Article Text

Abstract
Background M4112 is an oral, potent, and selective indoleamine 2,3-dioxygenase 1 (IDO1) and tryptophan 2,3-dioxygenase 2 (TDO2) dual inhibitor. Here, we report preclinical data and first-in-human phase I data, including safety, tolerability, pharmacokinetics, pharmacodynamics, and preliminary efficacy, of M4112 monotherapy in patients with advanced solid tumors.
Methods In preclinical studies, M4112 was administered to mice with IDO1-expressing tumors to determine tumor IDO1 and liver TDO2 inhibition. In the phase I trial, patients received doses of M4112 two times per day in 28-day cycles until progression, toxicity, or withdrawal of consent. The primary objective was to determine the maximum tolerated dose (MTD) and recommended phase II dose (RP2D). The primary endpoint was the incidence of dose-limiting toxicities (DLTs), treatment-emergent adverse events (TEAEs), and treatment-emergent changes in safety parameters. Other endpoints included pharmacokinetics, pharmacodynamics, and antitumor effects.
Results In mice, M4112 significantly decreased the kynurenine:tryptophan ratio in the liver and tumor. Fifteen patients received M4112 at five distinct dose levels (three patients per cohort: 100, 200, 400, 600, and 800 mg two times per day orally). Initially, all doses inhibited IDO1 ex vivo, but plasma kynurenine levels returned to or exceeded baseline levels after day 15. Despite initial changes in kynurenine, there was no significant reduction of plasma kynurenine at steady state. There was one DLT (grade 3 allergic dermatitis; 800 mg two times per day) and one grade 2 QT prolongation (800 mg two times per day), resulting in dose reduction (not a DLT). M4112 was well tolerated, and neither the MTD nor the RP2D was established. TEAEs included fatigue, nausea, and vomiting. The best overall response was stable disease (n=9, 60%).
Conclusions There were no serious safety concerns at any dose. Although M4112 inhibited IDO1 activity ex vivo, plasma kynurenine levels were not reduced despite achieving target exposure.
Trial registration number NCT03306420.
- drug evaluation, preclinical
- clinical trials as topic
- immunomodulation
- indoleamine-pyrrole 2,3,-dioxygenase
- immune evasion
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- drug evaluation, preclinical
- clinical trials as topic
- immunomodulation
- indoleamine-pyrrole 2,3,-dioxygenase
- immune evasion
Introduction
Immunotherapy with checkpoint inhibitors has shown considerable antitumor activity in patients with advanced cancer. However, not all patients respond, suggesting the existence of additional immunoregulatory pathways that control immunosurveillance.1 2 One such pathway is the catabolism of the essential amino acid tryptophan by indoleamine 2,3-dioxygenase 1 (IDO1) and the related enzyme tryptophan 2,3-dioxygenase 2 (TDO2). In normal physiology, IDO1 is expressed across many tissue types. TDO2 is expressed constitutively in the liver and is inducible by glucocorticoids and tryptophan in certain tumor types.3 4
Tryptophan metabolism significantly contributes to immunosuppression and helps tumor cells evade the immune system.5–7 In immune and tumor cells, IDO1 and TDO2 catalyze the first, rate-limiting step in the conversion of tryptophan to the immunosuppressive metabolite, kynurenine.8 9 Tryptophan catabolism can lead to an immunosuppressive tumor microenvironment through two mechanisms: (1) the depletion of tryptophan and (2) the production of kynurenine. The reduction of tryptophan can lead to autophagy and T cell anergy through the suppression of mammalian target of rapamycin complex 1 activity.10 Tryptophan depletion can also induce the stress-responsive kinase general control nonderepressible 2,5 which may in turn prevent T cell proliferation, promote de novo regulatory T cell differentiation, and enhance regulatory T cell activity.11 The production of kynurenine can also be immunosuppressive through the activation of the transcription factor aryl hydrocarbon receptor. Aryl hydrocarbon receptors inhibit the differentiation of naïve cluster of differentiation 4 T helper cells into proinflammatory T helper 17 cells and can promote a tolerogenic phenotype in dendritic cells.5
Both IDO1 and TDO2 overexpressions are associated with poor prognosis in various human cancer types.12–16 IDO1 is often overexpressed in hematologic cancers and solid tumors,17–20 whereas TDO2 is often overexpressed in glioma, breast cancer, lung cancer, colorectal cancer, and hepatocellular carcinoma.21–26 Although both enzymes are upregulated in several tumor types, their expression is distinct and not overlapping.21–26 This suggests that a dual inhibitor of IDO1 and TDO2 could provide an enhanced therapeutic benefit to patients with cancer.
M4112 is a first-in-class, potent, selective dual inhibitor of IDO1 and TDO2 that can be administered orally. Here, we present the results from a first-in-human phase I trial that evaluated the safety, tolerability, pharmacokinetics, pharmacodynamics, and preliminary efficacy of M4112. The preclinical evidence that provided the rationale for entering clinical development is also reported.
Methods
Preclinical investigations
Syngeneic and non-syngeneic murine models
The pharmacokinetic and pharmacodynamic properties of M4112 were investigated in preclinical models by assessing the concentration of kynurenine and tryptophan in mice. Female adult C57BL/6 and BALB/c mice were inoculated subcutaneously with IDO1-expressing syngeneic murine colon carcinoma MC38 cells (0.5×106 cells) and IDO1-expressing CT26-KSA cells (1×106 cells), respectively. The two cell lines did not express TDO2. When tumors reached an average volume of approximately 200–250 mm3, mice with MC38 tumors received a single, orally administered dose of M4112 (30, 75, or 200 mg/kg) or vehicle. When tumors reached an average volume of approximately 200–300 mm3, mice with CT26-KSA tumors received three doses of M4112 (100 mg/kg), selective IDO1 inhibitor MSC2574260 (100 mg/kg), or vehicle (0.1 mL). Mice were dosed at 0, 8, and 24 hours. The levels of M4112 and kynurenine, and the kynurenine:tryptophan ratio, in the plasma, liver, and tumors were then measured at 1, 2, 4, 8, and 24 hours after treatment using liquid chromatography with tandem mass spectrometry (LC-MS/MS).
Phase I trial design
This phase I, first-in-human, open-label, dose-escalation trial was conducted in patients with metastatic or locally advanced unresectable solid tumors. M4112 was given orally, two times per day in 28-day cycles, with three patients per cohort. The starting dose of 100 mg two times per day was determined from preclinical studies and pharmacokinetic/pharmacodynamic modeling of the predicted pharmacologic active dose range in humans. Patients continued M4112 treatment until disease progression, unacceptable toxicity, or withdrawal of consent.
The primary objective was to determine the safety and tolerability or, if observed, the maximum tolerated dose (MTD), and to define the recommended phase II dose (RP2D) of M4112 monotherapy in patients with solid tumors. The secondary objectives included M4112 pharmacokinetic characterization, evaluation of preliminary clinical activity (via Response Evaluation Criteria In Solid Tumors (RECIST) version 1.1), and assessment of potential M4112 concentration-related QT prolongation. Exploratory objectives included evaluation of the effect of M4112 on kynurenine and tryptophan in the plasma and on IDO1 activity in ex vivo stimulated whole blood from M4112-treated patients.
Patients
Adults with histologically or cytologically advanced or metastatic solid tumors refractory to standard therapies or tumors for which no effective treatment was available were eligible. Other key inclusion criteria were Eastern Cooperative Oncology Group performance status of 0 or 1, and an adequate hematologic and hepatic function. Key exclusion criteria included concomitant treatment with strong inhibitors or inducers of cytochrome P450 3A4. Full eligibility criteria are shown in the online supplementary information.
Supplemental material
Assessments
To assess the safety of M4112 (primary endpoint), adverse events (AEs), dose-limiting toxicities (DLT), physical examination findings, vital sign measurements, ECG results, and changes in clinical laboratory parameters (chemistry, hematology, and coagulation) were recorded. Safety parameters were assessed from the first dose to the end of treatment. AEs were graded and classified by the investigator according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03. The MTD was based on the incidence of DLTs during cycle 1 (days 1–28). DLTs were generally defined as any grade ≥3 AEs or immune-related AEs for non-hematologic toxicities and grade ≥4 AEs for hematologic toxicities during cycle 1 at any dose level.
Blood samples for pharmacokinetic analysis were collected on days 1 and 15 of cycle 1 (predose and 0.5, 1, 2, 3, 4, 6, and 8 hours postdose), day 8 of cycle 1 (predose), and on day 1 of cycle 2 (predose and 2 hours postdose) (online supplementary figure 1). Plasma concentrations were determined by LC-MS/MS. The pharmacokinetics of M4112 were characterized by non-compartmental analyses (Phoenix WinNonlin version 6.4, Certara USA, Princeton, New Jersey, USA) for calculation of maximum observed concentration (Cmax), time to Cmax (tmax), and area under the drug concentration–time curve from 0 to 8 hours postdose (AUC0–8). Accumulation (Racc) was calculated for day 15/day 1 for Cmax and AUC0–8 as well as for dose-normalized values (Cmax/dose and AUC0–8/dose). Predose concentrations (Cpre and Cpre/dose) were determined for day 1, day 8, and day 15, and for day 1 of cycle 2. Time-matched corrected QT values were extracted for planned concentration-QT analysis.
To evaluate the pharmacodynamic activity of M4112, plasma samples were collected on day 1 (predose and 1, 2, 4, and 6 hours postdose), day 8 (predose), day 15 (predose and 1, 2, 4, and 6 hours postdose), and day 1 of cycle 2 (predose and 2 hours postdose) (online supplementary figure 1). Concentrations of kynurenine and tryptophan were measured by LC-MS/MS and kynurenine:tryptophan ratios were calculated accordingly. In addition, to assess the activity of M4112 against IDO1, blood samples were drawn directly into TruCulture blood collection tubes (Myriad RBM, Austin, Texas, USA) and preloaded with 100 ng/mL lipopolysaccharide (LPS) to stimulate IDO1; whole blood samples were collected on day 1 (predose and 1, 2, 4, and 6 hours postdose), day 8 (predose), day 15 (predose and 1, 2, 4, and 6 hours postdose), and day 1 of cycle 2 (predose and 2 hours postdose). Tubes were incubated at 37°C for 24 hours before measuring kynurenine levels by LC-MS/MS. A predose blood sample was also collected into tubes without preloaded LPS as background. Inhibition of IDO1 was calculated by measurement of the decrease in kynurenine level compared with the predose level. All patients who received treatment with M4112 were included in the safety, pharmacokinetic, and pharmacodynamic analysis sets.
CT or MRI scans were performed at baseline and within 7 days prior to day 1 of cycles 3, 5, and 7, and every three cycles thereafter to assess tumor response. Objective response, best overall response, disease control rate, and progression-free survival were assessed by the investigator according to RECIST version 1.1.27
Statistical analysis
Decisions on dose escalation were made by the Safety Monitoring Committee based on recommendations from a Bayesian two-parameter logistic regression model. The model was updated with the number of evaluable patients and DLTs observed after completion of the DLT period for each cohort. Recommendations on the next dose level were based on the posterior distribution of toxicity. The primary analysis assessed the number and percentage of DLTs, performed on the DLT analysis set by dose level. The MTD was defined using a Bayesian two-parameter logistic regression model with a target toxicity of 30%. Descriptive statistics were used for baseline characteristics, safety assessments, pharmacokinetic and pharmacodynamic data, and efficacy endpoints. The 95% exact Clopper-Pearson CI was used for binary efficacy endpoints. The Kaplan-Meier method was used for survival endpoints. Dose proportionality of pharmacokinetic parameters was evaluated statistically using a power model based on the original parameters (ln[pharmacokinetic parameter]=α+β×ln[dose]).
Results
Preclinical studies
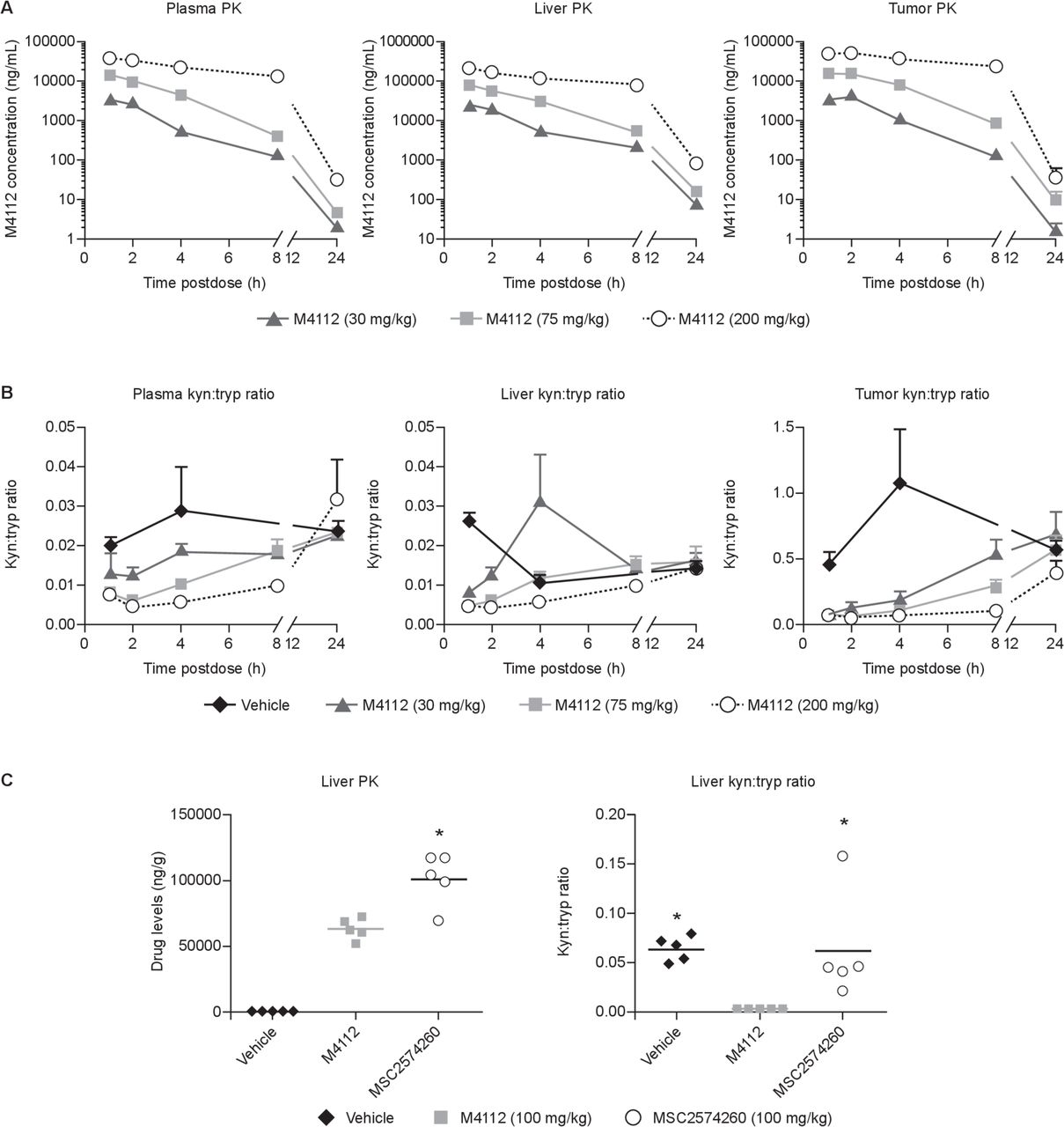
To assess the level of target engagement in vivo by M4112, kynurenine and tryptophan levels were measured in the plasma, liver, and tumors of mice. In mice with MC38 colon tumors expressing IDO1, the concentration of M4112 in the tumor peaked 1–2 hours post-treatment (figure 1A). By 24 hours post-treatment, the concentration of M4112 in the tumor decreased to baseline levels. A similar pharmacokinetic profile was also observed in the liver and the plasma. However, exposure to M4112 was approximately 10-fold higher in the liver compared with both the plasma and the tumor. In MC38 tumor-bearing mice, M4112 at all three dose levels (30, 75, or 200 mg/kg) significantly decreased the kynurenine:tryptophan ratio in the tumor 4 hours after M4112 treatment compared with vehicle (p<0.001) (figure 1B). A dose-dependent reduction in the kynurenine:tryptophan ratio was also often observed in the liver and plasma of mice that received M4112. In mice with IDO1-expressing CT26-KSA tumors, M4112 significantly decreased the kynurenine:tryptophan ratio in the liver after 2 hours compared with vehicle (p=0.0266) and MSC2574260 (p=0.0327) (figure 1C). Although exposure to MSC2574260 (the selective IDO1 inhibitor) was significantly higher in the liver compared with exposure to M4112 (p=0.0009), MSC2574260 failed to significantly modulate the kynurenine:tryptophan ratio.
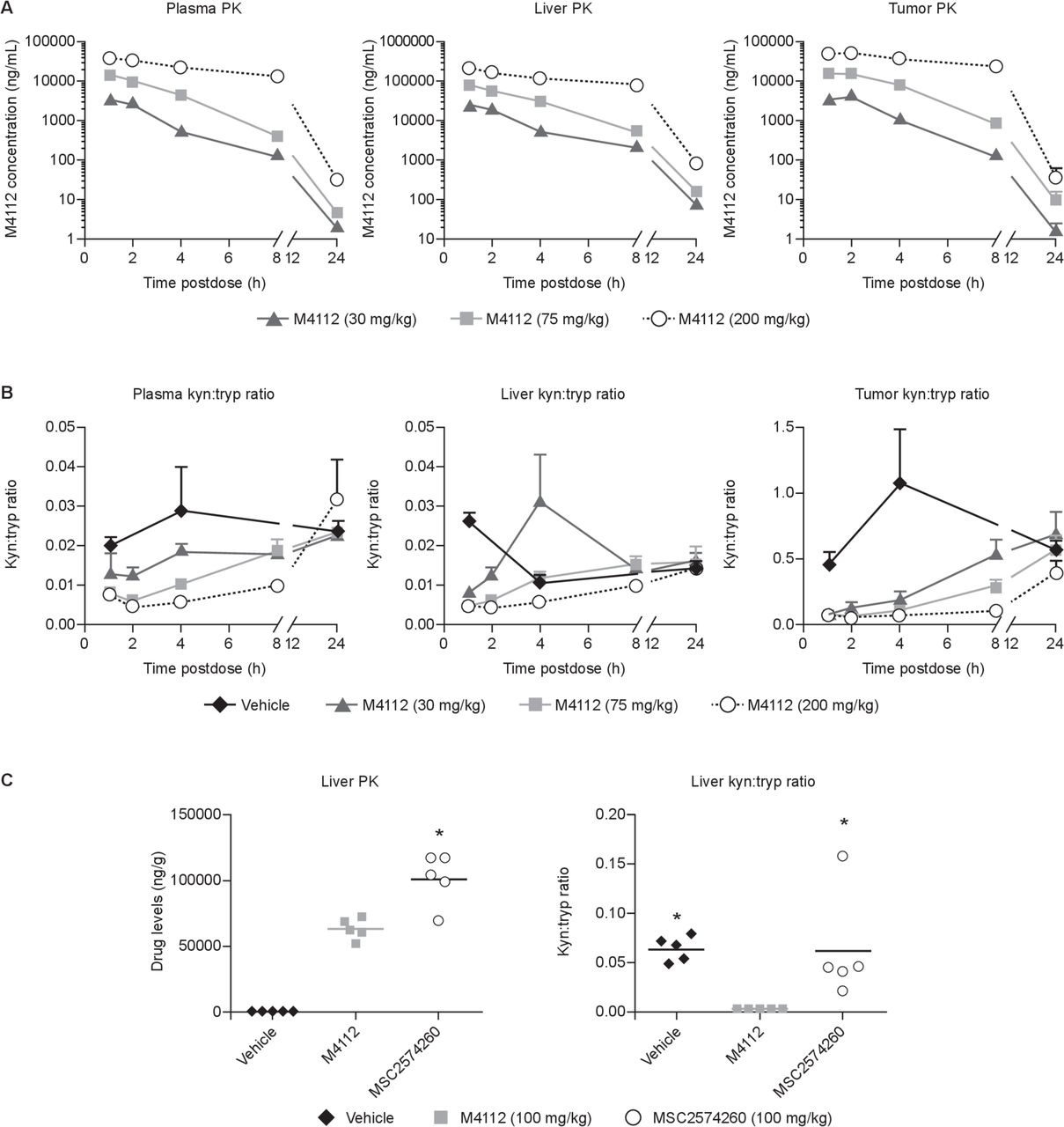
(A) M4112 levels and (B) the kyn:tryp ratio in mice bearing MC38 tumors treated with M4112 (30, 75, or 200 mg/kg) or vehicle. (C) Liver drug levels, kynurenine levels, and tryptophan levels in mice with IDO1-expressing CT26-KSA tumors treated orally with M4112 (100 mg/kg), IDO1-selective MSC2574260 (100 mg/kg), or vehicle (0.1 mL). For (A) and (B), plasma, liver, and tumor samples were collected at various timepoints. Data from vehicle-treated mice have been omitted for clarity. Also, for (A) and (B), levels were determined by LC-MS/MS, and data are presented as mean±SEM. The tumor kyn:tryp ratio after 4 hours was significantly different across all three dose levels compared with the vehicle group (p≤0.0001). For (C), liver drug levels and kynurenine and tryptophan levels were measured 2 hours after the last dose. In (C), the symbol ‘*’ represents a significant difference relative to M4112 (p<0.05). The drug level of MSC2574260 was significantly different from the drug level of M4112 (p=0.0009). The liver kyn:tryp ratio was significantly different in the M4112 group compared with the vehicle group (p=0.0266) and the MSC2574260 group (p=0.0327). Data are representative of two independent experiments. IDO1, indoleamine 2,3-dioxygenase 1; kyn, kynurenine; LC-MS/MS, liquid chromatography with tandem mass spectrometry; PK, pharmacokinetics; tryp, tryptophan.
Patient characteristics
Fifteen patients with advanced solid tumors were enrolled (table 1). Most patients were women (86.7%) and the median age was 57.0 years (range 37–77 years). All patients had received chemotherapy and eight (53.3%) patients had received radiotherapy. Patients (N=15) received one of the following doses of M4112: 100, 200, 400, 600, or 800 mg two times per day orally (three patients per cohort).
Demographic characteristics: safety analysis set
Safety
All 15 patients were included in the safety analyses. The median duration of M4112 exposure was 16.0 weeks (range 2.7–44.0 weeks). During cycle 1 (DLT assessment period), one patient in the 800 mg two times per day arm experienced a DLT (grade 3 allergic dermatitis). Another patient dosed at 800 mg two times per day developed a grade 2 QT prolongation, resulting in a dose reduction; this event did not meet the DLT criteria. Following the DLT in the 800 mg two times per day arm, the SMC recommended reducing the dose level of M4112 to 600 mg two times per day for the next patient cohort (n=3); no DLTs were observed at this dose level. Two patients in the 600 mg two times per day dose group had dose reductions due to AEs (one grade 3 diarrhea and one grade 3 rash after the DLT period). There were no deaths reported during the dose-escalation phase. The Bayesian model estimated the median probability of a DLT occurring at 800 mg to be 20.5%, with a 95% credible interval of 3.1%–62.2%. Given that the estimate was clearly below the target value of 30% and that the credible interval was quite wide, no MTD was defined. The RP2D was also not established.
The most common treatment-emergent AEs (TEAEs) that occurred in ≥2 patients (at any grade) are presented in table 2. The most frequently reported TEAEs were fatigue (33.3%), nausea (26.7%), and vomiting (26.7%). Three patients, one from each of the 400, 600, and 800 mg two times per day cohorts, experienced a serious TEAE during the trial; these included grade 3 disease progression (unrelated to treatment), grade 2 ascites (unrelated to treatment) and grade 3 allergic dermatitis (related to treatment). Two (13.3%) patients discontinued M4112 treatment due to TEAEs: one patient in the 400 mg two times per day cohort discontinued due to disease progression; and the patient on 800 mg two times per day who experienced a DLT discontinued due to allergic dermatitis.
TEAE occurring in two or more patients at any grade and SAE occurring in one or more patients (safety analysis set)
Pharmacokinetics
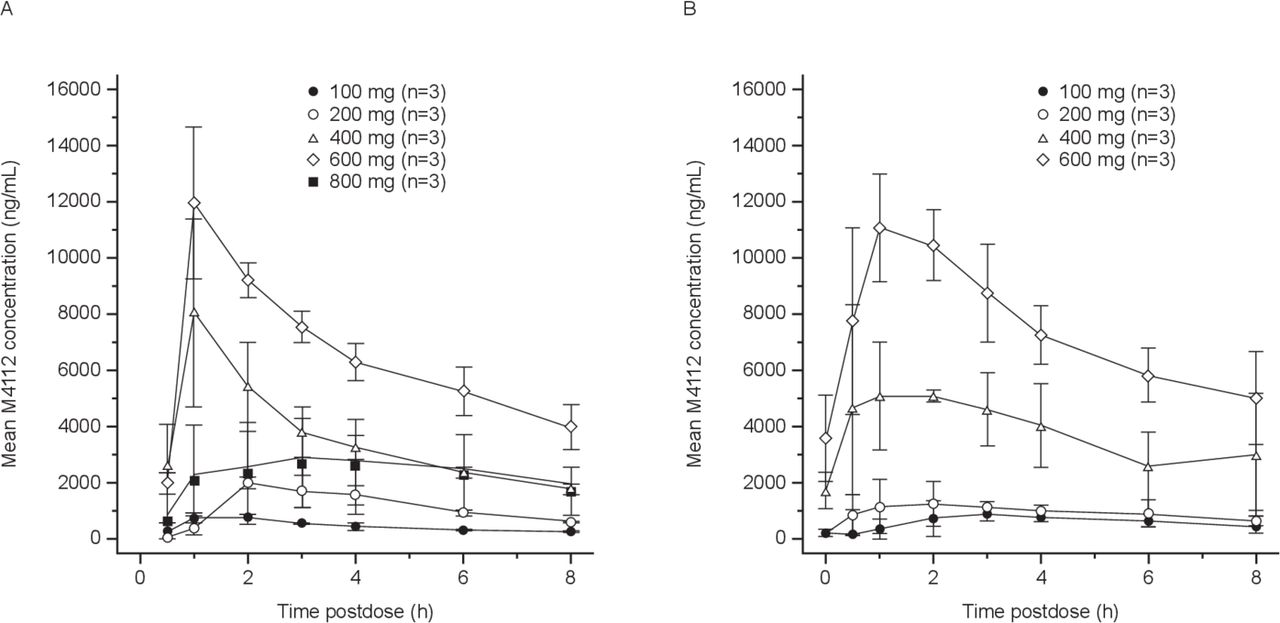
Pharmacokinetic evaluations were based on all 15 patients who received M4112 treatment. A summary of the mean pharmacokinetic measurements is presented in table 3. The pharmacokinetics of M4112 were characterized by a tmax of approximately 1–3 hours across all dose levels on both days 1 and 15. The mean M4112 plasma concentration curves on cycle 1 day 1 and day 15 are shown in figure 2. The mean exposure of M4112 increased in a greater than dose-proportional manner from 100 to 600 mg two times per day (slope estimate of 1.46–1.67) on day 15 (figure 2B). M4112 exposure for the 800 mg two times per day cohort was initially lower than that of the other dose groups (cycle 1, day 1). However, by day 15, exposure for the 800 mg two times per day dose group increased in the one patient that continued with this regimen. There was no appreciable accumulation or evidence of autoinduction following two times per day dosing over the 100–600 mg dose range.
Pharmacokinetic parameters

Mean M4112 plasma concentrations on cycle 1 (A) day 1 and (B) day 15, by dose cohort. Arithmetic mean values (±SD) are shown.
Pharmacodynamics
Following M4112 administration, a rapid onset of IDO1 inhibition in ex vivo stimulated blood was observed across all doses tested (figure 3A). Maximum inhibition was achieved 1–4 hours post-M4112 administration. There was a trend toward dose-dependent IDO1 inhibition at steady state. Approximately 90% of IDO1 activity was inhibited throughout the dosing interval at 800 mg two times per day.

Dotplots* of (A) baseline normalized kynurenine (% baseline) versus dose level on cycle 1, day 15 predose (steady state) in ex vivo stimulated whole blood, and (B) baseline normalized plasma kynurenine, tryptophan, and kyn:tryp ratio versus dose level on cycle 1, day 1, 6 hours postdose† and (C) on cycle 1, day 15 predose. *All data are shown with locally weighted scatterplot smoothing (lowess) lines. †One data point from the 800 mg two times per day group lies above the range of the axis. kyn, kynurenine; tryp, tryptophan.
A small yet consistent decrease in plasma kynurenine levels (12%–35%) was observed on day 1 of cycle 1 (up to 6 hours post-M4112 treatment) after the first dose was administered for all dose levels investigated (figure 3B). Plasma tryptophan levels increased on day 1 of cycle 1 following doses of 200 mg two times per day and above. The increase in plasma tryptophan levels was largely responsible for the decrease in the kynurenine:tryptophan ratio observed after 6 hours. However, on day 15 of cycle 1, kynurenine levels had returned to at least baseline levels (figure 3C). Based on these findings, it was decided to discontinue the clinical study due to the insufficient pharmacodynamic effect observed.
Preliminary antitumor activity
The best overall response was stable disease in 9 of 15 (60.0%) patients, and the median progression-free survival was 3.7 months. There were no objective responses. Figure 4 shows individual patient responses to treatment. There was no indication of a relationship between the dose of M4112 and disease control or duration of progression-free survival.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Swimmer plot showing best response and duration of M4112 therapy. *One patient was identified as having a synovial sarcoma of the right psoas muscle. BID, two times per day.
Discussion
The preclinical investigations demonstrated promising activity: M4112 significantly and dose-dependently reduced the kynurenine:tryptophan ratio in IDO1-expressing MC38 colorectal tumor-bearing mice. Furthermore, compared with a selective IDO1 inhibitor, M4112 demonstrated a greatly enhanced ability to reduce the kynurenine:tryptophan ratio in liver tissue. Since the liver is an organ with significant TDO2 expression, these findings support M4112 acting as a dual inhibitor of IDO1 and TDO2. Together, these data supported the phase I investigation of the dual IDO1 and TDO2 inhibitor, M4112.
This phase I trial is the first clinical investigation to evaluate a dual IDO1/TDO2 inhibitor. The primary endpoint of the trial was to evaluate the safety and tolerability of M4112 and to define the RP2D. M4112 did not result in any serious safety concerns at doses up to 800 mg two times per day in patients with advanced solid tumors. However, due to the early termination of the study, neither the MTD nor the RP2D was established. One patient treated with M4112 800 mg two times per day experienced a serious TEAE and DLT of grade 3 allergic dermatitis with fever and hypotension. This was considered an acute allergic drug reaction to M4112. Treatment was withdrawn and the DLT resolved within 6 hours. Serious TEAEs that occurred in two additional patients were not considered related to M4112 and no deaths were reported during the dose-escalation phase of the trial.
Mean exposure of M4112 increased in a greater than dose-proportional manner from 100 to 600 mg two times per day. Interestingly, exposure for the 800 mg two times per day group was initially much lower than for the other treatment cohorts. However, by day 15, exposure increased toward the expected range in the one patient who continued this treatment regimen. As patient numbers were small, further study is needed to explain this result.
With respect to pharmacodynamics, M4112 (at all dose levels tested) rapidly inhibited IDO1 activity in ex vivo whole blood stimulated with LPS. Moreover, in the 800 mg two times per day dose cohort, IDO1 activity was inhibited by approximately 90%. M4112 initially increased tryptophan (at or above 200 mg two times per day) and decreased kynurenine (following the first dose of M4112) in plasma. However, in contrast to IDO1-selective inhibitors such as epacadostat, the decrease of kynurenine in plasma was transient, and by day 15 of cycle 1 levels of kynurenine had returned to or exceeded baseline.28 It is unclear why plasma kynurenine levels did not significantly decrease at steady state despite inhibition of IDO1 activity in ex vivo stimulated whole blood. Potential explanations for this could be insufficient drug distribution to relevant IDO1-expressing and TDO2-expressing tissues, or the existence of unknown compensatory mechanisms. As there was no significant reduction of plasma kynurenine at steady state, it was decided not to continue the study.
Antitumor results showed a best overall response of stable disease, which was observed in nine patients (60%) and a progression-free survival of 3.7 months. It is worth reiterating that patients with advanced cancer were enrolled and a majority of these patients had tumor types that are typically unresponsive to immunotherapy.29 Also, IDO1 inhibition alone rarely exerts a strong antitumor response; for selective IDO1 inhibitors like epacadostat, indoximod, and navoximod, stable disease is also the best overall response reported in early phase I studies.28 30 31 One possible explanation is that tumors evade immune surveillance through multiple mechanisms. A dual inhibitor of the IDO1 and TDO2 pathway may thus be insufficient to provoke an antitumor response when administered as a single agent because of other inhibitory signaling pathways that are used by the tumor. The rationale for combination with other immuno-oncology agents will need to be explored to determine if this approach increases efficacy. Also, it is still unclear if kynurenine and tryptophan levels in the blood of patients do reliably and accurately represent target engagement of M4112 in tumors.
One limitation of the clinical study was that patient tumor biopsies were not obtained. Therefore, neither changes in IDO/TDO expression nor changes in the tumor microenvironment could be evaluated.
Further investigations regarding the pharmacodynamics, safety, and efficacy of M4112 in combination with checkpoint inhibitors are also warranted.
Acknowledgments
The authors would like to thank the patients and their families, investigators, coinvestigators, and study teams at each of the participating centers and at EMD Serono, a business of Merck KGaA, Darmstadt, Germany. They also thank IQVIA for supporting this clinical trial. The authors would also like to thank Markus Fluck (Merck KGaA, Darmstadt, Germany) for contributing to the pharmacodynamic data that are presented in this manuscript, and also Lu Li for contributing to the safety data that are presented in this manuscript. Medical writing assistance was provided by David Lester, Bioscript Science, Macclesfield, UK.
References
Footnotes
Twitter @AnaingMD
Contributors Conception and design: AN, CG and JM. Development of methodology: SZ. Acquisition of data (provided animals, acquired and managed patients, provided facilities, and so on): AN, JPE, SAP-P, KPP and SZ. Analysis and interpretation of data (eg, statistical analysis, biostatistics, computational analysis): AN, SZ, CH and JM. Writing, review, and/or revision of the manuscript: AN, JPE, SAP-P, CG, EH, SZ, VHi, VHo, CH, JM and KPP. Administrative, technical, or material support (ie, reporting or organizing data, constructing databases) and study supervision: CG.
Funding The trial was sponsored by Merck KGaA, Darmstadt, Germany. Medical writing assistance was also funded by Merck KGaA.
Competing interests AN has received research grants from Amplimmune, ARMO BioSciences, Atterocor, BMS, Calithera Biosciences, CytomX Therapeutics, EMD Serono, Healios Oncology Nutrition, Immune Deficiency Foundation, Incyte, Karyopharm Therapeutics, MedImmune, Merck, NCI, Neon Therapeutics, Novartis, Pfizer, Regeneron, and TopAlliance BioSciences. JPE has received consultancy fees from Roche/Genentech. SAP-P has received research funding from AbbVie, Alkermes, Aminex Therapeutics, BioMarin Pharmaceutical, Boehringer Ingelheim, Bristol Myers Squibb, Cerulean Pharma, Curis, Five Prime Therapeutics, FLX Bio, Genmab, GlaxoSmithKline, Helix BioPharma, Incyte, Jacobio Pharmaceuticals, Medimmune, Medivation, Merck Sharp and Dohme, Novartis Pharmaceuticals, Pattern Pharma, Pieris Pharmaceuticals, Pfizer, Principia Biopharma, Puma Biotechnology, Seattle Genetics, Taiho Oncology, Tesaro, TransThera Biosciences and XuanZhu Pharma. CG and CH are employees of Merck KGaA. EH, an employee of Nuventra, provided consultancy to EMD Serono. SZ, VHo and JM are employees of EMD Serono. KPP has received research funding from EMD Serono and Incyte.
Patient consent for publication Not required.
Ethics approval The trial was conducted according to Good Clinical Practice guidelines, the Declaration of Helsinki, and applicable regulations. Before the study could commence at a given site, the clinical study protocol had to be submitted together with its associated documents to the responsible Independent Ethics Committee or Institutional Review Board for its favorable opinion or approval, which will be filed in the Investigator Site File. This study was approved by the Western IRB (IRB tracking number: 20172687; board action date: May 1, 2018), IntegReview IRB (START2017.14; board action date: September 7, 2017) and The University of Texas MD Anderson Cancer Center IRB (MDACC protocol ID #: 2017-0549; official IRB approval date: September 28, 2017). Patients provided written, informed consent prior to enrollment.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. Any requests for data by qualified scientific and medical researchers for legitimate research purposes will be subject to Merck’s Data Sharing Policy. All requests should be submitted in writing to Merck’s data sharing portal (https://www.merckgroup.com/en/research/our-approach-to-research-and-development/healthcare/clinical-trials/commitment-responsible-data-sharing.html). When Merck has a co-research, co-development, or co-marketing or co-promotion agreement, or when the product has been out-licensed, the responsibility for disclosure might be dependent on the agreement between parties. Under these circumstances, Merck will endeavor to gain agreement to share data in response to requests.