Article Text

Abstract
Several human herpes viruses (HHVs) exert oncogenic potential leading to malignant transformation of infected cells and/or tissues. The molecular processes induced by viral-encoded molecules including microRNAs, peptides, and proteins contributing to immune evasion of the infected host cells are equal to the molecular processes of immune evasion mediated by tumor cells independently of viral infections. Such major immune evasion strategies include (1) the downregulation of proinflammatory cytokines/chemokines as well as the induction of anti-inflammatory cytokines/chemokines, (2) the downregulation of major histocompatibility complex (MHC) class Ia directly as well as indirectly by downregulation of the components involved in the antigen processing, and (3) the downregulation of stress-induced ligands for activating receptors on immune effector cells with NKG2D leading the way. Furthermore, (4) immune modulatory molecules like MHC class Ib molecules and programmed cell death1 ligand 1 can be upregulated on infections with certain herpes viruses. This review article focuses on the known molecular mechanisms of HHVs modulating the above-mentioned possibilities for immune surveillance and even postulates a temporal order linking regular tumor immunology with basic virology and offering putatively novel insights for targeting HHVs.
- antigen presentation
- immunity
- immune tolerance
- immunomodulation
- immunotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
The molecular mechanisms of virus-related malignant transformation of non-tumorous tissues are diverse and involve molecules encoded by viruses or induced on viral infection. Viruses are able to infect all types of life forms such as animals, plants, fungi, protists, and even microorganisms, like yeast, bacteria, and archaea.1–5 Viral infections can predispose a patient to various malignancies which might be mediated by oncogenic viruses like Epstein-Barr virus (HHV4, EBV), human herpes virus 8 (HHV8, Kaposi-Sarkom herpes virus (KSHV)), hepatitis B virus, and certain human papilloma viruses (HPV).
Viruses completely rely on the host cell machinery to propagate and lack an own metabolism and reproduction. The viral particles, also called virions, contain the genetic material (single-stranded/double-stranded (ss/ds) RNA or ss/dsDNA), a protein coat (capsid) and optional a lipid envelope. The space between the lipid envelope derived from the host cell membranes and the capsid is the tegument, which contains molecules from the infected host cell including, for example, proteins and non-coding RNA species. These molecules may originate from the host cell or by the viruses of the infected host cell and promote the generation of macromolecules required for the next infection cycle.6 Furthermore, the tegument contains fluids of the cytoplasm, the endoplasmic reticulum (ER) and/or of the Golgi apparatus.7 8
In ER, the assembly of the classical major histocompatibility complex (MHC) class Ia and non-classical MHC class Ib molecules consisting of the MHC class Ia/b heavy chain (HC), β2-microglobulin (β2-m), and a peptide derived from cellular proteins, which could theoretically also represent peptides derived from viral proteins or in the case of malignant transformed cells also from tumor antigens. The peptides are generated and processed by different components of the antigen processing and presentation machinery (APM) for their presentation to CD8+ cytotoxic T lymphocytes (CTLs) on the cell surface.9 10
The MHC class Ia molecules are physiologically expressed in nucleated cells with the exception of immune privileged tissues like cornea, brain, testis and chorion. In contrast, the MHC class Ib molecules, predominantly HLA-G and HLA-E, exert a physiologically restricted and tightly regulated expression exclusively in immune privileged tissues.11 12 Since MHC class Ib molecules represent potent ligands for inhibitory receptors of immune effector cells, the modulation of these molecules has functional relevance for immune responses.13 14
Independently of viral infections in solid and hematopoietic tumor diseases, classical MHC class Ia molecules are downregulated with high frequencies by equal strategies, while non-classical MHC class Ib molecules are induced, and even secreted or shedded into the local tumor microenvironment (TME) strongly contributing to the tumor immune escape.11
Next to antigen presentation toward CTLs, also other molecular mechanisms contribute to successful immune surveillance against viral infections. Despite primary virus infections could elicit host antiviral immune responses, these responses are often insufficient to eliminate the virus. Thus, the ability of viruses to persist suggests that the viruses could subvert antiviral immune responses. For example, the virus mediated reduced expression of NKG2D ligands (NKG2D-Ls) upon viral infection avoids antiviral immune responses of the host immune system.15–19
In addition, the secretion of certain cytokines and/or chemokines on herpes virus infection is altered, which can modulate a strong and effective antiviral immune response against infected cells and/or tissues. It is noteworthy that a number of inhibitory mechanisms mediated by certain herpes viruses have been identified, which prevent the secretion of proinflammatory cytokines or enhance the secretion of anti-inflammatory cytokines.20 Furthermore, the expression and the assembly of MHC class Ia/b and/or APM molecules and the NKG2D-Ls can be induced or reduced by different cytokines.
In this review, the known immune escape mechanisms of human herpes viruses (HHVs) are summarized. The family of dsDNA herpes viruses consists of the α-subfamily (herpes simplex virus, HSV-1, HSV-2, varicella-zoster virus (VZV)), the β-subfamily (cytomegalovirus (CMV), HHV6A, HHV6B, HHV7), and the γ-subfamily (EBV, KSHV), which exerts an oncogenic potential. This separation is based on their host range, genetic organization and replication strategies.21 These family members share a common viral morphology and approximately 40 conserved genes important for viral replication.22 Nevertheless, these pathogens differ in their pathogenicity. While the members of the α-subfamily have been identified to act as cofactors for some tumor malignancies resulting in elevated tumor incidences, the members of the γ-subfamily are causative inductors of solid and hematopoietic tumor diseases. Furthermore, CMV as a member of the β-subfamily may infect critical organs including the nervous system, hematological and vascular system, gastrointestinal system and therefore may be accompanied by severe disease outcome in apparently healthy individuals.23
Following a primary infection, it is characteristically for herpes viruses to persist in the host for an extended duration, considering the herpes viruses as highly successful pathogens.24 A contributing factor of the herpes viruses is their ability to adopt two different modes of life cycle: the latency and the lytic cycles. After such a primary productive infection, the herpes viruses switch to latency, a transcriptional and translational suppressed state. This latent state can be frequently interrupted by lytic episodes. During such latency, the latency-associated transcripts including coding transcripts resulting in viral peptides and proteins as well as non-coding transcripts like microRNAs (miRs)have been identified to contribute to immune evasion.24–27
Focusing on the MHC class I a/b molecules, APM components, NKG2D-Ls, cytokine and chemokine signaling on viral infection, this review article will highlight the known interfering molecular mechanisms of viral encoded or induced miRs, peptides and proteins. Exactly such processes are affected in tumor cells, also shaping the composition of the TME and therefore representing fundamental steps in immune surveillance or immune escape. Thus, the direct interactions between viral molecules of HHVs and host cell molecules next to indirect mechanisms leading to the induction/reduction of relevant host cell factors on viral infection will be addressed and discussed.
Viral proteins interfere with the peptide presentation of MHC class I molecules
Under physiologic conditions, the assembled trimeric MHC class I molecules consisting of the MHC class I HC, non-covalent bound β2-m, and processed peptides of 8–12 amino acids in length are transported via the trans-Golgi to the cell surface of nucleated cells and presented to CD8+ CTLs.11
After ubiquitination, cellular proteins are degraded into peptides by the multicatalytic proteasome with a correct C-terminus, but a relegated N-terminus. These peptides could be further trimmed by cytosolic or ER-resident aminopeptidases.28 The cytokine interferon (IFN)-γ induces the so-called immunoproteasome, which contains novel active subunits of the proteasome activator (PA)28 and the IFN-γ-inducible proteasomal β-subunits, the low molecular weight proteins (LMP)2, MECL1 and LMP7, replacing their constitutive homologs β1, β2, and β5 during proteasome assembly.29 During the initial viral infection, a rapid induction of the immunoproteasome is crucial, which leads to an altered peptide repertoire.30 Subsequently, the peptides are transported via the heterodimeric transporter associated with antigen processing (TAP)1/TAP2 into the ER, which forms a peptide loading complex with the chaperones calreticulin (CALR), tapasin (TPN) and the protein ERp57, thereby facilitating peptide loading onto MHC class I molecules non-cavalently bound to β2-m.
Reduced or impaired expression of any of these molecules can act as a bottleneck for proper MHC class I surface expression. For example, mutations or deletions of B2M or TAP subunits result in a complete absence of MHC class I molecules on the cell surface.31–34 Furthermore, downregulation of MHC class Ia surface levels on reduced expression of one or more APM components represents a potent mechanism for immune escape in tumor cells, but also of viral-infected cells to escape immune surveillance. However, in contrast to tumors, many viral proteins have been identified to interfere with the expression of components of the APM. A summary of such ‘immune evasions’ is listed in table 1.
Viral peptides/proteins as well as viral miRs targeting MHC class I and APM components
The human herpes virus (HHV) 1 (herpes simplex virus 1, HSV-1) produces the protein ICP47 that blocks the peptide loading on the MHC class I HC by direct binding to the TAP 1/2 heterodimer.35 36 In HHV-3 (VZV) infected cells, MHC class I complexes were hindered to pass through trans-Golgi to the cell surface, which is mediated by the VZV protein ORF66,37 while HHV-4 (EBV) encodes for the BNLF2a protein blocking the TAP 1/2 heterodimer even more efficient than ICP47 or US6.38 Furthermore, EBV encodes for the protein vIL-10, which downregulates TAP1 and LMP2 expression. Both genes are controlled by a bidirectional promoter.39 The EBV protein BILF1 reduces MHC class Ia surface expression,40 and the EBV protein BGLF5 as well as EBNA-1 interfere with the complete peptide generation.41 42 Interestingly, the late lytic BDLF3 gene product downregulates both MHC class I and class II molecules.43 The HHV-5 (cytomegalovirus, CMV) encodes for the US6 peptide, which is also able to interfere the peptide transport by blocking the TAP 1/2 heterodimer.44 Additionally, CMV encodes for the US3 protein that inhibits the TPN-mediated peptide loading and therefore retains HLA class I molecules within the ER.45 While the US10 protein directly binds to MHC class I HC and retaining it in the ER,46 the US11 and US2 protein directs the MHC class I HC toward proteasomal degradation.47 48 Furthermore, it has been shown that the combination of cytosolic and ER-resident aminopeptidases shapes the pool of antigenic peptides as shown for the immunodominant CMV pp65495-503 CTL epitope.49 It is noteworthy that CMV virions can even bind β2-m on the cell surface and might use it as receptor for virus entry.50
An immune evasion of the HHV-7 viruses is the inhibition of MHC class I presentation by the viral U21 protein.51 Furthermore, HHV-8 (KSHV) viruses express the K3 and K5 proteins leading to the downregulation of MHC class I molecules.52 The similar effect was also reported for LANA1.53
Viral microRNAs prevent immune surveillance by MHC class I molecules
Not only viral-encoded proteins can counteract immune surveillance by interfering with peptide processing and presentation of MHC class I molecules, also viral-encoded microRNAs (miRs) are reported to hinder the MHC class I mediated immune surveillance. miRs are small single-stranded non-coding RNAs of approximately 19–25 nucleotide (nt) in length54 binding sequence specifically to the 3′-untranslated region (UTR), but less frequent to the 5′-UTR and the coding sequence (CDS) of target mRNAs.55 The miR binding to the target mRNA results in translational inhibition leading to mRNA storage56 or in most cases to mRNA decay.57 Only the seed regions of the miRs between the second to seventh nt exert perfect complementary sequence homology to the target mRNA sequences, but the impact of their length and the resulting target repression is currently controversially discussed.58 Furthermore, miRs redundantly regulate mRNAs and one single miR may control the fate of many different target mRNAs.57
Some of the miRs affect tumor biologic relevant cellular functions, like cell proliferation, cell migration, invasion, angiogenesis, apoptosis inducibility, immune cell recognition and others. Therefore, based on their target genes, some miRs can be grouped into oncogenic, tumor suppressive or immune modulatory miRs.59
Interestingly, some of these miRs are functionally incorporated into the RNA-induced silencing complex (RISC) complex.60 So far, more than 250 viral miRs have been identified, predominantly encoded by herpes viruses, but also by polyomaviruses, ascoviruses, and adenoviruses.61 Herpes viruses encode and express not only own miRs binding to certain host mRNAs, they further alter the whole host cell miR transcriptome on infection61 and/or during the process of viral-induced malignant transformation.62 In the case of EBV, the viral-mediated induction of the oncogenic miR-155 in host B cells is one major molecular mechanism for immortalization and malignant transformation.63 64 On the other hand, the EBV-encoded miR-BART-1 targets the tumor suppressor gene PTEN, which is associated with tumor metastasis.65 The viral miR BART16 targets the transcriptional coactivator CREBBP mRNA and thereby inhibits type I IFN signaling and other target genes of this important transcriptional coactivator,66 affecting the whole host cell transcriptome for a putative shift toward immortalization and malignant transformation. If secreted factors, like hormones, cytokines, and chemokines, are affected on viral infection, an impact even on non-infected host cells/tissues/organs including immune effector cells could be possible. Indeed, KSHV downregulates the tumor suppressive miRs miR-221, miR-222, and the let7 family members of the infected host cell.67
The herpes viruses express miRs during the lytic phase and even during the latency.68 69 Currently, the tumor biologic and tumor immunologic targets of these viral miRs are identified. So far, there is no proof that the viruses do encode for additional miR processing enzymes or for additional RISC components, since the viral-encoded miRs use the miR processing machinery of the infected host cells.70
Furthermore, EBV targets the mRNA of TAP2 by miR-BART17 and miR-BHRF1-3.71 The CMV virus encodes for miR-US4-1 negatively regulating the ER-resident aminopeptidase ERAP1 involved in the peptide shaping/trimming for later presentation on MHC class I molecules.72 It is postulated that more herpes virus-encoded miRs will be identified impairing the function of the MHC class I APM in the future. All so far reported viral-encoded proteins and regulatory miRs affecting the MHC class I-mediated antigen presentation are summarized in table 1.
Viral proteins restrain the activity of immune effector cells
The current research does not only proof the inhibition of the MHC class I-dependent antigen presentation by viral proteins, but rather the disruption of the interaction between (infected/tumor) target cell and immune effector cells. Such disruption could be arranged by reduction of certain host cell proteins acting as ligands for activating receptors on immune effector cells or by enhancing the expression of viral and/or host molecules leading to the inhibition of immune effector cells. This includes an enhanced expression of immune modulatory molecules, like non-classical MHC class Ib molecules, in the host cell. Actually, the expression of non-classical MHC class Ib molecules is strongly induced in solid and hematopoietic tumor malignancies offering a strong mechanism for immune evasion.13
Indeed, HHV-1 induces HLA-G expression on infection in human neuronal cells.73 While HLA-G is a ligand for the inhibitory receptors ILT-2, ILT-4, and KIR2DL4, which are present on NK cells, CTLs, B cells, macrophages and dendritic cells,74 HLA-E binds to the inhibitory receptors CD94/NKG2A, -B and -C on NK cells and CTLs.75 For both immune modulatory molecules, a strictly controlled gene expression has been reported, which includes also miRs. Those HLA-G negative regulatoring miRs were also classified as tumor suppressive miRs13 76 77 and might also be downregulated on viral infections. Indeed, CMV infection interferes the regulation between miR-376a and HLA-E.71 Furthermore, the HCMV genome encodes for different NK cell modulators, like UL135, UL141, UL142, and UL148 thereby inhibiting NK cell activation and recognition.78 UL141 inhibits the expression of the activating ligands CD155 and CD112, the activating receptor CD226 (DNAM-1), TRAIL-R1 and -R2, while UL142 and UL148A target specifically MICA and UL148 the expression of LFA-3.
Next to the viral-mediated enhancement of MHC class Ib molecules, the checkpoint molecule programmed cell death1 ligand 1 (PD-L1) is induced on several viral infections including HIV, HCV, and HHV1.79 80 There exist more immune inhibitory molecules, which are directly or indirectly induced on viral infections. For example, HHV1 induces transforming growth factor-β (TGF-β) secretion of infected host cells.81 TGF-β induces among others HLA-G, but inhibits MHC class Ia and APM components gene expression.82 Such mechanisms are strongly required for the further processes of viral infection and immune evasion.
A second strategy is the reduced expression of molecules that act as ligands for activating immune cell receptors including among others different NKG2D-Ls, namely the major histocompatibility complex class I-related molecules (MIC) A and B as well as the UL16-binding proteins (ULBP) 1–6, also known as retinoic acid early transcript 1 (RAET1) proteins.83 84 In analogy to the tightly controlled cell surface expression of the MHC class Ia/b molecules, the NKG2D-Ls exert also strongly regulated cell surface expression to avoid respective immune responses by NKG2D expressing NK cells and CTLs.85 86 Various pathophysiological situations, such as viral infection, oxidative stress, genotoxic drugs, tissue damage, heat shock, inflammatory cytokines, and malignant transformation are known inductors for NKG2D-L surface expression.59
HHV-1-infected cells show reduced surface levels of MICA and ULBP2,87 as well as ULBP1 and −3.88 VZV-infected cells reported to show equal effects by downregulating ULBP2 and ULBP3.88 EBV infection decreased MICA, MICB, and ULBP4 surface levels.89 The many possibilities of the herpes viruses to downregulate NKG2D-L surface expression include viral-encoded proteins, binding to the NKG2D-Ls and causing a functional inhibition as well as viral miRs or a combination of them.
The CMV encodes for the UL16 protein, which is able to bind many ULBPs with the exception of ULBP3 and ULBP4.90 Even MICB can be bound and retained by UL16.91 Next to UL16, the viral UL142 protein binds and retains MICA as well as ULBP3.92 93 MICA is further targeted by the CMV gene products US18 and US20.94 The immune evasion by U21 of HHV7 not only inhibits MHC class I antigens, but it also contributes to the downregulation of MICA, MICB, and ULBP1.95 Furthermore, the KSHV protein K5 causes a downregulation of MICA and MICB.96 The viral proteins/peptides targeting NKG2D-L mRNAs are summarized in table 2.
Viral peptides/proteins as well as viral miRs and other mechanisms leading to downregulation of the NKG2D ligands
Viral-encoded miRs enable immune evasion by targeting NKG2D-L transcription
In analogy to the prevention of the antigen presentation by MHC class I molecules, the herpes viruses can also block the NKG2D-Ls via the expression of miRs. Indeed, current studies identified a number of viral miRs directly targeting the NKG2D-Ls or indirectly leading to a reduction of their surface levels. In addition, various host cell-encoded oncogenic miRs have been described, which can be induced after malignant transformation or as an indirect result of viral infection, such as miR-17–5 p, miR-20a, miR-93 directed against MICA, miR-10b against MICB, and miR-650 against ULBP1.59 The literature even lists more human miRs targeting NKG2D-Ls, but these miRs have not yet been classified as oncogenic based on their functional activity.
In addition to these host cell miRs, the viral miR EBV-miR-BART-6 directly targets Dicer in the host cells,97 which has an impact on the whole miR transcriptome in the host cell and even on the processing of viral encoded miRs themselves. HHV-1 encodes for miR-H8, which reduces the surface levels of ULBP2 and ULBP3.98 The EBV-miR-BART2-5p directly targets MICB,99while EBV-miR-BART7 targets MICA.100 MICB expression is also repressed by the CMV-miR-UL122101 and by KSHV-miR-K12-7.99 The viral encoded miRs targeting NKG2D-L mRNAs are summarized in table 2.
Herpes virus-mediated interference of the interaction between infected host cells and immune effector cells by targeting host cell cytokines and chemokines
For an early antiviral response, the secretion of certain proinflammatory cytokines is required, including among others IFN-γ, TNF-α, IL-1β, IL-2, IL-6, IL-12, IL- 18, and IL-23,102–105 leading to the activation of phagocytic cells like macrophages, but also to the activation of CTLs and NK cells. As a consequence of these proinflammatory cytokines, proinflammatory chemokines, like CXCL-8, CCL2 (MCP-1), CCL3, CCL4, CCL5 (Rantes), CCL11, CXCL10, are released and recruit other immune effector cells.106 A viral intervention at this point is crucial and is to be affiliated functionally and temporally before CTLs interact with MHC class I presented antigens or NK cells interact with the infected cell via NKG2D-Ls and/or other molecules. This putative intervention may include the block of proinflammatory cytokines/chemokines and/or the increased secretion of anti-inflammatory cytokines/chemokines. A respective summary of targeted cytokines and chemokines is listed in table 3.
Viral peptides/proteins as well as viral miRs and unknown mechanisms leading to modulation of host cell cytokines/chemokines
HHV-1 represses several proinflammatory cytokines, including IL-6, TNF-α, IFN-α/β, CCL5 (Rantes), and IL-12, IL-23 by the tegument localized proteins VP16, ICP4 and ICP27,107 which underlines the relevance of these molecules for an early immune evasion.
EBV inhibits the IFN-γ downstream signaling pathway by its immediate-early protein BZLF1.108 Furthermore, EBV lytic transactivator Zta was characterized as a potent suppressor of IFN-β production,109 while the EBV LMP1 protein inhibits TNF-α.110 The EBV protein LMP1 induces the secretion of the anti-inflammatory cytokine IL-10,111 whereas the EBV miR-BHRF1-2-5p blocks the proinflammatory IL-1 signaling.112
CMV disrupts multiple levels of the IFN-α signal transduction pathway113 and the IFN-β response with its US9 protein.114 Also targeting the TNF-α115 as well as the IFN-γ induced gene expression by the CMV-encoded protein UL23 has been reported.116
The β-subfamily and γ-subfamily of the herpes viruses encode for own viral chemokines and even viral chemokine receptors known to bind and interfere with the functions of the host cell chemokines.117 The CMV UL21.5 mRNA is also packed within the virion, and its protein binds and blocks the function of CCL5 (Rantes),118 whereas the CMV-encoded protein US28 blocks CCL5 function.119 KSHV expresses the viral chemokine vCCL2, which is a broad-spectrum chemokine receptor antagonist, which might impair the recruitment of antiviral immune cells to the site of infection.117 120 The KSHV infection is further accompanied by a reduced secretion of TNF-α and IL-1.121
Conclusion
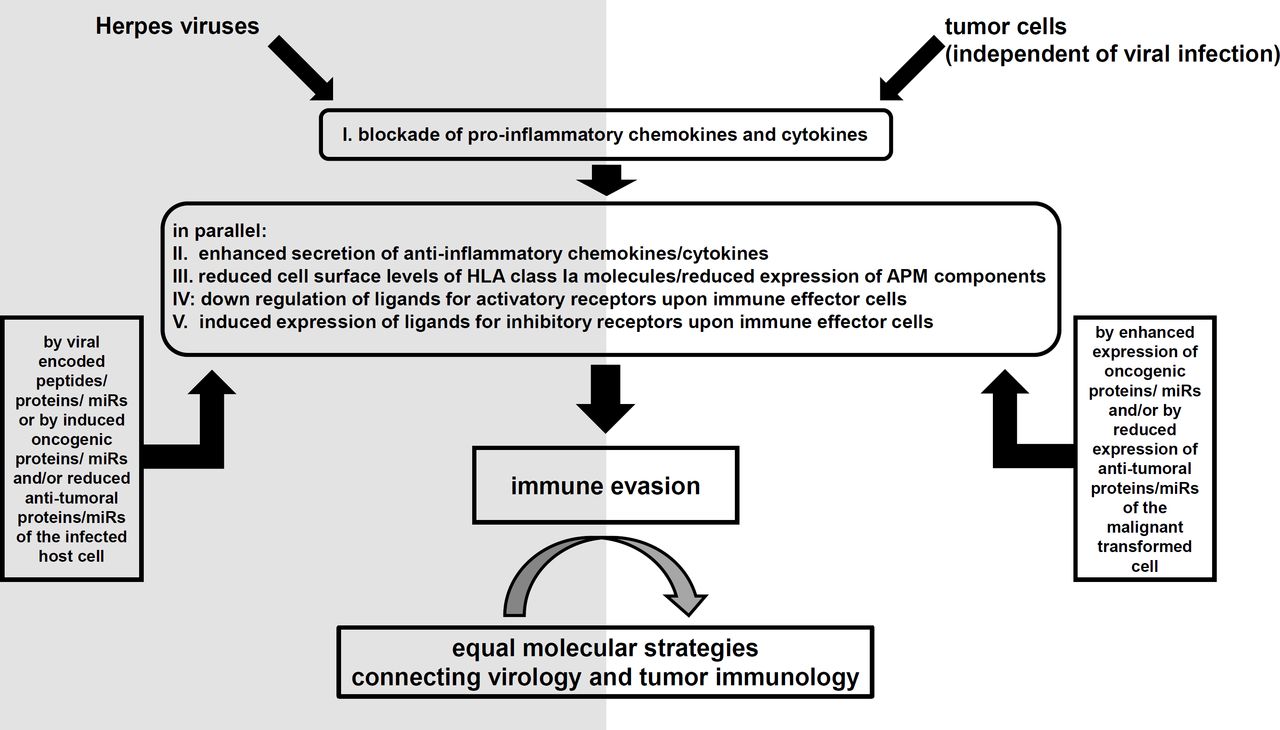
This review summarizes for the first time the combined mechanisms for immune evasion strategies of herpes viruses with focus on cytokine/chemokine signaling, thereby interfering the MHC class I-mediated antigen presentation and the interaction with immune effector cells via NKG2D-L (figure 1). While the cytokine/chemokine signaling and the NKG2D-based interactions reflect parts of the innate immunity also, the adaptive immunity is targeted by viral molecules including antigen processing and presentation via MHC class I molecules. It is noteworthy that also other subareas of the immune system are targeted by viral molecules encoded by HHVs that have not been addressed within this study.

Selected immune evasion strategies of herpes viruses in the cellular context of infected host cell and immune effector cells. APM, antigen processing and presentation machinery; CTLs, cytotoxic T lymphocytes; HLA, human leukocyte antigen; IL, interleukin.
The cytokine/chemokine signaling is a very early reaction to viral infection and might explain the presence of the respective inhibiting viral factors in the tegument. Such factors are present as protein encoding mRNAs or already as peptides/proteins. In the case of viral-based cytokine and chemokine inhibition, the involvement of respective viral miRs is not yet well investigated. In contrast, the involvement of viral miRs as well as peptides/proteins in the different steps of the complex MHC class I-mediated antigen presentation as well as in the NKG2D-Ls-based immune effector cell interaction is well studied and understood. A temporal order in the establishment of the strategies enabling the immune evasion on infection with herpes viruses is summarized in figure 2 and highlights the downregulation of proinflammatory cytokines and/or chemokines as first step based on the fact that already the virions contain molecules directly targeting a proper cytokine and chemokine signaling. Only after host cell infection and viral DNA transcription leading to viral miRs and after translation of viral mRNAs to viral peptides and/or proteins, other mechanisms of correct immune surveillance are targeted including inter alia MHC class I antigen presentation as well as NKG2D interaction.

{kind=link}
{kind=link}
Schematic summary of the highlighted mechanisms leading to immune evasion with postulated temporal order. APM, antigen processing and presentation machinery; HLA, human leukocyte antigen; miR, microRNA.
Unfortunately, only a little is known whether the reported molecular interactions between the viral miRs/peptides/proteins with immunological relevant targets in the host cell differ between primary infection, latency, reinfection, and so on, or whether they differ if different target cell types infected by the same virus, for example, EBV-infected B cells compared with infected epithelial cells of the respiratory tract. Further clinical studies are necessary to investigate and validate the clinical relevance of the reported in vitro studies.
Furthermore, it is important to consider the information of the viral-encoded immunomodulatory molecules in the context of coexpressed viral-encoded oncogenes. Already mentioned were the indirect mechanisms, like the induction of the human-encoded oncogenic miR-155 in host B cells by EBV. But, additionally, the herpes viruses are known to encode for genes exerting an oncogenic potential and such factors are not only limited to the strongly cancer-associated two members of the γ-subfamily of the human herpes viruses, namely, EBV and KSHV. In fact, even HSV-1 and HSV-2 are reported to act as cofactors for malignant transformation in several tumor malignancies including thyroid tumors, prostate cancer, and elevated incidences for melanoma, as well as cervical cancer in combination with HPV.122–125 As a major cause for a putative malignant transformation mediated by HSV-1/2, the oncogenic and antiapoptotic viral protein ICP10PK is discussed.126 VZV is also speculated to elevate the risk for certain malignancies mediated by its antiapoptotic IE63 protein.127 128 The oncogenic potential of EBV is well characterized. It is highly associated with the Burkitt’s lymphoma, Hodgkin’s lymphoma, and nasopharyngeal carcinoma. LMP1 was so far identified as the major EBV oncogene.129 The Roseoloviruses (HHV-6/7) were at least detected in some hematopoietic malignancies.130 131 In contrast, the KSHV is the causative inductor of the Kaposi’s sarcoma by inhibiting the apoptosis via the viral-encoded proteins LANA, viral Bcl-2, and K13.132 133 Many malignancies induced by the herpes viruses occur in combination with immune deficiencies. The ability of the herpes viruses to prevent complete elimination by the host immune system is therefore a possible neuralgic point for putative therapies.
All of the addressed mechanisms for immune surveillance are also targeted in tumor cells independently of viral infections and therefore the authors want to underline the strong connection between tumor immunology and virology.
Acknowledgments
The authors want to thank Maria Heise and Nicole Ott for secretarial assistance. Figure 1 was created with BioRender.
References
Footnotes
Contributors SJ-B and BS designed the review article. SJ-B, OM, and BS wrote the manuscript.
Funding This work was funded by the German-Israeli Foundation (GIF; I-37-414.11-2016) and Dr Werner Jackstädt Foundation.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.