Article Text

Abstract
Purpose To evaluate whether administration of the oral DNA hypomethylating agent CC-486 enhances the poor response rate of immunologically ‘cold’ solid tumors to immune checkpoint inhibitor durvalumab.
Experimental design PD-L1/PD-1 inhibitor naïve patients with advanced microsatellite stable colorectal cancer; platinum resistant ovarian cancer; and estrogen receptor positive, HER2 negative breast cancer were enrolled in this single-institution, investigator-initiated trial. Two 28 day regimens, regimen A (CC-486 300 mg QD Days 1–14 (cycles 1–3 only) in combination with durvalumab 1500 mg intravenous day 15) and regimen B (CC-486 100 mg QD days 1–21 (cycle 1 and beyond), vitamin C 500 mg once a day continuously and durvalumab 1500 mg intravenous day 15) were investigated. Patients underwent paired tumor biopsies and serial peripheral blood mononuclear cells (PBMCs) collection for immune-profiling, transcriptomic and epigenomic analyzes.
Results A total of 28 patients were enrolled, 19 patients treated on regimen A and 9 on regimen B. The combination of CC-486 and durvalumab was tolerable. Regimen B, with a lower dose of CC-486 extended over a longer treatment course, showed less grade 3/4 adverse effects. Global LINE-1 methylation assessment of serial PBMCs and genome-wide DNA methylation profile in paired tumor biopsies demonstrated minimal changes in global methylation in both regimens. The lack of robust tumor DNA demethylation was accompanied by an absence of the expected ‘viral mimicry’ inflammatory response, and consequently, no clinical responses were observed. The disease control rate was 7.1%. The median progression-free survival was 1.9 months (95% CI 1.5 to 2.3) and median overall survival was 5 months (95% CI 4.5 to 10).
Conclusions The evaluated treatment schedules of CC-486 in combination with durvalumab did not demonstrate robust pharmacodynamic or clinical activity in selected immunologically cold solid tumors. Lessons learned from this biomarker-rich study should inform continued drug development efforts using these agents.
Trial registration number NCT02811497.
- biomarkers, tumor
- combined modality therapy
- drug therapy, combination
- immunotherapy
- translational medical research
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- biomarkers, tumor
- combined modality therapy
- drug therapy, combination
- immunotherapy
- translational medical research
Background
Immune checkpoint inhibitors (ICIs) are approved for the treatment of many cancers, with the potential for promising durable responses in selected patients. However, objective response rates of these agents are low in most advanced solid tumors.1 Assessment for the presence of tumor-infiltrating lymphocytes (TILs) within the tumor microenvironment (TME) or the invasive margin of the tumor has been suggested to predict clinical response to ICI therapy.2 3 The abundance of TILs may classify a tumor as ‘hot’, T cell inflamed; or ‘cold’, T cell non-inflamed.4 ‘Hot tumors’ are more likely to respond to ICI, thus current efforts are focused on increasing the immunogenicity of ‘cold’ tumors to improve their responsiveness to immune checkpoint blockade.4 5
A potential therapeutic strategy proposed to improve antitumor immune response is the use of DNA hypomethylating agents (HMAs).6 7 The antitumor effects of HMAs have traditionally been attributed to their ability to reactivate aberrantly methylated tumor suppressor genes.8 Recently, our group and others have demonstrated novel antitumor effects of HMAs in activating the expression of repetitive elements such as human endogenous retroviruses (ERVs). This leads to the expression of thousands of previously unannotated transcription start sites and subsequent formation of double stranded RNA (dsRNA), which in turn activates an antiviral inflammatory immune response and thereby creating a state of ‘viral mimicry’.9–11 This ‘viral mimicry’ state is characterized by an increased expression of type I/III interferons and interferon-stimulated genes, triggering recruitment and activation of T cells,12 in line with a previously reported HMA-induced high viral defense signature.13 Moreover, this high viral defense signature expression in melanoma was associated with durable clinical response in patients treated with anti-CTLA4 therapy,9 and synergized with ICI therapy in preclinical epithelial tumor models.9 13–15 Taken together, this body of literature poses a strong rationale to combine HMAs with ICI to improve clinical efficacy.
Furthermore, recent studies demonstrate that the addition of physiological levels of vitamin C to HMAs results in enhancement of this viral mimicry phenomenon, including the upregulation of ERVs in the dsRNA form and the induction of viral defense pathways.16 17 At the molecular level, vitamin C can serve as a cofactor for ten-eleven translocation (TET) enzymes which are involved in active DNA demethylation.17 18 Given that vitamin C deficiency is common in patients with advanced cancers,19 coadministration of physiological levels of vitamin C with HMAs could potentially promote passive and active demethylation17 to enhance clinical response.
In this study, we hypothesized that CC-486 treatment, an oral HMA, would increase the immunogenicity of patients with advanced immunologically ‘cold’ tumors that typically do not respond to ICI monotherapy, specifically microsatellite stable colorectal cancer (MSS-CRC); platinum-resistant ovarian cancer (OC); and estrogen receptor positive, HER2 negative breast cancers (ER+HER2- BC). ICIs have shown minimal activity in patients with CRC and OC with the exception of mismatch repair deficient tumors that represent a small subset of all advanced CRC (3%–5%) and OC (2%–3%), respectively.20–23 The response rate in MMR-proficient or MSS-CRC and OC is minimal, even for high PD-L1 expressing tumors.24 25 Similarly, the overall response rate (ORR) to ICI monotherapy in ER+HER2- BC is modest at 2.8%–12%26 27; this subtype of BC being characterized by no significant immune cell infiltrates and low levels of PD-L1 expression.28–30 With the low expected responses to ICIs in these tumor types, any intervention that enhances immunosensitivity may lead to incremental clinical benefit that can be identified using a single arm study design combining HMA and ICI.
The open-label, phase II basket study of a hypoMEThylating Agent oral azacitidine CC-486 and DURvalumab (MEDI4736; anti-PDL1) in advanced solid tumors (METADUR) study was designed specifically to test whether HMAs can prime ‘cold’ tumors to become ‘hot’ tumors, in order to achieve therapeutic response to ICIs. Low dose CC-486, an oral formulation of azacitidine, was given in combination with an PD-L1 antibody durvalumab. In contrast to azacitidine which is given intravenously or subcutaneously, CC-486 has the advantages of flexible dosing and schedule, with responses demonstrated using extended dosing in hematological malignancies.31 Durvalumab is an approved anti-PD-L1 antibody for maintenance treatment of unresectable non-small cell lung cancer following concurrent chemoradiotherapy, and for previously treated advanced urothelial cancer. In METADUR, a fixed dose of durvalumab was given with two different dosing schedules of CC-486, administered with or without oral supplementation with physiological doses of vitamin C. Peripheral blood mononuclear cells (PBMCs) and tumor biopsies were collected before and after treatment for biomarker evaluations including genome-wide DNA methylation profiling, whole transcriptome analysis, and immune profiling by multiparametric flow cytometry.
We report here the clinical and correlative results of the METADUR trial. Overall, treatment was safe. Two different doses and treatment schedules of CC-486 were tested, however, no robust tumor DNA hypomethylation was observed. Consequently, no activation of the inflammatory ‘viral mimicry’ response, no TME modulation and no clinical activity was observed. Several other clinical trials are currently investigating the potential of HMAs in combination with other drugs, including ICIs. Our results highlight the need to profile tumor DNA methylation pre-HMA and post-HMA treatment in order to proper interpret the clinical results. Finally, encouraged by preclinical studies, drug development efforts to improve pharmacological efficacy of HMAs in solid tumors should continue.
Patients and methods
Study design
This was a single institution, multicohort, investigator-initiated phase II trial of an oral HMA azacitidine (CC-486) and durvalumab in advanced solid tumors (METADUR). The study recruited patients between September 20 2016 and July 16 2018. The data cut-off date was April 30 2019. Primary endpoint was ORR, defined as the proportion of patients with complete response (CR) or partial response (PR) based on Response Evaluation Criteria in Solid Tumors V.1.1 (RECIST 1.1). Secondary endpoints included safety and tolerability, disease control rate (DCR) defined as CR, PR or stable disease (SD) for ≥16 weeks, progression-free survival (PFS) and overall survival (OS).
Patient selection
Patients ≥18 years of age, with histologically or cytologically confirmed advanced MSS-CRC, irrespective of RAS or RAF mutational status (MSS-CRC); platinum resistant OC; or ER+HER2- BC who had progressed on or were intolerant of prior standard therapy were eligible for enrolment. Patients could not have received prior treatment with an ICI or epigenetic modifier such as HMAs or histone deacetylase inhibitors. Other key eligibility requirements were Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, adequate organ and bone marrow function and a life expectancy of ≥12 weeks. Patients were required to have RECIST measurable disease and at least one tumor lesion safely accessible for biopsy. Those with uncontrolled or symptomatic central nervous system metastases were excluded.
Treatment regimen
The initial treatment regimen included oral CC-486 300 mg once daily days 1–14 (cycles 1–3 only) in combination with durvalumab 1500 mg intravenous day 15, in 28 day-cycles (regimen A). However, 300 mg nce daily of CC-486 was poorly tolerated due to gastrointestinal (diarrhea, vomiting and nausea) and hematological toxicity (neutropenia), which are known to be associated with oral CC-486 (table 1).32 33 In preclinical models, lower doses of CC-486 at nanomolar concentrations are sufficient to induce the observed viral mimicry.11 Additionally, recent data suggest addition of vitamin C to HMA treatment protocols can enhance DNA demethylation and viral mimicry through the activation of TET enzymes.17 Therefore, a protocol amendment after the first 19 patients were enrolled included a reduced dose, increased duration, and altered schedule of CC-486 at 100 mg daily days 1–21 (cycle 1 and beyond), supplemented by continuous daily oral vitamin C 500 mg and maintained durvalumab 1500 mg intravenous day 15, in 28 day cycles (regimen B) (online supplementary figure 1). To improve tolerability, patients also received premedication with oral ondansetron 4–8 mg on days of CC-486 dosing in both regimens.
Supplemental material
Related adverse events (AEs) with frequencies ≥10%, by patient
Procedures
Adverse events (AEs) were assessed by Common Terminology Criteria for Adverse Events index V.4.03 and tumor response by RECIST 1.1 every two cycles. To evaluate pharmacodynamic effects, mandatory paired tumor biopsies were carried out at baseline (within 28 days of study treatment) and on treatment at cycle 2 day 12 (±2 days). Serial PBMCs were collected at baseline, day 1 and 15 of each cycle and at the end of treatment for immune-profiling and epigenetic analysis. All blood samples were collected prior to dosing. PD-L1 expression in tumor cells was assessed by immunohistochemistry using VENTANA SP263 assay (Roche Diagnostics), with positivity defined as staining in tumor cells >25%. Immune profiling to assess peripheral immune subsets was performed with multiparametric flow cytometry on serial PBMC samples following CC-486 treatment on cycle 1, day 15 (C1D15) and durvalumab cycle 2, day 15 (C2D15) treatment. DNA methylation status of tumor tissue and PBMCs was assessed using EPIC Methylation Array and LINE-1 methylation assay, respectively.
Sample handling and preparation
All tumor core biopsies and whole blood collected from patients were processed immediately after collection. Fresh core biopsies were pooled and minced into 2–4 mm2 fragments and subjected to enzymatic digestion using the human tumor dissociation kit, as per manufacturer’s direction (Miltenyi, catalog no.130-095-929) and gentle MACS dissociator (Miltenyi, catalog no. 130-093-235). PBMCs were isolated from whole blood by Ficoll density gradient centrifugation. Cell pellets were stored in −80 ºC prior to nucleic acid isolation. Both RNA and DNA were isolated from this frozen material using the AllPrep DNA/RNA Mini Kit (Qiagen, catalog no. 80204), allowing for simultaneous extraction of DNA and RNA from PBMCs or tumor biopsies.
DNA collected from tumor biopsies was used to determine genome wide DNA demethylation measured by Infinium Methylation EPIC BeadChip Kit (Illumina, catalog no. WG-317–1001). DNA collected from PBMCs was used to measure global DNA demethylation induced by CC-486 treatment by LINE-1 methylation assay by pyrosequencing. RNA collected from tumor biopsies was used for total RNA-sequencing to determine global gene expression changes.
LINE-1 bisulfite pyrosequencing
Quantification of DNA methylation levels of global LINE-1 retroelements was performed using pyrosequencing as previously described.34 In brief, a total of 500 ng DNA input was used for each sample with the PyroMark Q24 CpG LINE-1 kit (Qiagen, catalog no. 970012) to quantify methylation level of three CpG sites in positions 331–318 of LINE-1 (GenBank accession number X58075). Demethylation level of each of the three CpG sites were normalized against baseline methylation levels. The mean of all three CpG sites was calculated and plotted (along with the SD) as previously shown.35
DNA methylation profiling and analysis
A total of 250 ng of DNA was bisulfite converted using EZ DNA Methylation Kit (Zymo, catalog no. D5001), following the Illumina manufacturer’s protocol. The purified bisulfite converted DNA was then used as starting material for the Infinium MethylationEPIC bead chip, carried out by the Princess Margaret Genomics Centre, following manufacturer’s protocol. IDAT files for Illumina EPIC BeadChip data were processed using minfi R-package.36 Single sample Noob normalization was performed to yield normalized beta values that were used for further analysis. Limma R-package was used for statistical comparisons.37
Processed data were obtained from EBI array express for a previously published dataset of cell lines that were grown in the presence of azacitidine and profiled at multiple time-points on the Illumina 450 k platform.13 We computed beta-values for samples that had been grown in the presence of azacitidine for 7 days (beta-value >0.3, false discovery rate (FDR) <0.01). On the tumor biopsies, we identified all probes that were highly methylated (beta-value >0.8) at baseline and estimated beta-values for the same probes post-CC-486 treatment on the tumor biopsies.
RNA-sequencing and analysis
Library preparation and sequencing: for all samples, total RNA was quantified by Qubit RNA kit (Thermo Fisher Scientific, catalog no. Q32852) and quality assessed by Agilent Bioanalyzer instrument. All libraries (rRNA depleted, total, single stranded RNA) were prepared using TruSeq Stranded Total RNA Library Prep Gold (Illumina, catalog no. 20020599) following manufacturer’s protocol. All libraries were normalized, pooled together and loaded at 1.4 pM onto an Illumina NextSeq cartridge for cluster generation. Library preparation and sequencing was performed by the Princess Margaret Genomics Center on an Illumina Nextseq500 instrument using paired-end 75 bp protocol to achieve ~80 million paired-reads per sample.
RNA-seq data were processed using Kallisto 38 to yield quantitative estimates of Gencode transcripts and lncRNAs as well as consensus sequence repeats for RepBase repeats. Data were processed using the limma-trend 39 framework to perform all statistical comparisons. Signature scores were summarized using single sample Gene Set Enrichment Analysis scores through the GSVA R-package. Gene set enrichment analysis was performed using test statistics with the fgsea R-package.
Deconvolution was performed using CIBERSORTx 40 with unlogged counts per million whole transcriptome data. The LM22 reference matrix was used to estimate the abundance of 22 cell types, both as a relative fraction and as an absolute fraction, wherein the averages of marker genes for all infiltrating cell types and other cell types are taken into account. Linear modeling was used to examine whether there were concordant shifts from baseline to post-therapy samples for both the values themselves. Further analysis was performed by examining whether the post-therapy versus baseline fold changes varied by regimen to ascertain whether the therapeutic regimen made a difference to any changes in the TME.
Flow cytometry
Fresh PBMCs were stained with immune markers, and data were acquired using a 5-laser LSR Fortessa X-20 (BD, Mississauga, Ontario, Canada). Flow cytometry data were analyzed using FlowJo software V.10.6.1 (Treestar, Ashland, Oregon, USA). For flow cytometry panels, please refer to online supplementary table 2.
Supplemental material
Statistical analysis
Descriptive statistics, such as the mean, median, counts, frequency and proportion, were used to summarize the patient characteristics and AEs. Kaplan-Meier method was used for PFS and OS analysis. Estimated median and its 95% CIs were reported. For flow cytometry data, both paired t-test and non-parametric method were used. Results were consistent between the two methods; paired t-test results are indicated. Statistical analyzes were preformed using R V.3.5.2 (R Core Team 2014, Vienna, Austria).
The original planned sample size was 58 patients; 20 patients in the MSS-CRC cohort, 19 for both PR-OC and ER+HER2- BC cohorts. This sample would allow detection of a 20% difference in ORR from 0.05 to 0.25 in MSS-CRC and from 0.10 to 0.30 in both PR-OC and ER+HER2- BC with greater than 85% power at approximately 0.1 significance level. At least three responders would be required to claim a positive study in MSS-CRC, with at least four responders needed in PR-OC and ER+HER2- BC. Lastly, we observed zero response out of total of 28 patients, the probability of response rate being 0.25 or higher (alternative hypothesis) was calculated to be less than 0.001. Therefore, despite the fact that an interim futility analysis was not preplanned, the lack of antitumor activity seen after 19 patients on regimen A and 9 patients on regimen B led to early termination of this study.
Results
Patient characteristics and treatments
In total, 28 patients were enrolled and treated in the METADUR trial. Nineteen patients were treated on regimen A and nine patients were treated in regimen B. Fifteen patients had MSS-CRC, four PR-OC, and nine ER+HER2- BC. Median age was 56 (range 36–78), 68% were female and 75% ECOG 1. All patients had received ≥3 prior lines of therapy, with a median of 7 (range 3–12) prior systemic therapies, 100% and 64% of patients underwent prior surgery and radiotherapy, respectively. Patient baseline characteristics are shown in table 2 and online supplementary table 1.
Supplemental material
Demographics and patient characteristics
Safety
The most common treatment-related grade 1–2 AEs observed among all patients were fatigue (54%), diarrhea (46%), nausea (46%), vomiting (46%), anorexia (29%) and neutropenia (29%) (table 1). Toxicity was generally comparable between regimens, however the reduced dose and prolonged administration of CC-486 with vitamin C supplementation was associated with reduced hematological toxicity (neutropenia (11% vs 37%) and anemia (0% vs 16%), respectively). Grade ≥3 neutropenia was observed in 18% of patients across all cycles. Only one patient experienced febrile neutropenia. Other grade ≥3 AEs were rare. In regimen A, one patient experienced G3 AST/ALT increase and another patient experienced G3 anemia. In regimen B, one patient had asymptomatic G3 amylase and lipase elevation and two patients had G3 hyponatremia. Twenty-seven patients discontinued treatment due to disease progression, one patient discontinued due to an AE (G4 neutropenia on cycle 1, day 28). No patients required dose reductions of CC-486 in either regimen, however, on regimen A, 5% of patients experienced a 1-week delay on two occasions, 5% had three doses omitted and 16% did not receive durvalumab cycle 2, day 15. On regimen B, one patient did not receive cycle 1, day 15 durvalumab.
Efficacy
The median follow-up time was 4.7 months, median PFS was 1.9 (95% CI 1.5 to 2.3) months, and median OS was 5 (95% CI 4.5 to 10) months. No objective responses were observed as most patients experienced disease progression (online supplementary figure 2A), with a DCR of 7.1% (defined as SD for ≥16 weeks). For the platinum-resistant OC cohort, we measured cancer antigen 125 (CA125) in the blood, and all assessed patients experienced rise in CA125 levels during treatment (online supplementary figure 2B).
Supplemental material
Systemic DNA demethylation by CC-486
To address whether the lack of clinical response was due to low pharmacodynamic activity, we first sought to evaluate whether oral CC-486 in both of our treatment regimens were able to induce systemic DNA hypomethylation in PBMCs. Previous studies have reported correlation between global demethylation in whole blood and clinical response to DNA demethylating agents in myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) patients treated with HMAs.35 We performed global LINE-1 elements methylation assessment as a surrogate for genome-wide changes in DNA methylation.41 42
In most patients, CC-486 induced less than 10% demethylation of genome-wide LINE-1 repetitive sequences (figure 1). Regimen A was able to induce greater LINE-1 demethylation when compared with regimen B. Among the patients treated on regimen A, the maximum LINE-1 demethylation observed was 19.9% for ER+HER2- BC, 13.8% for MSS-CRC, and 9.2% for PR-OC. Among the patients treated on regimen B, the maximum LINE-1 demethylation observed was 6.8% for ER+HER2- BC, 6.6% for MSS-CRC, and 7.2% for PR-OC. Four patients in regimen A and none in regimen B achieved peak demethylation of at least 10% (figure 1). However, these patients did not experience clinical benefit, thus suggesting poor systemic pharmacodynamic activity of CC-486 in our study.
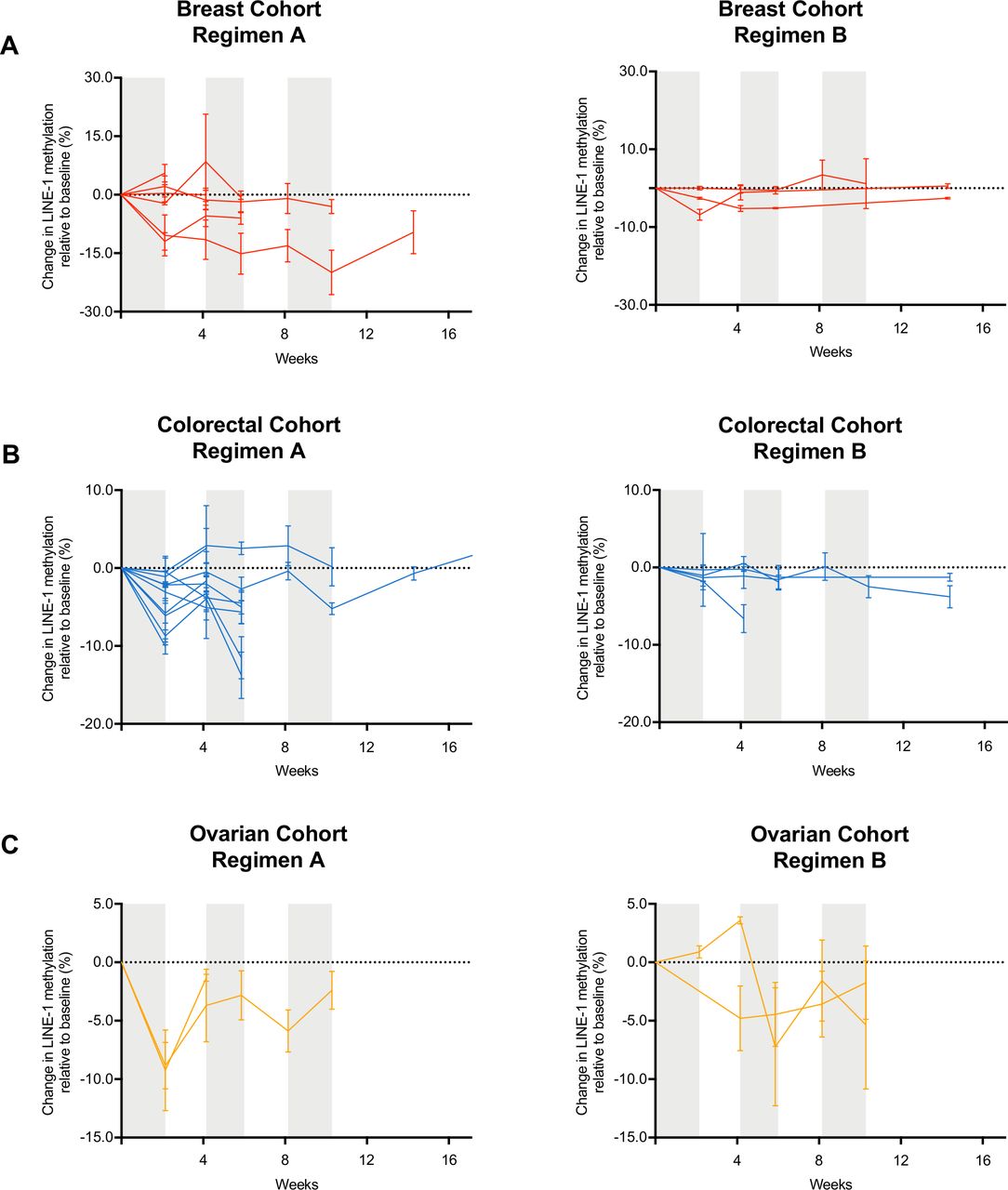
Demethylation levels of LINE-1 elements in PBMCs during CC-486 treatment. Targeted DNA methylation analysis for LINE-1 elements normalized to screening blood sample of (A) estrogen receptor positive HER2 negative breast cancer cohort, (B) microsatellite stable colorectal cancer cohort and (C) platinum-resistant ovarian cancer. Each line represents the serial methylation levels over time for one patient, in each cohort. PBMCs, peripheral blood mononuclear cells.
DNA demethylation levels in tumor biopsies by CC-486
To directly evaluate whether the two oral CC-486 regimens in METADUR were able to induce DNA demethylation in tumor samples, Illumina EPIC methylation array platform was used that surveys around 850 000 CpG sites genome-wide in matched pretreatment/post-treatment pairs. A total of four pairs for regimen A and seven pairs for regimen B was profiled (n=11 patient-matched samples). Most samples showed no or very weak global DNA demethylation (figure 2A, online supplementary figure 3). Altogether, these results suggest that the evaluated regimens of oral CC-486 were not able to induce comparable tumor demethylation as preclinical models, explaining the discrepancy between clinical and preclinical responses.
Supplemental material

Transcriptomic and epigenomic analysis of tumor biopsies during CC-486 treatment. Density distributions of average methylation (beta-values) comparing pre-HMA and post-HMA treatment from (A) cancer cell lines treated with azacitidine, as well as, tumor biopsies from patients that received CC-486 (regimen A) and that received CC-486 + vitamin C (regimen B). (B) Heatmap of fold change in expression of all annotated repetitive elements in the tumor biopsies of all cohorts, and CC-486 regimens. (C) CIBERSORT scores from RNA-sequencing of tumor biopsies of all cohorts, and CC-486 regimens. (D) Fold change of CIBERSORT infiltration scores from tumor biopsies of all cohorts, and CC-486 regimens.
To directly compare the levels of DNA demethylation in preclinical models and the tumor demethylation observed in the METADUR trial, we obtained in vitro demethylation estimates from a large panel of cancer cell lines (n=25 cell lines) grown in the presence of azacitidine and profiled in relation to mock-treated controls from a published source.13 A linear model fit on the delta-beta distribution from the patient biopsies reveal that tumor biopsies from regimen A or regimen B do not exhibit significant demethylation after controlling for baseline methylation levels of each patient, in sharp contrast to the preclinical models (figure 2A, p<2.2e-16).
CC-486 does not induce expression of repetitive elements
Next, we sought to rule out the possibility that despite a lack of robust demethylation in tumor biopsies, CC-486 could still induce the inflammatory ‘viral mimicry’ response. Therefore, we performed total RNA-sequencing on all assessable paired-tumor biopsies from regimen A (n=10 pairs) and regimen B (n=6 pairs). Then, we examined the change in expression of repetitive elements that may play a role in inducing viral mimicry response on preclinical HMA treatment.10 11 43 A simple comparison of baseline versus post-CC-486 treatment transcriptomes revealed there were no consistent changes in the expression of these putative immunogenic repetitive elements (figure 2B). These patterns were highly variable, and no repetitive elements were differentially expressed between the groups or individual patients. Moreover, we observed neither significant activation of anti-viral pathways nor interferon response as previously reported in preclinical models and in clinical samples,9 11 13 44 consistent with our results that tumor DNA demethylation was not achieved in this study (figure 2A, online supplementary figure 3).
Effects of CC-486 and durvalumab on the TME and peripheral immune cell subsets
Finally, we took advantage of the available data and available biospecimen to evaluate whether the combination of CC-486 and durvalumab had an impact on the TME and on peripheral immune cell subsets. Using the same RNA-sequencing data from paired tumor biopsies, we applied CIBERSORT to score the infiltration of 22 immune cell types.40 There were no large-scale changes in either regimen A or Regimen B, with no cell type reaching significance at FDR <0.1 (figure 2C). In addition, the fold change in infiltration scores comparing post-CC-486 samples versus matched baseline samples showed low variability (figure 2D).
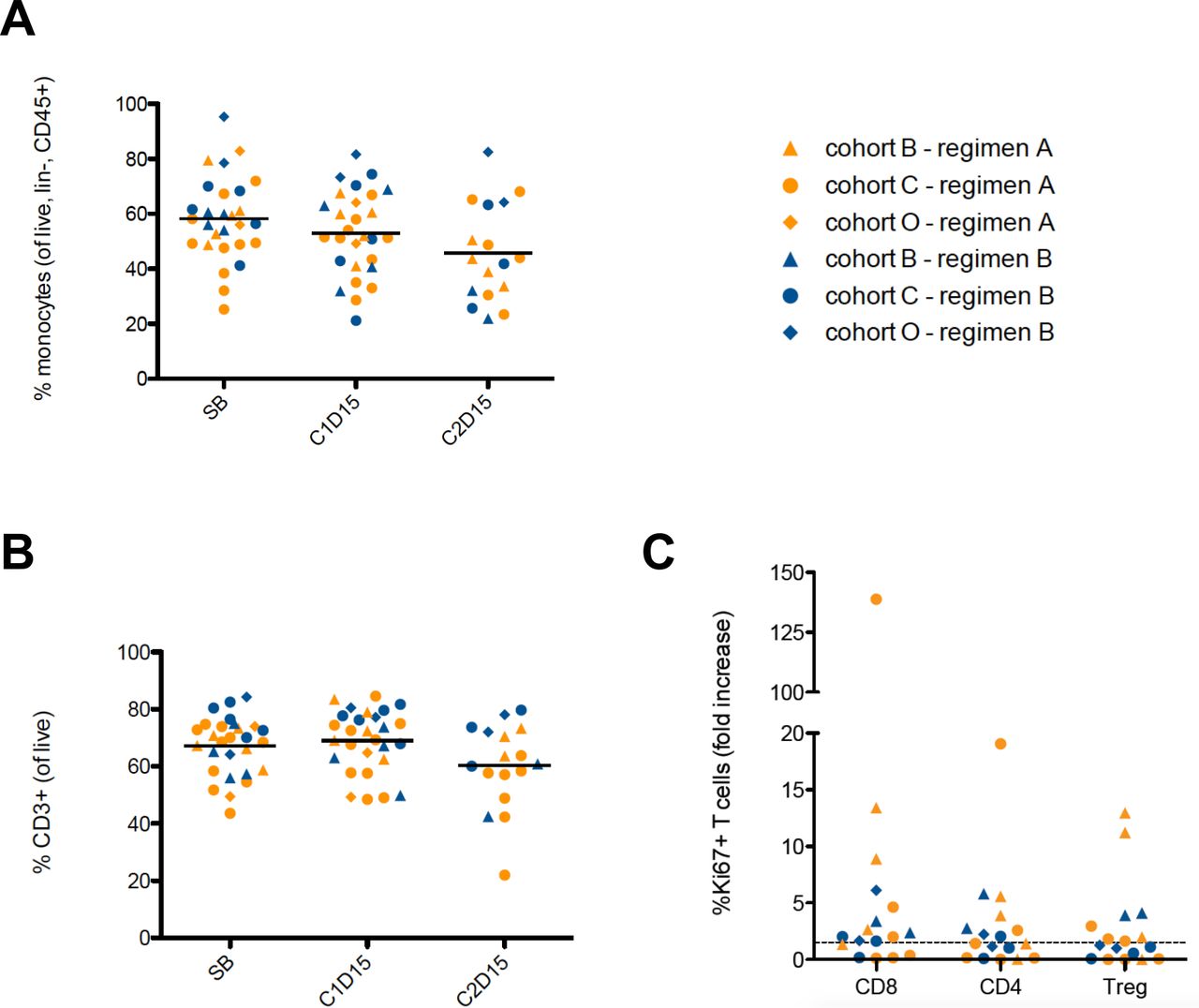
We also performed immunophenotyping by multiparametric flow cytometry (online supplementary table 2) in PBMC samples at baseline, cycle 1 day 15 (C1D15) and cycle 2 day 15 (C2D15) following CC-486 and durvalumab treatment. All evaluable patient samples, across all cohorts and treatment regimens were combined for our analysis. In comparison to baseline and C1D15, we observed a reduction in circulating monocytes (CD33+HLA-DR+CD14+) at C2D15 (p=0.09; vs baseline) (figure 3A), although not statistically significant. Similarly, when compared with baseline and C1D15, we observed a reduction in circulating CD3+ T cells at C2D15 (p=0.01, and p<0.01, respectively) (figure 3B). We did not observe changes in other peripheral immune subsets including CD3+CD4+ T cells, CD3+CD8+ T cells, regulatory T cells (Tregs; CD4+FOXP3+CD25+CD127-), γδ T cells and NK cells following CC-486 and durvalumab treatment online supplementary figure 4. Ki67, a marker of cell proliferation, was assessed in CD4+ and CD8+ T cells and in Tregs. Between C1D15 and C2D15, we observed a minimum 1.5-fold increase of Ki67 expression (figure 3C) in CD4+ T cells in 8/17 patients, in CD8+ T cells in 12/17 patients, and in Tregs of 8/17 patients.
Supplemental material

{kind=link}
{kind=link}
{kind=link}
Flow cytometry on serial PBMCs samples measuring circulating (A) percentage of monocytes (CD33+HLA-DR+CD14+) (p=0.09; C2D15 vs screening blood). (B) Percentage of CD3+ T cells at baseline, C1D15 and C2D15 following CC-486 and durvalumab treatment (p=0.01, C2D15 vs screening blood; and p<0.01, C2D15 vs C1D15). (C) Fold change expression levels of Ki67 expression in CD8+, CD4+ T cells and Tregs following CC-486 and durvalumab treatment. SB=screening blood, baseline. Cohort C represents the microsatellite stable colorectal cancers; cohort O represents the platinum resistant ovarian cancers; cohort B represents the estrogen receptor positive, HER2 negative breast cancers. PBMCs, peripheral blood mononuclear cells.
Discussion
Preclinical studies suggest that HMAs induce powerful antitumor responses through induction of viral mimicry leading to increased immune infiltration and activation of broad immune modulatory pathways. In particular, Gene Set Enrichment Analysis demonstrates significant upregulation of viral defense signature in tumors, which in turn sensitizes it to immune checkpoint therapy in multiple tumor models.9 45 In this investigator-initiated multicohort trial, we sought to test whether a similar viral defense signature would be upregulated in tumor biopsies from patients on treatment with CC-486 leading to increased responsiveness to immune checkpoint inhibition with durvalumab. The lack of response in the first 19 patients and the poor tolerance of CC-486 at 300 mg once daily (days 1–14) led to a protocol amendment to reduce the daily dose but prolong the delivery of CC-486 to 100 mg once daily (days 1–21) (regimen B). The protocol amendment also included the addition of a daily physiological dose of vitamin C, based on the literature evidence in support of its role in promoting passive and active demethylation to potentially enhance clinical response when combined with HMAs.16 17 Deficiencies in vitamin C levels is common in patients with advanced cancers,19 therefore, vitamin C supplementation was also included in regimen B. Overall, no clinically meaningful responses to CC-486 plus durvalumab were observed with either regimen.
The lack of clinical response observed in this study may be due to the lack of relevant levels of DNA demethylation being induced by the combinations used. The current study is unable to confirm or refute whether the inflammatory ‘viral mimicry’ response to HMAs is sufficient to induce anti-tumor response in solid tumors directly or by enhancing response to ICIs.46 This is of stark contrast to human and murine preclinical models. Alternatively, since most in vivo preclinical models use systemic administration of HMAs, it is possible that oral and subcutaneous formulation exhibits poor or no demethylation activity in solid tumors and, therefore, not able to induce the inflammatory ‘viral mimicry’ response. This alternative hypothesis would explain the discrepancy between our current clinical results and the published preclinical results.9 11 13 15 45 47 Also, the patients enrolled in the current study were typical for trial enrollees in early phase clinical trials, with heavy pretreatment history (all patients had >3 prior lines of therapy), such that the threshold for induction of viral mimicry may not be biologically achievable due to therapeutic resistance.
Low demethylation of LINE-1 elements in the PBMCs of assessed patients indicate weak systemic pharmacodynamic activity of CC-486. In a previous trial in AML and MDS patients treated with the second generation HMA, guadecitabine, responders were characterized by a decrease in LINE-1 methylation of more than 10% relative to baseline, and a median of 25% LINE-1 demethylation in peripheral blood.35 In METADUR, the majority of patients did not meet these thresholds. Only four patients exhibited a minimum of 10% LINE-1 demethylation in PMBCs (with none reaching 25% demethylation), with no observed clinical benefit. These results warrant the design of future trials aimed to identify the level of demethylation necessary to induce antitumor response and activation of immune responses in solid tumors. In addition, CC-486 pharmacodynamic activity was not detected at the intended tumor site, even with the supplementation of vitamin C, evidenced by the lack of demethylation measured by EPIC array in tumor biopsies. The lack of pharmacological activity of CC-486 at the tumor site explains the absence of gene expression changes in the viral mimicry signature that were expected and have been consistently demonstrated with HMAs in multiple preclinical9 11 13 45 and clinical studies.44
While we did not find changes in immune composition in the tumor biopsies by in silico analysis, we detected modest changes in the immune composition in PBMC samples by flow cytometry analysis. However, it is worth mentioning that peripheral blood immune phenotype does not necessarily reflect tumor immune phenotype, as recently shown by our group.48 Additionally, the results of serial PBMC samples have some limitations such as small sample size, multiple pathologies, different treatment regimens and the potential effects of CC-486 alone, and in combination with durvalumab. The observed reduction in the proportions of circulating CD14+ monocytes and CD3+ T cells may be due to the cytotoxic effects of CC-486.32 Modest increases in Ki67 expression in T cells in 47% (CD4+ T cells and Tregs) and 70% (CD8+ T cells) of patients following 1 cycle of durvalumab suggests that the addition of a PD-L1 inhibitor—in the presence of CC-486—can lead to increased peripheral T cell proliferation in a subset of patients. However, increased T cell proliferation as measured by Ki67 expression has been previously reported in patients receiving anti-PD-1 monotherapy.49–51 Taken together, our results indicate that the CC-486 regimen tested did not induce sufficient demethylation to induce viral mimicry and alter the immune microenvironment sufficiently to sensitize to a PD-L1 inhibitor.
The rationale of combining HMAs with ICI has been demonstrated in multiple preclinical studies,11 52 53 but the combination of CC-486 and durvalumab did not produce meaningful pharmacodynamic effects or clinical benefits in the current study. Given the lack of demethylation in tumor biopsies, future trials should consider targeted delivery of HMAs with more potent pharmacodynamic activity or the development of novel small molecule inhibitors of DNMT1, with potentially better half-life. The patient cohorts tested in METADUR had immunologically ‘cold’ tumors with a high disease burden, and thus a higher threshold of demethylation by HMAs may need to be reached in order to achieve clinical efficacy. Conversely, patients with earlier stage disease or other tumor types with a more immunologically ‘hot’ microenvironment may derive greater benefit from the combination of HMAs plus ICIs. Nonetheless, the METADUR study contributes to the existing body of knowledge, confirms overall safety and tolerability of this combination and can help inform the design of future studies, where tumor DNA methylation need to be monitored in order to interpret clinical results, and supports drug development efforts to improve pharmacological efficacy of HMAs in solid tumors.
Conclusions
The combination of CC-486 and durvalumab, with or without vitamin C supplementation, is deemed safe at the dosages delivered. However, clinical activity was not observed. Global DNA methylation of LINE-1 elements in peripheral blood cells and genome-wide DNA methylation analysis in tumor biopsies revealed insufficient DNA demethylation. Encouraged by preclinical studies, efforts to improve clinical efficacy of HMAs in solid tumors should continue.
Acknowledgments
The authors would like to acknowledge AstraZeneca Canada Inc. (Ontario, Canada) who provided durvalumab and clinical trial funding and Celgene (New Jersey, USA) who provided CC-486. We would also like to acknowledge the Princess Margaret Cancer Foundation (PMFC), the Schneider Family Foundation and the Ontario Institute for Cancer Research (OICR) for their support of the correlatives studies. We wholeheartedly thank the patients who have made this research possible by participating in the trial.
References
Footnotes
Twitter @kirstt_13, @looloohlemon, @lillian_siu, @decarvalho_lab
KT and HLY contributed equally.
Presented at AACR Annual Meeting 2019; Atlanta, GA Abstract CT190: A Phase II basket study of hypomethylating agent oral cc-486 and durvalumab in advanced solid tumors (METADUR)
Contributors LS and DDC codesigned the clinical trial and the correlative studies. LS supervised the clinical work. DDC supervised the experimental work. KT, HLY, LS and DDC contributed to writing of the manuscript. HLY, SYS and IE contributed to acquisition, and interpretation of data and intellectual content of manuscript. CI provided critical feedback for intellectual content of manuscript. AC performed all bioinformatic analysis. TP contributed to genomic data analysis and interpretation. LW provided biostatistics support. PLB, AAR, ARH, AS, DC, MOB, AMO, SL and NS contributed to enrolment and management of study subjects. BVA contributed to trial data coordination and regulatory operations. SB-H contributed to clinical trial nursing of support for study subjects. BW performed all cytometry experiments and analysis. PO contributed to immune-profiling data analysis and interpretation. All authors participated in review of data and manuscript, and approval of final manuscript.
Funding LS holds the BMO Chair in Precision Cancer Genomics. DDC is supported by the Gattuso-Slaight Personalized Cancer Medicine Fund at the Princess Margaret Cancer Centre. DDC is supported by the Canadian Institutes of Health Research (CIHR PJT – 165986, CIHR FDN 148430 and CIHR New Investigator Salary Award 201512MSH-360794-228629), OICR Ovarian Cancer TRI from the province of Ontario, Canada Research Chair (950-231346) and the Helen M Cooke Professorship from Princess Margaret Cancer Foundation.
Competing interests PLB: Consultant for: Bristol-Myers Squibb (compensated), Sanofi (compensated), Pfizer (compensated). Grant/Research support from (Clinical Trials for aarinstitution): Bristol-Myers Squibb, Sanofi, AstraZeneca, Genentech/Roche, Servier, GlaxoSmithKline, Novartis, SignalChem, PTC Therapeutics, Nektar, Merck, Seattle Genetics, Mersana, Immunomedics, Lilly. AAR: Honoraria: Boehringer Ingelheim. Consultant for: Lilly (compensated), Merck (compensated), Boehringer Ingelheim (compensated). Grant/Research support from (Clinical Trials): CASI Pharmaceuticals, Boehringer Ingelheim, Lilly, Novartis, Deciphera, Karyopharm Therapeutics, Pfizer, Genentech/Roche, Boston Biomedical, Bristol-Myers Squibb, MedImmune, Amgen, GlaxoSmithKline, Blueprint Medicines, Merck, Abbvie, Adaptimmune. ARH: Advisory/Consulting/Research for Genentech/Roche, Merck, GSK, Bristol-Myers Squibb, Novartis, Boston Biomedical, Boehringer-Ingelheim, AstraZeneca, Medimmune. AS: Consultant for (Advisory Board): Merck (compensated), Bristol-Myers Squibb (compensated), Novartis (compensated), Oncorus (compensated), Janssen (compensated). Grant/Research support from (Clinical Trials): Novartis, Bristol-Myers Squibb, Symphogen AstraZeneca/Medimmune, Merck, Bayer, Surface Oncology, Northern Biologics, Janssen Oncology/Johnson & Johnson, Roche, Regeneron, Alkermes, Array Biopharma. DC: Consulting/Advisory: Agendia, AstraZeneca, GSK, Merck, Novartis, Pfizer, Puma, Roche, Dynamo Therapeutics. Grant/Research support from (Clinical Trials): GlaxoSmithKline, Merck, Pfizer. Intellectual Property: Biomarkers for TTK inhibitors (assigned to institution). MOB: Consulting for: Bristol-Myers Squibb, Novartis, Merck, GlaxoSmithKline, EMD Serono, Sanofi, Immunocore. Grant/Research support from (Clinical Trials): Merck, Takara Bio. AMO: Consultant for: AstraZeneca, MERCK, Clovis Oncology, TESARO (all non-compensated). SL: Consultant for: Merck (compensated), AstraZeneca (compensated), GlaxoSmithKline (compensated), Roche (compensated). PO: Consulatnt for: Providence (compensated), Symphogen (compensated), Tessa (compensated), Research support from: EMD Serono. LS: Consultant for: Merck (compensated), Pfizer (compensated), Celgene (compensated), AstraZeneca/Medimmune (compensated), Morphosys (compensated), Roche (compensated), GeneSeeq (compensated), Loxo (compensated), Oncorus (compensated), Symphogen (compensated), Seattle Genetics (compensated), GSK (compensated), Voronoi (compensated), Treadwell Therapeutics (compensated), Arvinas (compensated), Tessa (compensated), Navire (compensated). Grant/Research support from (Clinical Trials for institution): Novartis, Bristol-Myers Squibb, Pfizer, Boerhinger-Ingelheim, GlaxoSmithKline, Roche/Genentech, Karyopharm, AstraZeneca/Medimmune, Merck, Celgene, Astellas, Bayer, Abbvie, Amgen, Symphogen, Intensity Therapeutics, Mirati, Shattucks, Avid. Stockholder in: Agios (spouse). DDC: Research support from Pfizer and Nektar therapeutics.
Patient consent for publication Not required.
Ethics approval The study was approved by the Research Ethics Board at the University Health Network, conducted in accordance with the principles of Good Clinical Practice, the provisions of the Declaration of Helsinki.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. For data availability, GEO access number GSE147537.