non-loaded DCs. This effect was not observed when CD8 +T cells were stimulated with DCs loaded with a control wild type tissue lysate (figure 2B), demonstrating that mesothelioma lysate-loaded DCs can generate tumor-antigen specific T cells reactive to antigens also expressed by pancreatic cancer cells. In these in vitro assays, CD8 +T cells from vaccinated mice also responded better than those from untreated mice to DCs loaded with B16F10 melanoma lysate (online supplementary figure S4), suggesting induction of immunity to shared tumor antigens across KPC3, AE17 and B16F10 as listed in online supplementary table S1.
Supplemental material

Tumor lysate DCs outperforms non-loaded DCs. (A) Tumor volume measured over time as individual tumor outgrowth curves and per group, and tumor size at day of sacrifice (day 21) of treated and untreated animals. (B) In vitro efficacy assay; relative production of CD107a, granzyme B, IFNγ and TNFα by CD8 +splenocytes of three treatment groups after stimulation with DCs loaded with autologous tumor lysate or control lung lysate, normalized for untreated mice. n=8 per group. Significance was determined using the non-parametric Mann-Whitney U test. Data presented as the mean±SEM. *P<0.05,**P<0.01. DC, dendritic cell; IFNγ, interferon-γ; TNFα, tumor necrosis factor α.
CD40-agonistic antibody treatment sensitizes established pancreatic tumors to DC vaccination and improves efficacy
As DC therapy generated systemic antitumor immune responses capable of stalling tumor growth when given prophylactically, we set out to test its capacity to control established KPC3 tumors. Although our pilot study demonstrated that tumor lysate-loaded DCs are capable of inducing systemic changes in T-cell subsets, as a single therapy it was unable to increase intratumoral T cells or delay tumor growth (online supplementary figure S5). The lack of increased T-cell infiltration found in established tumors in the presence of a systemic immune response suggested that the PDAC TME might physically obstruct T cells from infiltrating the tumor. CD40-agonistic antibodies have previously been found to allow T-cell infiltration due to TME-reorganization in pancreatic cancer, offering a treatment rationale for combination therapy with DC vaccination.16 As CD40 is also highly expressed on the tumor lysate-loaded DCs (online supplementary figure S2), administering the antibody early following DC transfer might offer additional synergy between these treatments (online supplementary figure S6a). Interestingly, αCD40 combined with DC vaccination resulted in significant tumor growth control when compared with untreated mice while monotherapy DC or αCD40 did not (online supplementary figure S6b). αCD40 monotherapy was able to induce systemic and intratumoral responses (online supplementary figure S6c–f). To show that the efficacy of this combination therapy was not limited to pancreatic cancer or the C57BL/6 mouse strain, we performed a comparable experiment in a mesothelioma tumor model (CBA/J background) yielding similar results (online supplementary figure S7).
Supplemental material
Supplemental material
Supplemental material
As treatment at day 5 after tumor cell injection still reflects minimal disease burden, we aimed at treating larger tumors (day 10) using an intensified treatment schedule (figure 3A). In this experimental setup, tumor growth and survival of mice treated with monotherapy DC vaccination or αCD40 also did not significantly differ from untreated tumor-bearing mice (figure 3B–D and online supplementary figure S8). The combination therapy, however, significantly delayed tumor growth (figure 3B), led to significantly smaller tumor volumes and improved survival (figure 3C,D). In order to elucidate the immunological prerequisites of therapeutic efficacy and to demonstrate if the observed antitumoral response is T-cell dependent, mice were depleted of CD4+ and CD8+T cells before receiving treatment (online supplementary figure S9). Antitumoral efficacy was retained in αCD4 and isotype-treated mice receiving DC vaccination and αCD40 (figure 3E–G). However, therapeutic responses were mitigated in mice depleted for CD8 +T cells. Importantly, we assessed the efficacy of combination therapy in an orthotropic mouse model, in order to examine if our results could be replicated in a more translational model mimicking the anatomical location and phenotypic features of PDAC (figure 3H). Strikingly, 56% (5/9) of all combination therapy-treated mice were macroscopically free of tumor at the day of analysis (figure 3I and online supplementary figure S10). In contrast, all untreated or monotherapy-treated mice bore tumors and tumor sizes were significantly larger compared with the remaining combination therapy-treated mice with tumor (figure 3J).
Supplemental material
Supplemental material
Supplemental material
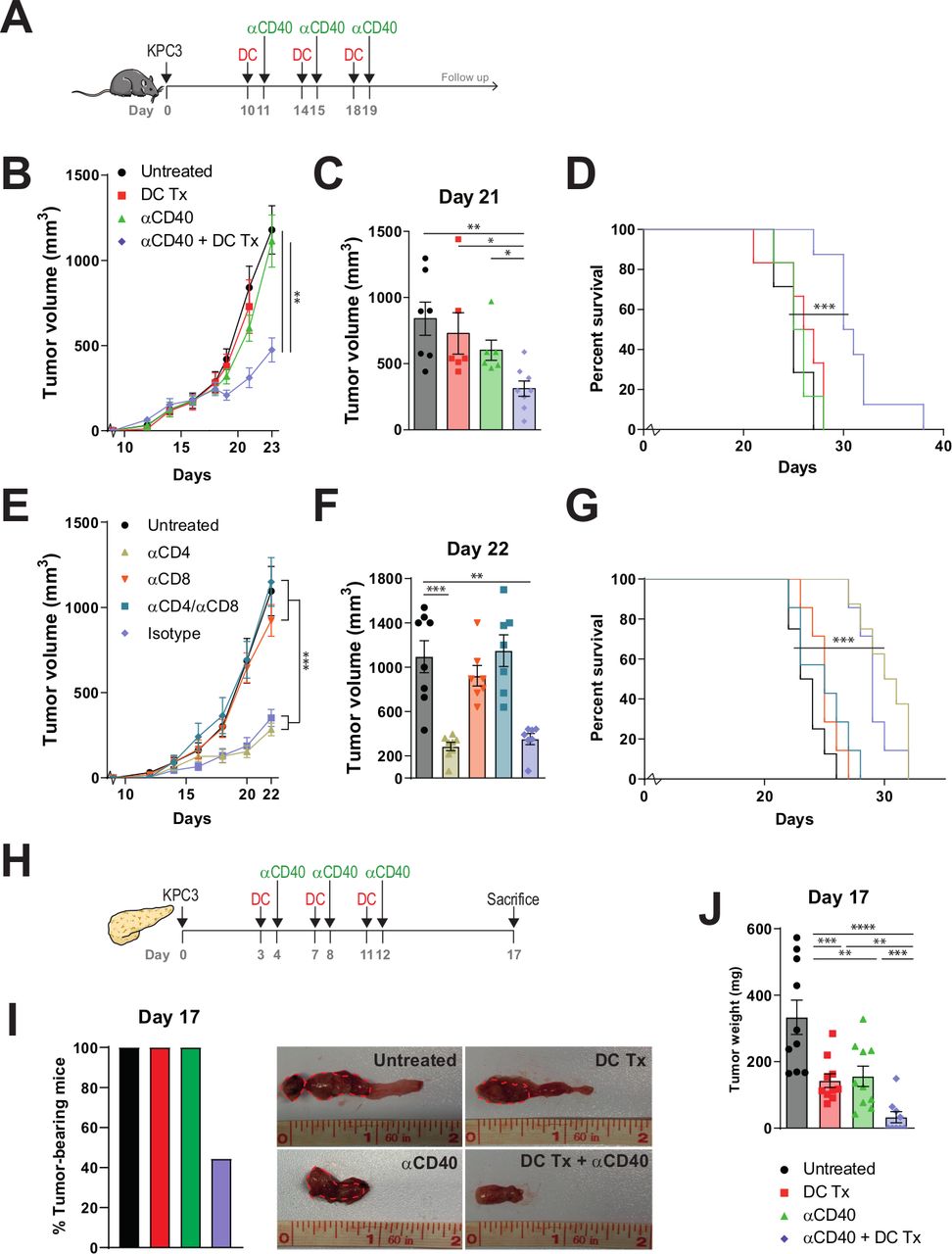
DC vaccination-αCD40 combination therapy improves survival of tumor-bearing mice. (A) Subcutaneous tumor model study setup. Mice were treated with AE17-lysate DCs and FGK45. (B) Tumor volumes measured over time. (C) Tumor size at day 21. (D) Kaplan-Meier analysis of treated and untreated animals. (E) Tumor volumes measured over time. (F) Tumor size at day 22. (G) Kaplan-Meier analysis of treated and untreated animals. (H) Orthotopic tumor model study setup. (I) Percentage of tumor bearing mice. (J) Tumor weight on day 17. n=8–10 per group. Significance was determined using the non-parametric Mann-Whitney U test or log-rank test. Data presented as the mean±SEM. *P<0.05, **P<0.01, ***P<0.001. DC, dendritic cell.
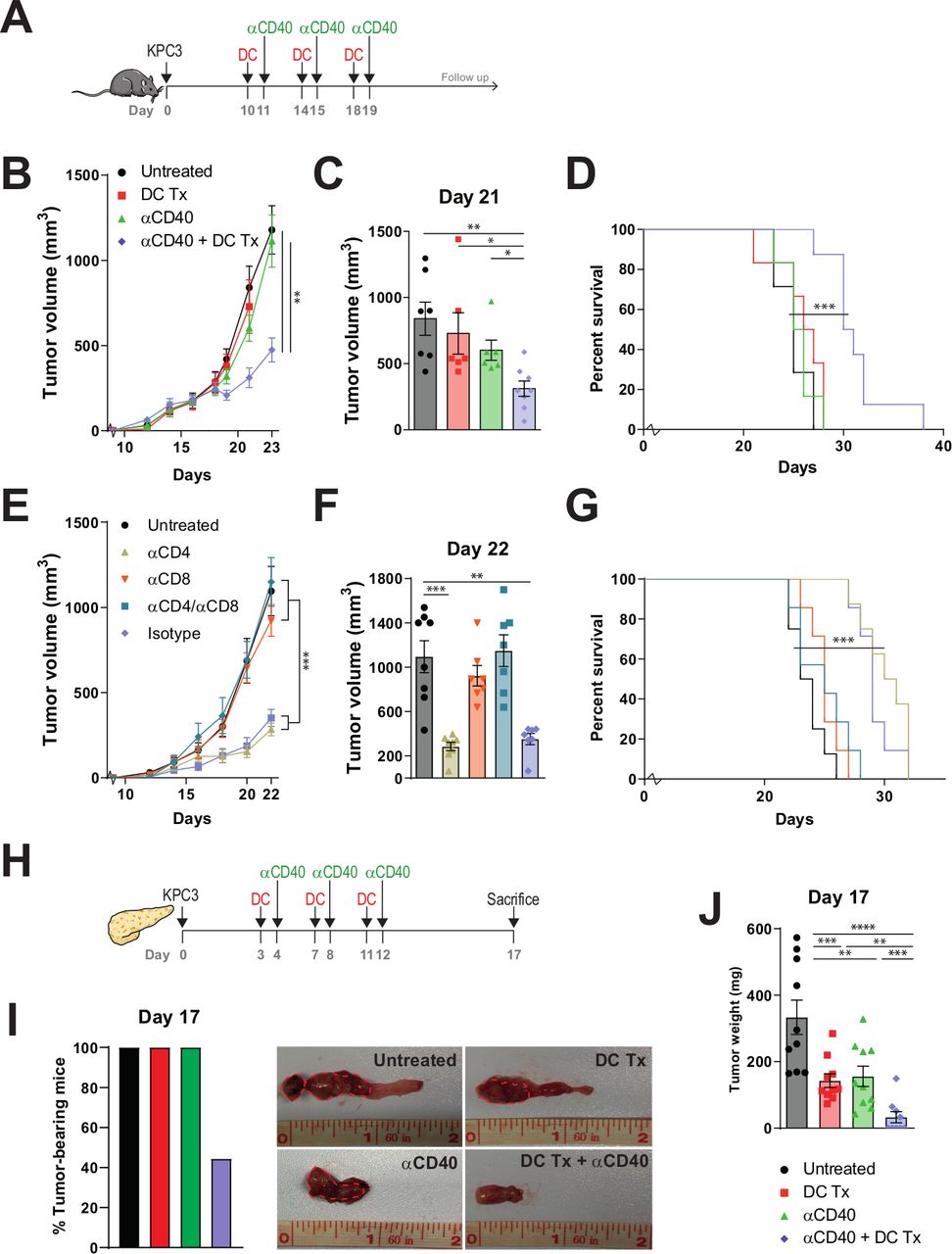
Interim peripheral blood analysis demonstrated that both monotherapy DC vaccination and αCD40 treatment induced higher frequencies of CD69+, Ki-67+ and PD-1 +T cells. However, this effect was more confined to CD4 +T cells when mice were only treated with DC vaccination, and to CD8 +T cells for monotherapy αCD40 (figure 4A). Combination therapy induced higher frequencies of CD69+, Ki-67+ and PD-1+ for both CD4+ and CD8+T cells. Enrichment over time of Ki-67+ and PD-1 +T cells was detected in mice treated with either combination therapy or αCD40 monotherapy. Furthermore, CD44 +CD62L- effector memory T cells were significantly increased after both monotherapies and combination therapy (figure 4B). Over time, mice treated with combination therapy yielded the highest frequencies of effector memory T cells compared with mice treated with monotherapy or untreated mice. This was observed for both CD4+ and CD8+T cells. The enrichment of effector memory T-cell frequencies was less prominent after single DC vaccination and subsequent αCD40 treatment (online supplementary figure S6d), promoting the role of multiple vaccinations.

Immune activation in peripheral blood following AE17-lysate DC and FGK45 treatment. (A) Percentage of CD69+, Ki67+ and PD-1+ subsets of CD4+ and CD8+ circulating T cells at day 14 and 22. (B) Percentage of CD44 +CD62L− effector memory subsets and memory status of CD4+ and CD8+ circulating T cells at day 14 and day 22. n=8 per group. Significance was determined using the non-parametric Mann-Whitney U test. Data presented as the mean±SEM. *P<0.05, **P<0.01, ***P<0.001. DC, dendritic cell.
Combining DC vaccination and αCD40 remodels the TME, including T-cell exhaustion markers
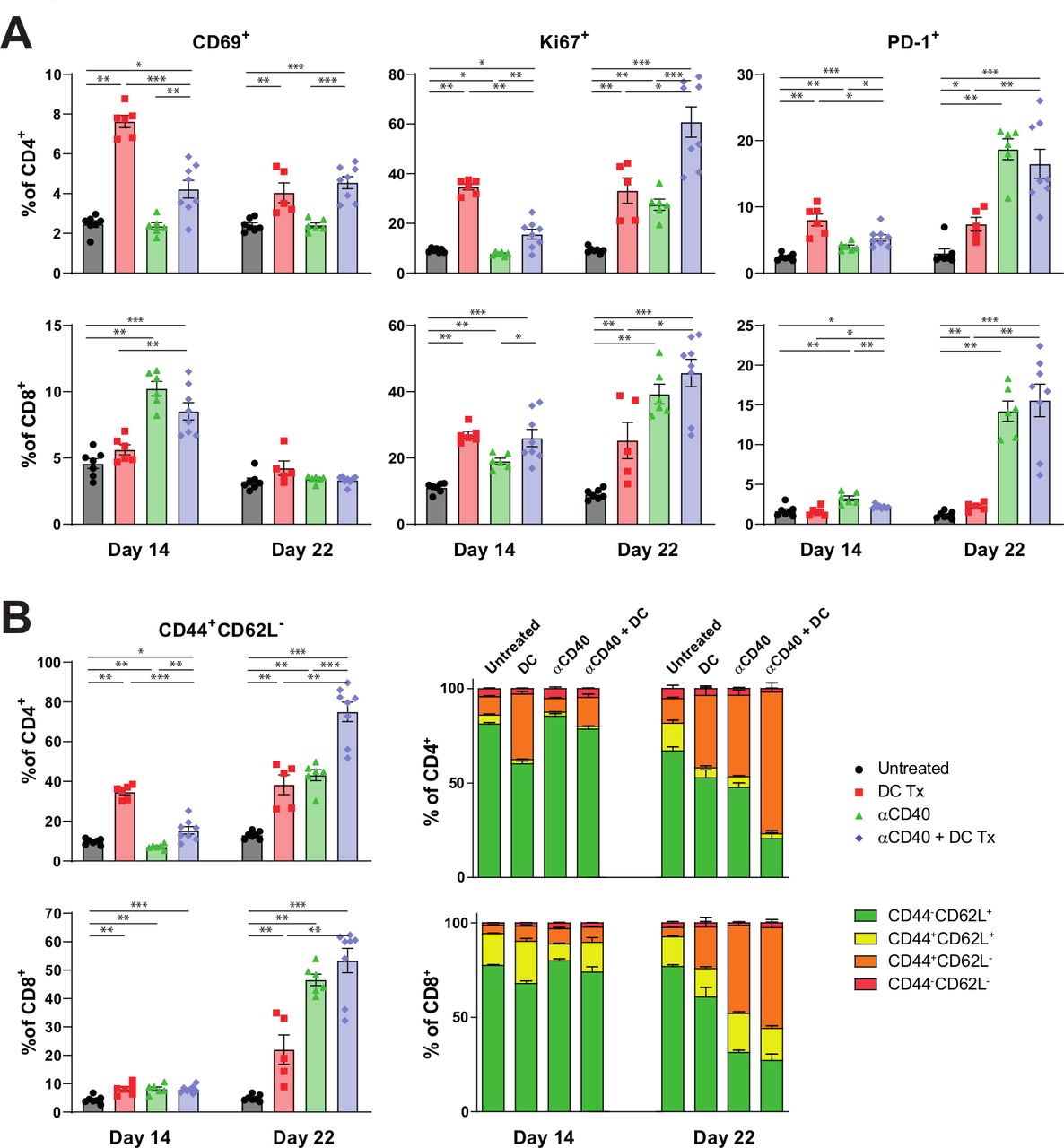
To further assess the mechanistic underpinnings of combination immunotherapy, we performed extensive analysis on the tumor and intratumoral immune cells, both numerically and phenotypically, using gene expression analysis and multicolor flow cytometry. Intratumoral analysis revealed increased T-cell numbers in treated mice (figure 5A). No distinct changes in myeloid subsets could be found (online supplementary figure S11). However, DC therapy did induced a PD-L1 rich TME (figure 5B). To get a more profound insight in the intratumoral immune changes induced by combination therapy, we applied NanoString gene-expression technology on tumors of treated and untreated mice. Unsupervised clustering of significantly different immune-related genes revealed that tumors of untreated or monotherapy-treated mice displayed a distinct gene expression profile as compared with mice treated with the combination therapy (figure 5C,D and online supplementary figure S12), indicating a unique remodeling of the TME.
Supplemental material
Supplemental material

Tumors of combination therapy-treated mice displayed a distinct gene expression profile compared with tumors of untreated or monotherapy-treated mice (A) number of CD3+, CD4+, CD8+, CD19+ and CD335+TILs per Mg tumor at end-stage disease. (B) PD-L1 MFI of non-myeloid (CD45-), monocyte (CD45 +F4/80-CD11b+Ly6C+Ly6G-), granulocyte (CD45 +F4/80-CD11b+Ly6C-Ly6G+), cdc1 (CD45 +F4/80-CD11b+CD11b-CD11c+MHCII+ CD103+), cdc2 (CD45 +F4/80-CD11b+CD11c+MHCII+), MDSC (CD45 +F4/80-CD11b+Ly6CintLy6Gint) and TAM (CD45 +F4/80+CD11b+) population, and percentage of PD-L1+ subset of cDC2s, MDSCs and TAMs. (C) Unsupervised clustering of genes significantly different between groups. Downregulated genes are marked blue and upregulated genes are marked yellow. (D) t-SNE clustering of individual tumor samples based on genes significant different between groups. n=5–10 per group. Significance was determined using the non-parametric Mann-Whitney U test. Data presented as the mean±SEM. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. DC, dendritic cell; MDSCs, myeloid derived suppressor cells; TAMs, tumor-associated macrophages.
Mice treated with combination therapy had consistently lower transcript amounts of a wide range of inhibitory receptors, including Pdcd1 (PD-1), Ctla4, Entpd1 (CD39), Vsir (VISTA), Cd244 (2B4), Havcr2 (Tim-3) and Tigit, compared with monotherapy-treated mice (figure 6A). Remarkably, both monotherapies induced higher expression of various effector molecules like Prf1 (perforin), Gzma and Gzmb (Granzymes) and Ifng. Differential gene expression analysis between monotherapy groups and the combination therapy confirmed significantly higher transcript levels of both inhibitory receptors and effector molecules in tumors of monotherapy-treated mice (online supplementary figure S13).
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
DC vaccination is able to reduce hallmarks of T-cell exhaustion. (A) Heatmap illustrating the average transcript expression of the indicated genes, grouped by function. Rows represent averaged z-scores. (B) Number and percentage of PD-1+, Tim-3+, VISTA+, CD39+ and NKG2A+ subsets of CD8 +TILs. (C) Number and percentage of IFNγ+, granzyme B+, IL-10+ and Ki67+ subsets of CD8 +TILs. n=7–8 per group. Significance was determined using the non-parametric Mann-Whitney U test. Data presented as the mean±SEM. *P<0.05, **P<0.01 DC, dendritic cell; IFNγ, interferon-γ; TILs, tumor infiltrating lymphocytes.
Although the findings of increased effector molecules in the absence of clinical benefit may seem counterintuitive, similar cells displaying high levels of coinhibitory and effector molecules were recently found to be consistent with a terminally exhausted T-cell phenotype.24–28 In line with these findings, gene-set enrichment analysis (GSEA) revealed that gene transcripts associated with T-cell exhaustion were significantly enriched in tumors of αCD40 therapy treated mice compared with combination therapy treated animals (online supplementary figure S14a). This was not observed for DC therapy-treated mice. As increased levels of coinhibitory receptors and effector molecules may be linked to a more exhausted T-cell phenotype, we interrogated markers associated with this state. High expression of Tbx21 (T-Bet) and Klrg1 was found in both monotherapies while high expression of the transcription factor Eomes was only found in aCD40 treated mice (figure 6A). Interestingly, combination therapy induced higher expression of Sell (L-selectin) and the chemokine receptor Cxcr5 in the tumor compared with other groups. Furthermore, we also found lower expression of genes related to various collagen markers and ‘M2’ phenotype macrophages after αCD40 therapy indicating TME remodeling. In order to confirm αCD40-induced stromalysis, histochemical staining were performed. Tumors of both αCD40 monotherapy as combination therapy-treated mice showed decreased collagen content (online supplementary figure S15). Strikingly, high mRNA expression of genes related to glycolysis were detected in tumors after combination therapy as compared with αCD40 monotherapy (figure 6A). A glycolysis GSEA indeed revealed higher activity in the combination therapy treated mice compared with αCD40 treated mice (online supplementary figure S14b). Combination therapy was also able to significantly upregulate expression of Vegfa, adm and Flt1 compared with αCD40 treated mice (online supplementary figure S13). This is indicative for angiogenesis and vascular formation and may promote immune cell infiltration into the tumor. When immunohistochemically stained for the endothelial marker CD31, tumors of combination therapy-treated mice did express more CD31 compared with untreated or monotherapy-treated mice (online supplementary figure S16).
Supplemental material
Supplemental material
Supplemental material
As gene expression analysis was performed on whole tumor material, inhibitory markers and effector molecules were further validated and quantified at the protein level on both CD4+ and CD8+TILs (figure 6B,C and online supplementary figure S17). Untreated and αCD40 treated mice had the highest frequencies of CD8 +TILs expressing various inhibitory receptors (ie, PD-1, Tim-3, VISTA, CD39, NKG2A) (figure 6B). However, only αCD40 treated mice had the highest number of CD8 +TILs expressing coinhibitory receptors. DC therapy was able to reduce the frequencies of PD-1+, Tim-3+, VISTA+, CD39 +TILs. A similar trend was also observed when coexpression of multiple inhibitory receptors was assessed (online supplementary figure S17c‒d). In addition, DC vaccinated and combination therapy treated mice had the highest frequencies of PD-1/Tim-3 double negative TIL, which have been described to exhibit the highest effector potential, whereas PD-1/TIM-3 double positive T cells are known to be severely dysfunctional.26 αCD40 mediated induction of IFNγ+ and granzyme B+TILs came at the expense of increased numbers of cells producing interleukin-10 (IL-10) (figure 6C). Both the mRNA and protein-expression data point to a preferential induction of effector T-cells expressing less multiple coinhibitory receptors in the combination immunotherapy treated, as compared with CD40-agonistic monotherapy-treated mice.
Supplemental material
Recently, targeting NKG2A on T cells has been described as a novel approach to promote antitumor immunity and has been linked to T-cell dysfunction.29 30 Interestingly, αCD40 induced the highest numbers of NKG2A+CD8+TILs compared with combination therapy arm (figure 6B). Moreover, although αCD40 therapy increased TIL numbers, the frequencies of proliferating TILs were lower compared with untreated mice suggesting that this is not explained by local expansion, but enhanced infiltration (figure 6C). Altogether, these findings offer an explanation for the observed efficacy of DC-CD40-agonist combination therapy where an influx of T cells exhibiting low levels of co-inhibitory checkpoints is associated with restricted tumor growth.
Discussion
Our data highlight that rationally combining immunotherapies in pancreatic cancer can lead to synergistic improvements in antitumor T-cell immunity and clinical responses. For these studies, we used immune-competent mice-bearing PDAC tumors obtained from KrasG12D/+;Trp53R172H/+;Pdx-1-Cre (KPC) mice. This model mimics (immune) phenotypic features and the aggressiveness of human pancreatic adenocarcinoma.16 31 We mainly focused on DC-based therapy to strengthen the tumor-specific effector T-cell response. Previous trials in pancreatic cancer patients used single peptide or autologous tumor as a lysate source for DC therapy. We assessed the possibility of loading DCs with mesothelioma lysate based on the rationale that cross-reactive T cells would be generated due to expression of a number of shared TAAs by both mesothelioma and pancreatic cancer. The use of an allogeneic-tumor lysate offers an off-the-shelf approach which is not dependent on the identification of immunodominant epitopes and can be used irrespective of the patient’s HLA type and exploits a broad spectrum of TAAs.32 We found that mesothelioma-lysate DC therapy was able to delay pancreatic tumor growth, generate KPC-reactive T cells and induce TIL influx, confirming cross-reactivity. Although some efficacy was observed with non-loaded DCs, possibly by an unspecific inflammatory response that activates bystander T cells or during culture phagocytosed bovine serum proteins, the use of tumor lysate-loaded DCs had significant higher efficacy against the tumor in vivo and in vitro (figure 2). Interestingly, cross-reactivity could also be found in vitro when B16F10 lysate was used to load DCs but not to non-loaded DCs (online supplementary figure S4), indicating the involvement of shared antigens (online supplementary table S1).
We also investigated if targeting CD40 is able to control tumor growth in established disease. CD40 can be found on B cells, DCs and macrophages and ligation leads to activation.33 αCD40 therapy may, therefore, also activate endogenous DCs that present tumor antigens and contribute to a monotherapy effect. Also, Schoenberger and Bennett et al demonstrated that CD40-activated APCs might replace the requirement for CD4 +T helper-mediated licensing, thereby lowering the threshold for CD8 +effector T-cell priming. This could explain why CD4 +T cell depletion prior combination immunotherapy did not affect efficacy.34 35 Alternatively, CD40 ligation may also license delivered DCs, thereby enhancing their capacity to prime CD8 +T cells.36 37 Indeed, when bone marrow-derived DCs were treated with αCD40, increased IL-12 production could be detected (online supplementary figure S18). Furthermore, αCD40 therapy can also directly modulate the TME: targeting CD40 on macrophages can lead to phenotypic polarization from immunosuppressive ‘M2’ into inflammatory ‘M1’ macrophages, the latter being tumoricidal and capable of ablating tumor stroma.16 38 In line with this thought, our mRNA expression data and histochemical staining on tumors confirmed decreased collagen content after αCD40 mono and combination therapy (figure 6A). Also, lower expression of mRNA levels related to M2 macrophages was found in tumors of mice treated with αCD40 (figure 6A). Furthermore, it was shown that M2 macrophage-derived granulin contributes to CD8 +T cell exclusion and that this process is driven by colony-stimulating factor-1 (CSF-1). It has been found that CSF-1 inhibition leads to desmoplasia depletion and sensitizes pancreatic cancer to immune checkpoint blockade therapy.39 We were able to show lower Csf1 mRNA levels in tumors after αCD40 therapy and combination therapy (figure 6A). A recently reported combination therapy involving αCD40 and αPD-1 therapy showed promising results in preclinical PDAC models, and demonstrated that therapy reprograms the TME resulting in the increase of DCs and decrease of granulocytic-MDSCs.40 As we now focused on the T-cell phenotype responsible for slowing tumor progression following combination treatment, further in-depth studies immediately following αCD40 therapy are likely required to formally dissect its spatiotemporal roles on macrophages and DCs in promoting antitumor immune responses.
Supplemental material
Interestingly, despite the absence of clinical responses in monotherapy-treated animals in the established tumor model, both monotherapy and combination therapy-treated mice were able to increase total CD3 +TIL numbers. The effect of DC therapy was most pronounced on CD4 +T cells and less on CD8 +T cells whereas αCD40 treatment displayed an inverse pattern. DC and αCD40 treated mice showed improved survival and increased both CD4+ and CD8+ intratumoral T-cell numbers. However, we demonstrated that the clinical response was primarily driven by CD8 +T cells (figure 3E–G). The sensitizing role of DC vaccination may be mainly priming of MHC class I-restricted cytotoxic T lymphocytes. Even though CD40 agonistic antibodies significantly increased T-cell infiltration in established pancreatic tumors, clinical efficacy was lacking, prompting further phenotypic analysis of these cells. We observed high expression of various inhibitory receptors and effector molecules on TILs of αCD40 monotherapy-treated mice when compared with the other treatment groups. Studies only recently published have associated this phenotype with that of terminally exhausted T cells in both solid cancer and chronic viral infection settings.25 41 Although our mRNA expression data also demonstrated the expression of various inhibitory receptors in DC monotherapy-treated mice, mRNA analysis was performed on whole tumor tissue, challenging the interpretation of our data as we were not able to assign specific markers to individual immune cell subsets. However, lower amounts of mRNA of various stimulatory receptors (ie, CD28, ICOS, GITR, CD137, OX-40) and high expression of Tbx21 (T-Bet) and Eomes found in αCD40 monotherapy-treated mice suggest that this phenotype is primarily restricted to these tumors. In addition, KLRG1hiIL7rlo CD8 +T cells have previously been described as dysfunctional.42 We found that monotherapy with αCD40 induced higher expression of Klrg1 but not Il7r, whereas DC vaccination increased the levels of both Klrg1 and Il7r. In accordance with the aforementioned phenotype, Sell (L-selectin), a marker associated with naïve-like memory T-cells and T-cell homing, was particularly induced in mice treated with combination therapy.24 25 28 Finally, the chemokine receptor CXCR5 has been recently found to mark a specific T-cell population capable of responding to PD-1 checkpoint blocking antibodies, which expresses lower levels of coinhibitory receptors and effector molecules as compared with their CXCR5-negative counterparts.25 26 41 We found that combination DC and αCD40 therapy indeed induced higher Cxcr5 expression in the tumor compared with other groups.
Flow cytometry analysis confirmed the reduced expression of various inhibitory markers on CD8 +TILs derived from combination therapy-treated mice compared with αCD40-treated mice. In addition, the lower proliferation rate as evidenced by ex vivo measurements of Ki-67 in combination therapy treated animals also matches with an improved T-cell phenotype, as others have previously found these cells to persist in culture longer compared with their Ki-67-high counterparts.25 As human cancers grow at a considerably slower pace than most murine tumor models, it is conceivable that longer T-cell persistence is crucial for durable tumor control.
The presence of low amounts of glycolysis-related gene transcripts following αCD40 monotherapy fits with a more exhausted, terminally differentiated memory T-cell state, as has been proposed by others.43 44 Glut-1 (Slc2a1) was found to be essential for T-cell activation and Slc2a1 was highly expressed in combination therapy treated mice.45 However, as gene expression was performed on whole tumor material, it is unclear whether glycolysis-related transcripts originated from tumor cells or immune infiltrates. Further functional studies on our combination treated T-cell phenotype are needed to truly assess which factors determine their superior antitumor efficacy.
DCs loaded with allogeneic mesothelioma-tumor cell lysate have already proven to be feasible, with clinical efficacy in the absence of toxicity in patients with mesothelioma.22 Following this, a phase II clinical trial examining whether this holds true for macroscopically disease-free, postresection PDAC patients is currently being conducted (REACtiVe Trial; Netherlands Trial Register NL7432). However, as the majority of pancreatic patients presents with irresectable or metastatic disease, rational and safe treatment combinations are needed to offer perspective for this group of patients too. Currently, several studies with combination strategies incorporating CD40 agonists in PDAC patients are ongoing and recruiting (NCT03214250; NCT02588443; NCT03329950). We have shown that DC-therapy pretreatment allows for proper CD40-agonist efficacy by precluding the formation of T-cells associated with an exhaustion phenotype when administered alone. The lack of DC-therapy toxicity in patients is of particular importance since CD40-agonistic antibodies are associated with serious adverse events leading to premature termination of treatment in some patients.16 46 To assess the feasibility and safety of our combinatory approach we are currently in the process of initiating a trial involving DC-CD40-agonist combination therapy in metastatic disease. Since DC vaccination also induced a PD-L1 rich TME, future combination strategies with immune checkpoint blockers are warranted.
In conclusion, we have found pancreatic cancer and mesothelioma lysate-loaded DCs to be effective in restraining immunologically cold pancreatic tumors when administered prophylactically. In established tumors, effective intratumoral immunity was achieved when DC vaccination was combined with CD40-agonistic antibodies, generating non-redundant immunological effects capable of restraining tumor progression.
Methods
Mice
C57BL/6 and CBA/J mice were purchased from Charles River Laboratories and Janvier, respectively. All mice were housed in individually ventilated cages, maintained under specific pathogen-free conditions and used at 8–10 weeks of age. All mouse experiments were controlled by the animal welfare committee (IvD) of the Leiden University Medical Center (Leiden) or Erasmus University Medical Center (Rotterdam) and approved by the national central committee of animal experiments (CCD) under the permit numbers AVD116002015271 and AVD101002017867, in accordance with the Dutch Act on Animal Experimentation and European Union (EU) Directive 2010/63/EU.
Mouse tumor cell lines
The pancreatic cancer KPC3 cell line is derived from a primary tumor of a female KPC mouse.31 AE17 and AC29 cell lines are derived from mesothelioma tumors in C57BL/6 and CBA/J mice and kindly provided by Bruce W.S. Robinson (Queen Elizabeth II Medical Center, Nedlands, Australia) and Peter D. Katsikis (Erasmus Medical Center, Rotterdam, The Netherlands), respectively. KPC3, AE17 and AC29 tumor cell lines were cultured in RPMI 1640 containing glutamax-I (Gibco), 50 µg/mL gentamicin (Invitrogen), and 8% fetal bovine serum (FBS) (Gibco) at 37°C in a humidified atmosphere containing 5% CO2. Cell lines were assured to be free of rodent viruses and Mycoplasma by regular PCR analysis. Low passage number cultures from stock vials were used for all experiments. Transcriptomes of KPC3, AE17 and B16F10 cells from stock vials were analyzed by Macrogen NGS Services (Macrogen, Seoul, Republic of Korea). Illumina platform was used with TruSeq Stranded Total RNA LT Sample Prep Kit (Human Mouse Rat) Library. MC38 transcriptome data were previously published47 and downloaded from Sequence Read Archive SRX6812144.
Generation of DC vaccination
Bone marrow-derived cells seeded in 100 mm Petri dishes (day 0) and cultured in 10 mL DC culture medium: RPMI 1640 containing glutamax-I (Gibco), 50 µg/mL gentamicin (Invitrogen), 5% FBS (Gibco), 50 mol/L mercaptoethanol (Sigma-Aldrich) and 20 ng/mL recombinant murine granulocyte macrophage-CSF (kindly provided by B. Lambrecht, VIB Ghent, Belgium). Cells were cultured at 37°C in a humidified atmosphere containing 5% CO2. At day 3 and 6, fresh DC culture medium was added. Tumor cell lysate was prepared by freeze-thawing and subsequent sonication for 3×10 s with an amplitude of 10 mm, using a Soniprep 150 ultrasonic disintegrator equipped with a microtip (Sanyo Gallenkamp). After 9 days of culture, tumor cell lysate was added to the DC cultures, to the equivalent of three tumor cells per DC. After 8 hours, 10 g/mL CpG (ISS-ODN 1668, Invitrogen) was added to the culture to allow complete maturation while incubated overnight. The next day, DCs were harvested and washed three times in PBS. The quality of the DC preparation was determined by cell counting, morphology and cell surface marker expression by flow cytometry, as previously described.48
In vivo experiments
Cultured tumor cells were harvested at 70% confluency. The pancreatic cancer model was generated by injecting 100 000 KPC3 cells in 100 ul PBS/0.1% BSA subcutaneously in the flank of the mice or by injecting 10 000 KPC3 cells in 20 ul PBS/0.1% BSA orthotopically in the pancreas. The mesothelioma model was generated by injecting 20×106 AC29 cells in 200 µL PBS intraperitoneally. Subcutaneous tumors were measured 3–7 times a week in three dimensions using a caliper. Mice were treated with DC immunotherapy at day −7 (7 days before tumor injection) or day 5. Repeated DC vaccination occurred at day 10, 14 and 18 in mice with subcutaneous pancreatic tumors and at day 3, 7 and 11 in mice with orthotopic pancreatic tumors. One day after DC vaccination, FGK45 or isotype IgG2a was administered intraperitoneally (BioXCell, 70 µg/dose). Mice with AC29 tumors were DC vaccinated on day 10 followed by FGK45 on day 10, 12 and 14. For CD4+ and CD8+T cell depletion, mice were injected i.p. 2 days before treatment and every 6 days onward with GK1.5 and/or 2.42 or isotype IgG2b (BioXCell, 100 µg/dose). Peripheral blood samples for interim analysis were collected 4 days after DC vaccination and were immediately stained (see the flow cytometry section). Mice were sacrificed at the predefined experimental endpoint (figures 1–3H–J) or when tumors reached a volume of 1000 or 1500 mm3.
Cell preparation and flow cytometry
Whole blood or single-cell suspensions of spleen and tumor were prepared for flow cytometry. Spleens were passed through a 100 µm mesh with RPMI 1640 containing glutamax-I (Gibco) and collected through centrifugation. Lymph nodes were excluded during tumor collection and tumors were dissociated using a validated tumor dissociation system (Miltenyi Biotec). To assess cytokine production, lymphoid cells were stimulated for 4 hours at 37°C using PMA and ionomycin supplemented with GolgiStop (BD Biosciences). Intracellular cytokine and transcription factor staining was performed using PFA/Saponin protocol and Foxp3 Transcription Factor Staining Buffer Kit (eBioscience), respectively. Cell surface staining was performed after blocking Fc II/III receptor using antimouse 2.4G2 antibody (kindly provided by L. Boon, Bioceros, Utrecht, the Netherlands) by incubating cells with fluorescently conjugated mAbs directed against murine CD3e (145–2 C11), CD4 (GK1.5), CD8a (53-67), CD11b (M1/70), CD11c (N418), CD19 (1D3), CD25 (PC61), CD40 (1C10), CD44 (IM7), CD45 (30-F11), CD62L (MEL-14), CD69 (H1.2F3), CD80 (16-20A2), CD86 (GL1), CD103 (2E7), CD107a (1D4B), CD335 (29A1.4), F4/80 (BM8), FoxP3 (FJK-16s), Granzyme B (NGZB), IFNγ (XMG1.2), IL-2 (JES6-5H4), IL-10 (JES5-16E3), Ki-67 (SolA15), LAG-3 (eBioC9B7W), Ly6C (AL-21), Ly6G (RB6.8C5), MHCII (M5/114.15.2), NKG2A (16a11), PD1 (J43) PDL1 (MIH5), TIM3 (8B.2C12), TNFα (MP6-XT22), VISTA (MH5A). Cells were in addition stained for viability using fixable LIVE/DEAD aqua cell stain (Thermo Fisher Scientific). Data were acquired using an LSR-II flow cytometer (BD Biosciences) and analyzed by FlowJo v10.0.7 (Treestar).
In vitro experiments
Tumor antigen-specific T-cell detection assay: Dissected subcutaneous tumors from treated mice and lungs from wild-type C57BL/6 mice were beads homogenized in 150 µL Milli-Q for four cycles of 1 min. A Bradford assay was performed in order to assess the protein concentration. Bone marrow-derived DCs were generated as described above, and loaded with 70 µg tumor lysate or 200 µg lung lysate/mL DC suspension. Tumor cell line lysate loaded DCs were prepared as described above. Tumor loaded DCs were in in vitro cocultured with paired splenocytes at a ratio of 1:10 for 4 hours at 37°C supplemented with GolgiStop (BD Biosciences). After 4 hours, intracellular cytokine expression was assessed by flow cytometry as described above.
IL-12p40 detection: Bone marrow-derived DCs were cultured as described above. At day 9, FGK45 (BioXCell, 30 µg/mL) or isotype IgG2b (BioXCell, 30 µg/mL) was added to the DC culture. After 24 hours, supernatant was collected and a sandwich ELISA assay was performed as previously described.37
(Immuno)histochemistry
Immunohistochemistry was performed with an automated, validated and accredited staining system (Ventana Benchmark Discovery ULTRA, Ventana Medical Systems, USA) using Omnimap antirabbit or mouse and the universal DAB detection Kit. In brief, following deparaffinization and heat-induced antigen retrieval the tissue samples were incubated according to their optimized time with CD31 (Abcam; polyclonal). Incubation was followed by hematoxylin II counter stain for 8 min and then a blue coloring reagent for 8 min according to the manufactures instructions. Tonsil tissue was used as positive control. Thrichome blue was stained using optimized protocol provided in the fully automated Ventana Benchmark Special staines system. Sirius Red was stained by hand, in brief, following deparaffinization slides were rehydrated by passage through decreasing ethanol series, 5 min predifferentation step using 0.2% fosformolybdeen-acid followed by 45 min incubation with 0.1% Sirius Red solution. Slides were analyzed using polarization method.
mRNA expression analysis
NanoString nCounter Technologies was applied on 120 µm of Tissue-Tek(Sakura)-embedded fresh frozen tumor samples using the PanCancer IO 360 Panel. To identify the differentially expressed genes, raw data was normalized using the values of the most stable 15 housekeeping genes selected by applying the geNorm algorithm. Unsupervised clustering of normalized gene expression values (row Z-scores) was performed using the complete linkage method with Euclidean distance measure or standard Principal Component Analysis (PCA)/T- distributed Stochastic Neighbor Embedding (t-SNE) functions in R (through RStudio V.1.1.463). For the volcano plots, Mann-Whitney U test was conducted to compare the normalized count values in two groups (ie, monotherapy vs combination therapy) for each of the 750 markers. The original p values were adjusted for multiple testing using Benjamini-Hochberg procedure. All calculation and the volcano plots were done in program R.
GSEA was performed by ranking all genes based on difference of means scaled by the standard deviation (signal-to-noise).49 Previously reported gene sets M9480 and M5937 were used for exhausted phenotype and glycolysis enrichment analysis,50 respectively. The false-discovery rate adjusted p values (q-value) was considered significant when <0.05.
Statistical analysis
Difference between groups of interest were statistically analyzed with the non-parametric Mann-Whitney U test. Data are displayed as means with the SE of the mean and analyzed using GraphPad Prism software (Graphpad, V.7.0a). Survival data were plotted as Kaplan-Meier survival curves. The non-parametric log-rank test (Mantel-Cox test) was used to compare the survival distribution of groups of mice. In all cases a p value of 0.05 and below was considered significant (*), p<0.01(**), p<0.001 (***) and p<0.0001 (****) as highly significant.
Acknowledgments
We thank R. van der Breggen, D. Ruano and N.F. de Miranda of the Immunogenenomics group of the department of Pathology, LUMC for their help with NGS analysis of the KPC3, B16 and AE17 tumor cell lines. We also want to thank A. Nigg, T.P.P. van den Bosch and the PARTS team for their help with the (immune)histochemistry staining.
References
Footnotes
Contributors SPL designed all laboratory studies, performed experiments, analyzed the data and wrote the manuscript. PK, ML, LK, MvN, MvG and CG performed murine experiments. JD performed the NanoString experiments and DAMM provided help in the analysis. YL, AS, KAM and RS contributed to bioinformatic and statistical analyses. HV and NvM participated and oversaw design of all laboratory studies, and data analysis and interpretation. NvM, SHvdB, JA, TvH, FD and CHJvE oversaw design of all laboratory studies, data analysis and interpretation, and wrote the manuscript. All authors contributed to the editing of the final report. All authors agreed to all of the content of the submitted manuscript.
Funding This work has been supported by the Survival with Pancreatic Cancer Foundation under project numbers OVIT-17-06 and 17-04.
Competing interests JA: Stock or other Ownership: Amphera. Consulting or Advisory Role: Eli-Lilly, MSD Oncology, Bristol-Myers Squibb, Roche Speakers Bureau: AstraZeneca, Takeda, Boehringer Ingelheim.
Patient consent for publication Not required.
Ethics approval All mouse experiments were controlled by the animal welfare committee (IvD) of the Leiden University Medical Center (Leiden) or Erasmus University Medical Center (Rotterdam) and approved by the national central committee of animal experiments (CCD) under the permit numbers AVD116002015271 and AVD101002017867, in accordance with the Dutch Act on Animal Experimentation and EU Directive 2010/63/EU.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as online supplementary information. All data are available on reasonable request. Please contact CHJvE.