Article Text

Abstract
Background Although programmed cell death-1/programmed death-ligand 1 (PD-L1) inhibitors show remarkable antitumor activity, a large portion of patients with cancer, even those with high PD-L1-expressing tumors, do not respond to their effects. Most PD-L1 inhibitors contain modified fragment crystallizable region (Fc) receptor binding sites to prevent antibody-dependent cellular cytotoxicity (ADCC) against PD-L1-expressing non-tumor cells. However, natural killer (NK) cells have specific antitumor activity in the presence of tumor-targeting antibody through ADCC, which could enhance NK cell-induced cytotoxicity. We evaluated the antitumor efficacy of ADCC via anti-PD-L1 monoclonal antibodies (mAbs) and NK cells against several PD-L1-positive cancer cell lines.
Methods Various cancer cell lines were used as target cell lines. Surface PD-L1 expression was analyzed by flow cytometry. IMC-001 and anti-hPD-L1-hIgG1 were tested as anti-PD-L1 mAbs with ADCC and atezolizumab as an anti-PD-L1 mAb without ADCC. NK cell cytotoxicity was measured by 51Cr-release assay and CD107a degranulation assay. Also, live cell imaging was performed to evaluate cytotoxicity in a single-cell level. NK-92-CD16 (CD16-transduced NK-92 cell line) and peripheral blood mononuclear cells from healthy donors, respectively, were used as an effector cell. FcγRIIIa (CD16a)-V158F genotyping was performed for healthy donors.
Results We demonstrated that the cytotoxicity of NK-92-CD16 cells toward PD-L1-positive cancer cell lines was significantly enhanced in the presence of anti-PD-L1 mAb with ADCC. We also noted a significant increase in primary human NK cell cytotoxicity against PD-L1-positive human cancer cells when cocultured with anti-PD-L1 mAb with ADCC. Moreover, NK cells expressing a FCGR3A high-affinity genotype displayed higher anti-PD-L1 mAb-mediated ADCC lysis of tumor cells than donors with a low-affinity genotype.
Conclusion These results suggest that NK cells induce an ADCC response in combination with anti-PD-L1 mAbs, which helps promote ADCC antitumor activity against PD-L1-positive tumors. This study provides support for NK cell immunotherapy against high PD-L1-expressing tumors in combination with ADCC through anti-PD-L1 mAbs.
- killer cells
- natural
- immunotherapy
- cytotoxicity
- immunological
- lung neoplasms
- head and neck neoplasms
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Immune evasion mechanisms are a survival strategy for cancer. Binding between programmed cell death-1 (PD-1) and programmed death-ligand 1 (PD-L1) provides immune evasion through which cancer can escape a host immune response.1–4 PD-1/PD-L1 blockade therapy has shown promising clinical results for antitumor activity in several cancer types, especially PD-L1-positive tumors.5 6 Although PD-L1 expression can be used as a predictive biomarker for PD-1/PD-L1 blockade therapy, its efficacy remains low, with response rates of approximately 20% in non-small-cell lung cancer (NSCLC) and head and neck squamous cell carcinoma (HNSCC),6 7 including high PD-L1-expressing tumors. Therefore, new strategies are needed to increase the efficacy and response rates for anti-PD-L1 monoclonal antibodies (mAbs) in PD-1/PD-L1 therapy.8
Most anti-PD-1/PD-L1 mAbs contain modified Fc receptor (FcR) binding sites to prevent antibody-dependent cellular cytotoxicity (ADCC) against PD-L1-expressing immune or non-tumor cells. However, avelumab is a fully human IgG1 anti-PD-L1 mAb contained wild-type FcR that can induce ADCC. Avelumab has been used in various phase I and II clinical trials with toxicity and efficacy similar to those of FcR-modified anti-PD-1 or PD-L1 mAbs.9 ADCC induction by directly targeting PD-L1 using a fully human antibody could therefore be safe and effective in patients.
The antitumor capabilities of natural killer (NK) cells have been extensively studied in cancer immunotherapy.10 NK cells play a major role in cancer immunotherapy, possessing specific antitumor activities in the presence of tumor antigen-targeting mAbs through ADCC.11 The common FcR on NK cells (CD16 or FcRγIII) recognizes the Fc portion of tumor-bound antibodies, which activates NK cells for tumor cell lysis by releasing cytotoxic factors and cytokines that recruit and activate other immune cells.
Patients with NK cells expressing the high-affinity CD16 valine (V) allele experience better clinical outcomes than those with the lower affinity phenylalanine (F) allele and F/F genotypes.12 Several clinical studies showed the ADCC of NK cells and FcγRIIIa polymorphism are related to clinical responses in rituximab (CD20, in follicular lymphoma Blood),13 trastuzumab (HER2, in breast cancer),14 cetuximab (EGFR, in colorectal cancer),15 and avelumab (PD-L1 in lung cancer).16 These studies suggest that ADCC and PD-1/PD-L1 blockade could be another immunotherapeutic strategy.
Our aim for this study was to explore NK cell-induced ADCC cytotoxicity against PD-L1-expressing cancer cell lines when cocultured with anti-PD-L1 mAbs. We hypothesized that NK cells induce the ADCC response in combination with anti-PD-L1 mAbs to promote ADCC and enhance the antitumor response against high PD-L1-expressing tumors. To investigate the antitumor efficacy of NK cells and anti-PD-L1 mAbs to generate ADCC against several PD-L1-positive cell lines, we evaluated ADCC activity and NK cell cytotoxicity across various PD-L1-expressing cell lines.
Materials and methods
Cell lines and cell cultures
Various human cancer cell lines were used in this study, including those of HNSCC and NSCLCs. SNU-1066, SNU-1041, and SNU-1076 cell lines were purchased from the Korean Cell Line Bank (Seoul, Korea). Detroit 562 (CCL-138), FaDu (HTB-43), NCI-H1975 (CRL-5908), and NCI-H1650 (CRL-5883) cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, Virginia, USA). The HN31 cell line was kindly granted by John F. Ensley, Wayne State University. SNU-1066, SNU-1041, SNU-1076, HN31, NCI-H1650 and NCI-H1975 cell lines were cultured in Roswell Park Memorial Institute Medium (RPMI 1640); Gibco, Invitrogen) supplemented with 10% heat‐inactivated Fetal Bovine Serum (FBS; Gibco) and 10 µg/mL gentamycin. Detroit 562 and FaDu cell lines were maintained in Eagle’s Minimum Essential Medium (EMEM) supplemented with 10% heat‐inactivated FBS and 1% penicillin/streptomycin (Gibco).
Allogeneic NK cell lines engineered to express the high-affinity CD16 allele were used. NK-92-CD16 cells (purchased from the ATCC, PTA-8836) were maintained in alpha minimum essential medium supplemented with 25% FBS, 0.2 mM inositol, 0.1 mM 2mercaptoethanol, and 0.02 mM folic acid. NK-92-CD16 cells were cultured with recombinant human interleukin (IL)-2 (200 U/mL).
All cell lines were cultured for less than 6 months and tested negative for mycoplasma using a detection kit (iNtRON Bio Technology, Seongnam, Korea) prior to experiments. The human cancer cell lines were authenticated by CosmoGenetceh (Seoul, Republic of Korea) using short-tandem repeat profiling.
Preparation of peripheral blood NK cells and activation
Peripheral blood mononuclear cells (PBMCs) of healthy donors were isolated from leukoreduction system chambers using Ficoll density gradient sedimentation. PBMCs were rested overnight in RPMI 1640 medium (Gibco) and activated by recombinant human IL-15 (PeproTech, Rocky Hill, New Jersey, USA) 1 ng/mL with the RPMI1640 medium for 3 days before an in vitro ADCC assay.
Reagents and antibodies
To perform mAb-mediated ADCC in vitro experiments, the IgG1 isotype kappa control from human myeloma plasma was used (Sigma-Aldrich, St. Louis, Missouri, USA). The following mAbs were used for anti-PD-L1 mAb. Atezolizumab (Selleck Chem, Houston, Texas, USA) is IgG1 mAb with a modified Fc region designed to limit ADCC. IMC-001 (kindly provided by ImmuneOncia, Yongin, Korea) is a fully human PD-L1 recombinant IgG1 mAb that can induce ADCC. An anti-hPD-L1-hIgG1 (InvivoGen) is atezolizumab-based wild-type IgG1 that displays ADCC.
Cells were stained with fluorescent-labeled mAbs for the following cell-surface markers: anti-CD274-PE (clone MIH1, RRID:AB_647198), isotype control-PE (MOPC-2, RRID:AB_396091), anti-CD3-FITC (UCHT1, RRID:AB_395739), anti-CD56-APC (B159, RRID:AB_398601), anti-CD107a-PE (H4A3, RRID:AB_396135), anti-CD107a –APC (H4A3, RRID:AB_1645722) (BD Biosciences, San Jose, California, USA); anti-CD16-APC (CB16, RRID:AB_2016663), anti-CD16-PE-Cy7 (CB16, RRID:AB_10714839) (Thermo Fisher Scientific, Waltham, Massachusetts, USA); anti-CD56-PE-Vio770 (REA196, RRID:AB_2726091; Miltenyi Biotech, Bergisch Gladbach, Germany). For CD107a degranulation, a protein transport inhibitor, Golgi-Stop (#554724, BD Biosciences) was used. Fixable viability dye eFluor 506 (#65-0866-14, Thermo Fisher Scientific) was used to exclude dead cells. A purified anti-human CD16 antibody (3G8, RRID:AB_314202 BioLegend) was used for blocking assay.
Immunofluorescence analysis by Flow cytomet
Surface molecule expression was measured by flow cytometry. Cells were incubated with fluorochrome-conjugated antibodies for 20 min at 4°C. Data were acquired by a FACS Calibur or FACS Canto II (BD Biosciences) and analyzed with FlowJo software (Tree Star, Ashland, Oregon, USA). We presented mAb-stained cells as percentage of positive cells or mean fluorescence intensity (MFI). Analyzing PD-L1 expression on the surface of tumor cell lines, the MFI values of isotype control vary slightly between cell lines. Therefore, we defined the difference between control and PD-L1-stained cancer cell lines as delta mean fluorescence intensity (ΔMFI) and use high PD-L1 expression according to the ΔMFI rather than the absolute MFI.
In vitro ADCC assay
Various cancer cell lines were used as target cells, including HNSCC and lung cancer cell lines. Standard 51Cr-release and CD107a degranulation assays were performed to evaluate the in vitro ADCC efficacy of three groups: control group, anti-PD-L1 mAb without ADCC (atezolizumab) group, and anti-PD-L1 mAbs with ADCC group (IMC-001 and anti-hPD-L1-hIgG1 (atezolizumab with wild-type FcR binding site, hPD-L1mAb)).
51Chromium-release assay
51Cr-release assays were performed to evaluate cytotoxicity induction of NK-92-CD16 cells via ADCC. The method is described in a previous study.17 Cancer cell lines evaluated as targets are indicated in the figures as appropriate. Target cells (1×106) were labeled with 50 µCi for 1 hour at 37°C, then 2.5×105 cells were washed twice using growth media. Target cells were cultured with 10 µg/mL of the indicated antibodies for 30 min at 37°C and washed again to avoid contact interference between NK and target cells. Target cells were seeded at 5×103 cells/well into U-bottom 96-well plates and coincubated with effector cells at the indicated effector-to-target (E:T) ratios for 4 hour at 37°C in triplicate. Following co-culture, 75 µL supernatant from each well was transferred to 96-well PP 1.2 mL cluster tubes for gamma counting. A WIZARD2 Automatic Gamma Counter (PerkinElmer, Waltham, Massachusetts, USA) was used to calculate radioactivity.
CD107a degranulation assay
CD107a expression is commonly used to determine NK cell functional and cytotoxic activity.18–20 Thus, a CD107a degranulation assay was performed using PBMCs from healthy donors with an E:T ratio of 1:1. PBMC was activated by IL-15 for 3 days. PBMC and target cell densities were adjusted to 2×106 cells/mL. After washing target cells, 5×105 cells were cultured with 10 µg/mL IgG1 isotype control, atezolizumab (anti-PD-L1 mAb without ADCC), or IMC-001 or anti-hPD-L1-hIgG1 (anti-PD-L1 mAbs with ADCC) for 30 min at 37°C and 5% CO2. Cells were then washed with growth media to eliminate excess antibody. For 1:1 E:T ratio, 2×105 cancer cells and PBMCs were incubated for 1 hour at 37°C in U-bottom 96-well plates. Afterwards, 10 µL CD107a antibody was added to each PBMC and incubated for 4 hours at 37°C. Target cells and PBMCs were centrifuged at 15 g for 3 min at room temperature. After incubating for 1 hour, 5 µL/well of GolgiStop solution was added to each well. Whole samples were transferred to FACS tubes then stained with fluorescently labeled mAbs for NK cell surface markers. Stained cells were maintained at 4°C and protected from light until flow cytometry acquisition.
Genotyping of FCGR3A polymorphism (FcγRIIIa-158 V/F polymorphism)
Genotyping was performed using PCR. First, an Exgene Cell SV Kit (Geneall Biotechnology, Seoul, Korea) was used to extract genomic DNA (gDNA) from PBMCs. gDNA was amplified with specific primers (5′-ATA TTT ACA GAA TGG CAC AGG-3′, 5′- GAC TTG GTA CCC AGG TGG AA-3′) and Green 2X premix (Applied Biosystems, Waltham, Massachusetts, USA) using the GeneAmp PCR System (Applied Biosystems). The PCR program consisted of an initial 5 min denaturation step at 95°C followed by 35 cycles for 30 s at 94°C, 30 s at 56°C, 1 min at 72°C, and 7 min at 72°C. Images were captured with the Gel Logic 200 Imaging System (Kodak, Rochester, New York, USA). PCR products were purified using the PCR Purification Kit (Invitrogen, Carlsbad, California, USA) and sequenced by bidirectional Sanger sequencing using specific primers (5′-ATA TTT ACA GAA TGG CAC AGG-3′, 5'-ATG CTG CAG AGT GAA TGA CAC-3').
Live cell imaging for cytotoxicity assay
HN31 cells were seeded on gelatin-coated coverslips and placed in a cell culture incubator for 12 hours to allow cells to adhere and spread on the surfaces. Then, the HN31 cells on coverslips were treated with various antibodies (10 µg/mL) for 30 min in the cell culture incubator, washed three times with cell culture media, and loaded in a magnetic chamber (Chamlide CF, Live Cell Instrument, Korea) for live cell imaging. The magnetic chamber was mounted on a microscope stage equipped with a Chamlide TC incubator system (Live Cell Instrument), which maintains a cell culture condition (37℃, 5% CO2), and NK-92-CD16 cells were added to the chamber. Time-lapse imaging was initiated 15 min after the addition of NK-92-CD16 cells. Differential interference contrast (DIC) images were acquired every 5 min for 4 hour. A modified Olympus IX 83 epifluorescence microscope with a ×40 (UPlanFLN, NA=1.30) objective lens and an ANDOR Zyla 4.2 sCMOS camera was used for imaging experiments. The microscope was automatically controlled by Micro-manager. Acquired images were analyzed and processed with Image J.
Statistical analysis
Data are shown as mean±SD. Data were analyzed by GraphPad Prism software V.7.0 (GraphPad Software, La Jolla, California, USA). Statistical significance in multiple groups was compared by one-way analysis of variance. Paired groups were compared by a paired two-tailed Student’s t-test. Two-sided p<0.05 was considered significant.
Results
PD-L1 expression in human cancer cell lines
To find target cells with high PD-L1 expression, we screened PD-L1 mRNA expression levels in cancer cell lines using the Cancer Cell Line Encyclopedia (https://portals.broadinstitute.org/ccle/)21 database (figure 1A). PD-L1 protein expression at the cell surface in different tumor types and human cancer cell lines was then analyzed by flow cytometry (figure 1B,C). High PD-L1 cell lines were defined by ΔMFI≥5. SNU-1076, FaDu, HN31, and NCI-H1975 cell lines were classified as high PD-L1 cell lines, and SNU-1041, SNU-1066, Detroit 562, and NCI-H1650 cell lines were classified as low PD-L1 cell lines.

Surface PD-L1 expression in various cancer cell lines. (A) PD-L1 mRNA expression in various cancer cell lines was determined by Affymetrix expression data in the Cancer Cell Line Encyclopedia database. Bars are colored by cancer type: DLBCL and RCC. (B) Expression levels of surface PD-L1 measured by flow cytometry. Bar graphs show ΔMFI averaged from three independent experiments. (C) Representative histogram showing isotype control (gray shaded) and staining with an anti-PD-L1 antibody (blue line). Data shown are mean±SD. ΔMFI, delta mean fluorescence intensity; DLBCL, diffuse large B-cell lymphoma; HNSCC, head and neck squamous cell carcinoma; NSCLC, non-small-cell lung cancer; PD-L1; programmed death-ligand 1; RCC, renal cell carcinoma.
Expression of PD-L1 in cancer cell lines is positively correlated with NK-92-CD16 cytotoxicity mediated by anti-PD-L1 mAb for ADCC
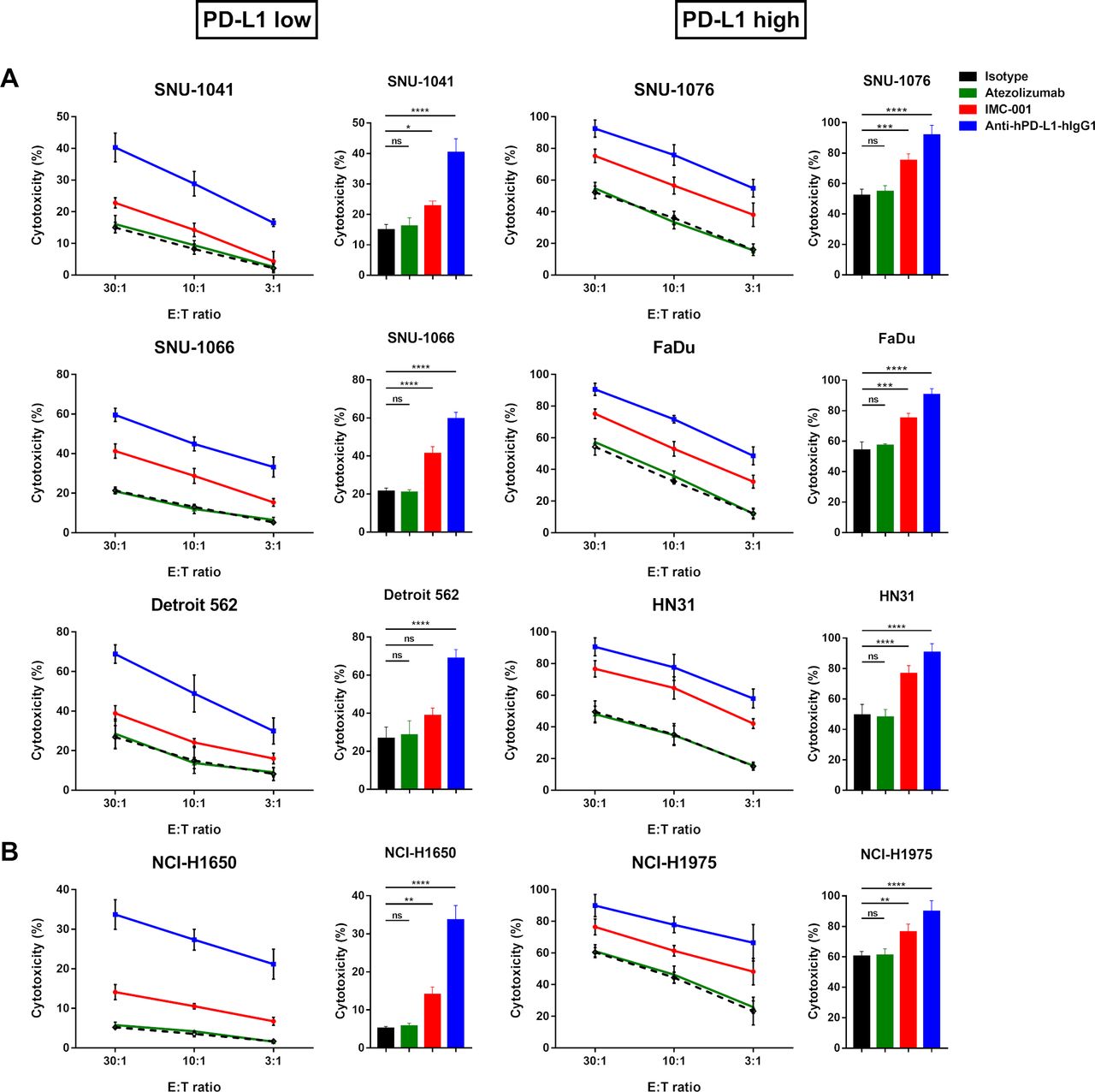
We performed the a standard 51Cr-release assay to measure the ADCC-mediated cytotoxicity of NK-92-CD16 cells against HNSCC (figure 2A) or lung cancer cell lines (figure 2B) in the presence of anti-PD-L1 mAbs. To distinguish NK-92-CD16-induced cytotoxicity based on PD-L1 expression in target cells, HNSCC and NSCLC cell lines were grouped as ‘PD-L1 low’ or ‘PD-L1 high’.

Anti-PD-L1 mAbs-mediated ADCC in human cancer cell lines. NK-92-CD16 cytotoxicity against tumor cells was measured by a standard 51Cr-release assay with various E:T ratios (30:1, 10:1, and 3:1). Bar graph represents cytotoxicity of NK-92-CD16 cells against cancer cell lines at 30:1 E:T ratio. All cancer cell lines were treated with 10 µg/mL of IgG1 isotype control (black dotted lines and bar), atezolizumab (green), IMC-001 (red), and anti-hPD-L1-hIgG1 (blue). (A) Head and neck squamous cell carcinoma cell lines. PD-L1 low (left) includes SNU-1041, SNU-1066, and Detroit 562, and PD-L1 high (right) includes SNU-1076, FaDu, and HN31 cells. (B) For non-small-cell lung cancer, NCI-H1650 cells are PD-L1 low (left), and NCI-H1975 cells are PD-L1 high (right). All experiments were performed three times independently. Statistical significance across groups 6 was determined by one-way analysis of variance. All data are shown as mean±SD. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. ADCC, antibody-dependent cellular cytotoxicity; E:T, effector-to-target; mAb, monoclonal antibody; NK, natural killer; ns, not significant. PD-L1; programmed death-ligand 1.
NK-92-CD16-induced cytotoxicity was greater against PD-L1 high cell lines than PD-L1 low cell lines. In combination with IMC-001, an anti-PD-L1 mAb with preserved ADCC, NK-92-CD16 cytotoxicity was slightly increased in PD-L1 low cell lines, except for Detroit 562 cells, in which IMC-001 had no beneficial effect on ADCC with NK-92-CD16 cells. Interestingly, the other anti-PD-L1 mAb anti-hPD-L1-hIgG1 demonstrated significant cytotoxicity improvement for NK-92-CD16 cells against HNSCC and NSCLC cell lines, irrespective of PD-L1 expression.
To evaluate the contribution of anti-PD-L1 mAbs to ADCC-mediated NK cell cytotoxicity against HNSCC and NSCLC cell lines, we generated two groups: a control group (isotype and atezolizumab) and an anti-PD-L1 mAb with ADCC group (IMC-001 and anti-hPD-L1-hIgG1) (figure 2). For the control group, we found no significant increase in NK-92-CD16 cytotoxicity mediated by ADCC. However, NK-92-CD16-induced cytotoxicity significantly improved in the anti-PD-L1 mAbs with the ADCC group for PD-L1-positive cell lines.
Together, these results imply that PD-L1 expression in cancer cell lines correlated with NK cell cytotoxicity against tumor cells and that even low PD-L1 expression could be targeted for NK-92-CD16 cytotoxicity with anti-PD-L1 mAbs.
Primary human NK cells in combination with anti-PD-L1 mAbs cause ADCC-mediated cytotoxicity in PD-L1-positive cell lines
We confirmed that NK cell-induced cytotoxicity could be enhanced by ADCC using anti-PD-L1 mAbs in PD-L1-positive cancer cell lines. Next, we aimed to extend these findings using primary NK cells in the PBMCs. PBMCs consist of subsets of immune cells such as monocytes, immature neutrophils, macrophages, T cells and B cells, in addition to NK cells. Some of these immune cells can mediate antibody dependent cytotoxicity, such as antibody-dependent cellular phagocytosis (ADCP). ADCP initiated by macrophages must be differentiated from monocytes with macrophage colony-stimulating factor for a week.22 Unlike ADCP, it is known that ADCC classically requires NK cells. Therefore, we used PBMCs but not purified NK cells. The reason why we used PBMCs but not purified NK cells for ADCC assay was that the proportion of NK cells in PBMC is 5-10%, not much. In addition, it is only necessary to confirm the reaction within the same donor-derived NK cells. For NK cells, CD3 and CD56 molecular markers were stained to confirm NK cell-mediated ADCC analysis of NK cells. The gating strategy employed for this study is described in online supplementary figure S2, and the percentage of NK cells in the PBMCs is stated in online supplementary table 1.
Supplemental material
Similar to previous experiments, we evaluated ADCC in vitro using NK cells in combination with anti-PD-L1 mAbs against human cancer cell lines classified by PD-L1 expression level. Figure 3A illustrates NK cell cytotoxicity against PD-L1 low cell lines. For the PD-L1 low cell line NCI-H1650, ADCC sensitivity using IMC-001 was not observed in combination with resting or activated NK cells. However, other PD-L1 low cells, including SNU-1041 and Detroit 562, showed increased ADCC in both resting and activated NK cells induced by IMC-001 and anti-hPD-L1-hIgG1 (figure 3A). Moreover, cytotoxicity induced by NK cells was increased significantly when PD-L1 high cancer cell lines were opsonized with anti-PD-L1 mAbs with preserved ADCC (figure 3B). Interestingly, FaDu cells (high PD-L1) treated with IMC-001 showed no statistical difference in NK cell-induced cytotoxicity when cocultured with resting cells, likely because of biological variation between NK cell donors.

Enhanced cytotoxicity of NK cells against human cancer cells through anti-PD-L1 mAb-mediated ADCC. Cytotoxicity of primary NK cells mediated by anti-PD-L1 mAb ADCC was measured in human head and neck squamous cell carcinoma and non-small-cell lung cancer cell lines using a CD107a degranulation assay. PBMCs from healthy donors were used for effector cells both in resting (black) and activated (blue) states (n=8). PBMCs were cultured overnight or activated with 1 ng/mL interleukin-15 for 3 days. Cancer cell lines were pretreated with anti-PD-L1 mAbs for 30 min. After washing, human cancer cell lines were cocultured with effector cells 1 hour at ratio of 1:1. (A) Results of CD107a degranulation assay in PD-L1 low-expressing and (B) PD-L1 high-expressing cancer cell lines. Statistical significance across groups was determined by one-way analysis of variance. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. ADCC, antibody-dependent cellular cytotoxicity; mAb, monoclonal antibody; NK, natural killer; ns, not significant; ns, not significant; PBMC, peripheral blood mononuclear cell; PD-L1; programmed death-ligand 1.
NK cell-induced cytotoxicity in PD-L1-positive human cancer cell lines treated with anti-hPD-L1-hIgG1 improved regardless of PD-L1 expression and to similar degrees, except for NCI-H1650 (low PD-L1) with resting primary NK cells. Taken together, these findings provide crucial insights into ADCC of activated NK cells and support the previous results. NK cells could enhanced the cytotoxicity through ADCC induced by anti-PD-L1 mAb in PD-L1-positive human cancer cell lines treated with anti-PD-L1-preserved ADCC. The enhanced ADCC of NK cells occurs in a CD16-dependent manner (online supplementary figure S4).
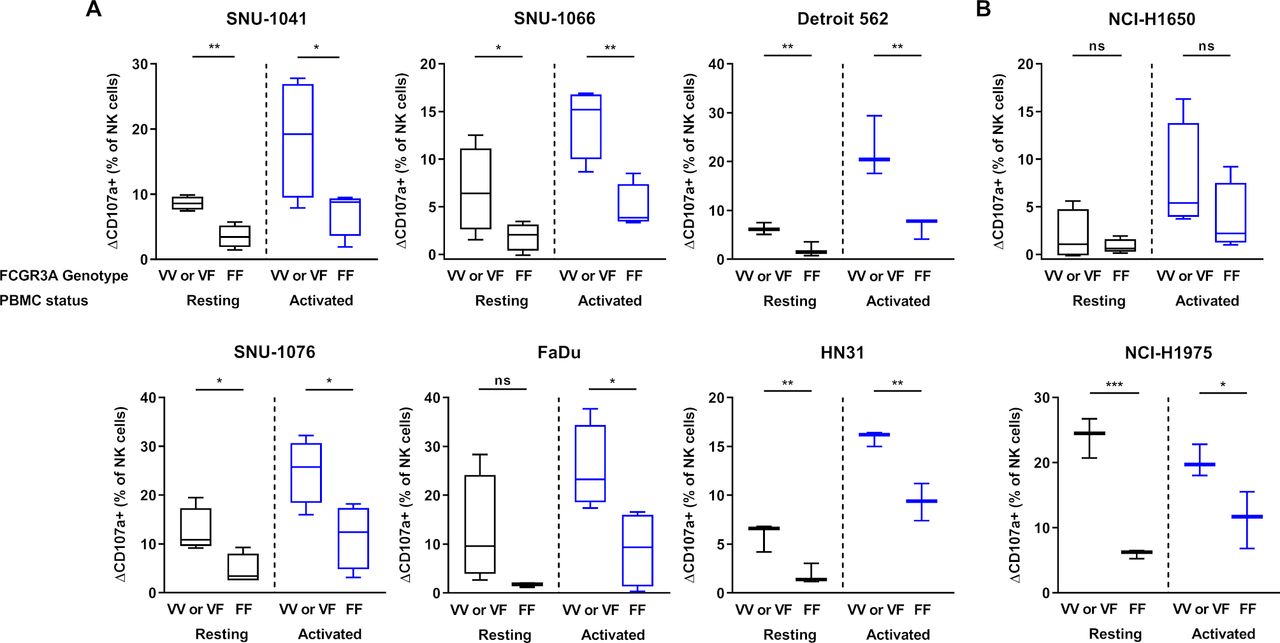
FCGR3A high-affinity (158 V/V) genotype displays higher anti-PD-L1-mediated ADCC lysis of tumor cells than F/F genotype donors
To confirm the effects of NK cells FCGR3A gene polymorphism on ADCC induced by IMC-001, one of the anti-PD-L1 mAb groups, we revisited the results of the CD107a degranulation assays (figure 3). NK cells from six or more healthy donors were genotyped for FCGR3A polymorphism by PCR-based Sanger sequencing. We showed that IMC-001 mediated ADCC with NK cells of FCGR3A genotyped donors (online supplementary figure S3).
We compared the cytotoxicity of NK cells with V/V or V/F donor and F/F genotyped donor toward the HNSCC (figure 4A) or NSCLC cell lines (figure 4B). For the comparison, we defined the difference between control and IMC-001 percent of CD107a positive (CD107a+) NK cells in value as delta CD107a positive (ΔCD107a+ (% of NK cells)). The one-tailed unpaired t-tests were used to determine the significant differences. The figure 4 shows that the IMC-001-mediated ADCC of primary NK cells appeared to be affected by the presence of the FCGR3A high-affinity allele (V/V or V/F genotype; valine (V) phenylalanine (F)).

Effect of FCGR3A polymorphism on IMC-001-mediated antibody-dependent cellular cytotoxicity with NK cells. To compare the cytotoxicity of NK cells between high-affinity genotyped (V/V or V/F genotype) with low affinity genotyped (F/F genotype), we defined y-axis as ΔCD107a+ percentage of NK cells subtracting control from IMC-001 CD107a+ percentage of NK cells. Healthy donors were genotyped for FCGR3A-158 polymorphisms by PCR-based Sanger sequencing (n=6 or 8). Human cancer cell lines were used as targets with high-affinity (n=4; Detroit 562, HN31 and NCI-H1975 n=3) or low-affinity NK cell genotypes. Human (A) head and neck squamous cell carcinoma and (B) non-small-cell lung cancer cell lines classified by expression level of PD-L1 (PD-L1 high-expressing (right) and low-expressing (left) cancer cell lines). The status of primary NK cells are grouped as resting primary (black) and activated (blue) cells. A one-tailed unpaired Student’s t-test was used to compare statistical significance. *P<0.05, **P<0.01, ***P<0.001. NK, natural killer; ns, not significant; PBMC, peripheral blood mononuclear cell; PD-L1; programmed death-ligand 1.
Except for FaDu and NCI-H1650 that did not experience increased ADCC, the cytotoxicity of resting primary NK cells with the FCGR3A high-affinity genotype demonstrated significantly increased ADCC-mediated cytotoxicity. We next assessed the cytotoxicity of NK cells activated by IL-15. The cytotoxicity of FCGR3A high-affinity genotype NK cells which was activated by IL-15 revealed much higher anti-PD-L1 mAb-mediated ADCC lysis of tumor cells than donors with the F/F genotype, except for NCI-H1650 (figure 4A,B).
In summary, these results indicate that primary NK cells with FCGR3A high-affinity allele (V/V or V/F genotype) affected the sensitivity of ADCC through IMC-001. Primary NK cells with the FCGR3A high-affinity genotype showed a more pronounced ADCC increase in the presence of IMC-001.
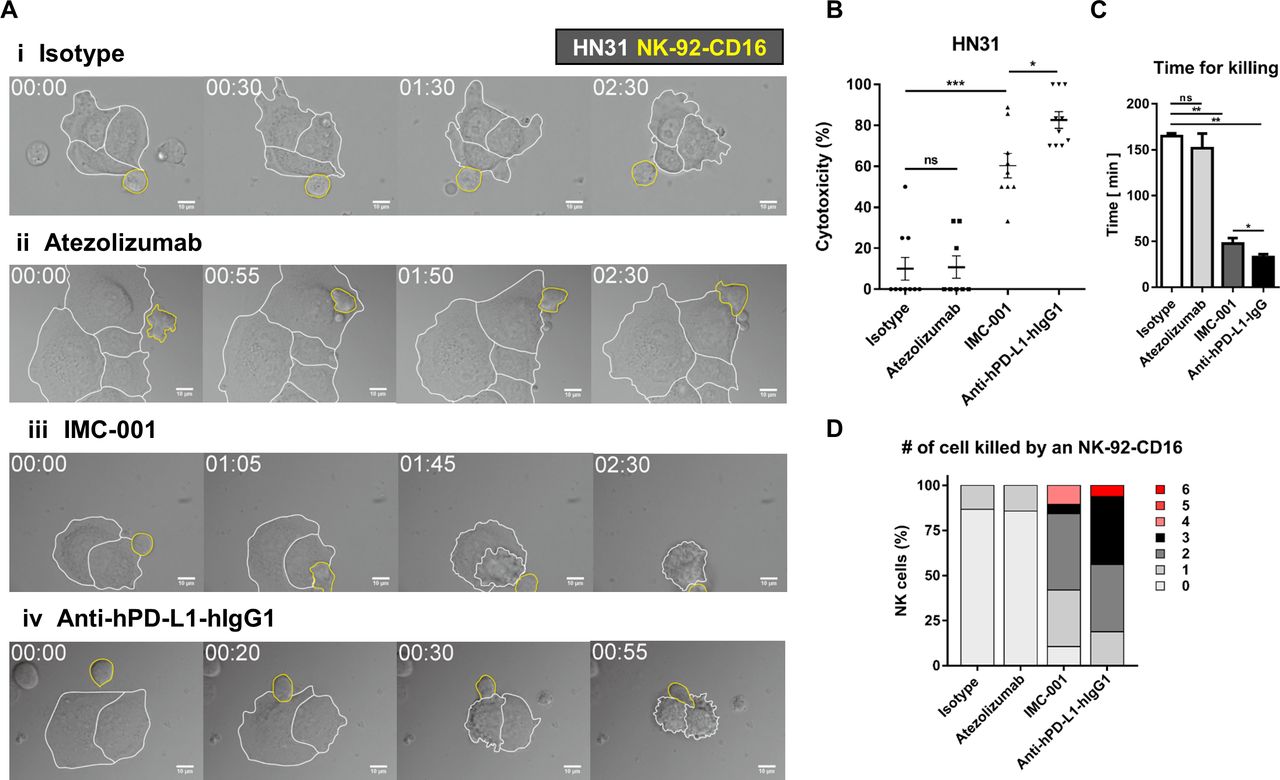
Evaluation the in vitro ADCC efficacy based on live cell imaging
Live cell imaging was performed to directly observe ADCC of NK cells and quantitatively evaluate cytotoxicity in a single-cell level.23 24 NK-92-CD16 cells were added to HN31 cells treated with various antibodies and observed by time-lapse imaging (figure 5A and online supplementary videos S1–4). HN31 cells (marked with white boundaries in figure 5A) and NK-92-CD16 cells (marked with yellow boundaries in figure 5A) exhibited distinct behaviors: HN31 cells spread on the substrates with minimal translocation, whereas NK-92-CD16 cells dynamically migrated and attached on HN31 cells. Some HN31 cells contacted by NK-92-CD16 cells detached from the substrates and rounded, meaning the HN31 cells were killed by the NK-92-CD16 cells.24 Only a fraction of NK cells contacting isotype-treated or atezolizumab-treated cancer cells exerted cytotoxicity, whereas the majority of NK cells contacting IMC-001- or anti-hPD-L1-hIgG1-treated cancer cells did. Of note, NK cells contacting atezolizumab-treated cancer cells undergo extensive morphology changes compared with NK cells contacting isotype-treated cancer cells, indicating NK cells recognized Fc region of atezolizumab but showed no cytotoxicity.
Supplementary video
Supplementary video
Supplementary video
Supplementary video

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Live cell imaging-based analysis of NK cell cytotoxicity. (A) Representative time-lapse images of interaction between NK-92-CD16 cells (yellow lines) and HN-31 cells (white lines) treated with various antibodies. (B–D) Effects of antibody treatment on overall cytotoxicity (B), time for killing (C), and number of cancer cells killed by an NK cell (D). Mann-Whitney test was used. *P<0.05,**P<0.01, ***P<0.001. NK, natural killer; ns, not significant.
Next, we quantitatively analyzed cytotoxicity of NK cells in each case. Overall cytotoxicity was measured by evaluating the percentage of dying/dead HN31 cells throughout the time-lapse imaging (figure 5B). For cancer cells undergoing apoptosis, membrane blebbing proceed detachment from the substrates; thus, cancer cells exhibiting membrane blebbing were counted as a dying cells.23 25 The overall cytotoxicity of atezolizumab group was comparable to that of isotype group. In contrast, the overall cytotoxicity of IMC-001 group was ~5 times higher than that of isotype group, and anti-hPD-L1-hIgG1 group exhibited the highest overall cytotoxicity. These results agree well with the 51Cr-release assay that measures bulk population-level cytotoxicity. To assess cytotoxicity in a single-cell level, time for killing, the duration between NK cell engagement to cancer cells and the onset of cancer cell blebbing, was measured for each NK cell successfully killed cancer cells (figure 5C). For isotype and atezolizumab groups, time for killing was ~150 min, whereas for IMC-001 and anti-hPD-L1-hIgG1 groups, time for killing was <40 min (figure 5C). These results mean ADCC-mediated by anti-PD-L1 accelerate cancer cell killing rate of NK cells. Accelerated tumor cell killing mediated by ADCC frequently lead to serial killing of cancer cells, which means one NK cell kills many cancer cells. To assess serial killing capability of NK cells, the number of cancer cells killed by an NK cell was measured (figure 5D). More than 80% of NK cells in isotype or atezolizumab groups failed to kill any cancer cells or killed 0 cancer cell. In contrast, the majority of NK cells in IMC-001 or anti-hPD-L1-hIgG1 groups killed more than two cancer cells. Of note, some NK cells in an anti-hPD-L1-hIgG1 group killed up to six cancer cells. Taken together, single-cell level cytotoxicity assay revealed that anti-PD-L1-mediated ADCC of NK cells substantially accelerated cancer cell killing and enhanced serial killing.
Discussion
In this study, we demonstrate that NK cells contribute to ADCC-mediated cytotoxicity against PD-L1-positive cancer cell lines when cocultured with anti-PD-L1 mAbs containing preserved FcR binding for ADCC. The present study suggests that NK cells have the potential to improve anti-PD-L1 mAb therapeutic efficacy through ADCC.
Prior studies have queried the ability of NK cells to participate in ADCC through anti-PD-L1 mAb.16 26 The ability of avelumab to mediate ADCC tumor cell lysis has been evaluated,16 and avelumab-mediated ADCC through NK cells was significantly enhanced in triple-negative breast cancer cells.26 Our results are consistent with these previous studies16 26 and confirm the role of ADCC-mediated NK cell cytotoxicity. The current study expands our understanding of ADCC and NK cell cytotoxicity to additional anti-PD-L1 mAbs against diverse PD-L1 expressing cancer cell lines.
Tumor cells often harbor a loss of major histocompatibility complex (MHC) class I molecules on their surface,27 which is considered a primary or acquired immune escape mechanism for PD-1/PD-L1 blockade therapy.28 MHC class I loss has been frequently observed in PD-L1-positive tumors.29 Because NK cell-mediated cytotoxicity can occur in the absence of MHC class I, NK cell therapy in combination with anti-PD-L1 mAbs for ADCC could be a beneficial strategy to overcome resistance.
We assessed the ADCC induction of NK-92-CD16 using51Cr-release assays and evaluated ADCC of primary NK cells by either a CD107a degranulation or 51Cr-release assay. The CD107a degranulation and 51Cr-release assays showed a similar pattern (online supplementary figure S5). In the presence of IMC-001, NK cell-induced cytotoxicity was significantly increased against PD-L1 high-expressing cancer cell lines. In particular, anti-hPD-L1-hIgG treatment showed a remarkable increase in NK cell cytotoxicity, regardless of PD-L1 expression in target cancer cell lines. We also performed live cell imaging and revealed that anti-PD-L1-mediated ADCC of NK cells were enhanced in a single-cell level.
Previous studies have shown a role for FCGR3A polymorphism engagement in avelumab-mediated ADCC.16 However, little research has been conducted to demonstrate the effect of FCGR3A polymorphisms in other anti-PD-L1 mAbs with ADCC in various cancer cell lines. We confirmed that the FCGR3A high-affinity genotype (V/V or V/F) in NK cells correlated with increased ADCC in some human cancer cell lines. As seen in figure 4, NK cells with the FCGR3A high-affinity genotype displayed higher anti-PD-L1 mAb-mediated ADCC than those with the low-affinity F/F genotype. Since the percentage (%) of NK cells in the PBMC were different from each donor, the differences in the activity of ADCC may be interpreted as due to the number of NK cells in the assay but not the activity of NK cells. Additional studies with larger sample sizes and patient homogeneity are needed to confirm this correlation between FCGR3A polymorphisms and NK cell cytotoxicity.
Figure 3 shows the enhanced antitumor effect of primary NK cell through anti-PD-L1 mAbs via ADCC. We found no significant increase in primary NK cell cytotoxicity with IMC-001-treated FaDu (PD-L1 high) or NCI-H1650 (PD-L1 low) cell lines, while cytotoxicity increased for activated NK cells. There are several possible explanations for such a result. First, NCI-H1650 cells have low PD-L1 expression and consequently no interaction with anti-PD-L1 mAbs. Second, NK cell cytotoxicity requires an established and maintained immunological synapse, a dynamic interface for the interaction between NK cells and their target cells.30 31 This interaction could be diverse, depending on the target cancer cell line, causing differing susceptibilities to cytotoxicity. The exceptional result of the NCI-H1650 or FaDu cell line may be due to its low susceptibility to NK cells compared with other cancer cell lines. Third, there are characteristic differences between the NK-92-CD16 cells and primary NK cells as effector cells. 17 NK-92-CD16 cells displayed higher cytotoxicity against cancer cells than primary NK cells because NK-92 cells expressed no KIRs.32–35We confirmed the phenotype of NK-92-CD16 cells, including the expression of CD107a, CD18, CD11a, CD11b, CD11c, CD2, DNAM1, TIGIT, NKG2D, killer immunoglobulin-like receptor (KIR)2DL1,2,3, KIR3DL1, PD-1, PD-L1, 4-1BB, TIM3, CD66a, and CD66 through previous studies that used the same NK-92-CD16 cells as ours. 17 Therefore, the cytotoxicity of NK-92-CD16 is stronger than the primary NK cell against IMC-001-treated cancer cell lines.
In the anti-PD-L1 mAbs with ADCC groups, treated with anti-hPD-L1-IgG Ab mediated ADCC was higher efficacy than IMC-001 (figures 2 and 3). These findings indicate that an anti-hPD-L1-hIgG1 may display a higher binding affinity with NK cells than IMC-001.
One limitation of this study is that the experiments were conducted only in vitro. In the actual tumor microenvironment, many other factors, such as NK cell infiltration and the activity of other immune cells, can contribute to NK cell therapeutic efficacy in solid cancers36 and others. Therefore, in vivo or ex vivo studies are necessary. However, our results provide crucial insights into the potential for NK cells to drive effective ADCC in combination with anti-PD-L1 mAbs. This study suggests that PD-L1 may be meaningful as a target molecule for ADCC and serve as a prognosticator for ADCC as a promising antitumor therapeutic.
In conclusion, NK cells display ADCC-mediated cytotoxic activity against PD-L1-positive cancer cell lines when cocultured with anti-PD-L1 mAbs with preserved ADCC. This study provides rationale for NK cell immunotherapy in PD-L1-positive tumors in combination with ADCC-preserved anti-PD-L1 mAb. Combining NK cell therapy and anti-PD-L1 mAb with preserved ADCC offers a valuable immunotherapeutic strategy for cancers that express high levels of PD-L1 but do not respond to anti-PD-1 or PD-L1 mAbs.
References
Footnotes
Contributors Conception and design: J-EP and BK. Development of methodology: J-EP, S-EK, H-RP, BK, SK and TMK. Acquisition of data (eg, provided cells and provided facility): J-EP, S-EK, H-RP and SK. Analysis and interpretation of data: J-EP, S-EK, BK, H-RP, SK, TMK and JD. Study supervision: SK, BK, MK, TMK, JD, D-WK and DSH. Writing, review and/or revision of the manuscript: all authors.
Funding This study was supported by a grant of the Korea Health Technology R&D Project Strategic Center of Cell and Bio Therapy for Heart, Diabetes & Cancer through the Korea Health Industry Development Institute, funded by the Ministry of Health and Welfare, Republic of Korea (grant number : HI17C2085).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. Contact email: bhumsuk@snu.ac.kr.