Article Text

Abstract
Melanoma treatment has been revolutionized over the past decade. Long-term results with immuno-oncology (I-O) agents and targeted therapies are providing evidence of durable survival for a substantial number of patients. These results have prompted consideration of how best to define long-term benefit and cure. Now more than ever, oncologists should be aware of the long-term outcomes demonstrated with these newer agents and their relevance to treatment decision-making. As the first tumor type for which I-O agents were approved, melanoma has served as a model for other diseases. Accordingly, discussions regarding the value and impact of long-term survival data in patients with melanoma may be relevant in the future to other tumor types. Current findings indicate that, depending on the treatment, over 50% of patients with melanoma may gain durable survival benefit. The best survival outcomes are generally observed in patients with favorable prognostic factors, particularly normal baseline lactate dehydrogenase and/or a low volume of disease. Survival curves from melanoma clinical studies show a plateau at 3 to 4 years, suggesting that patients who are alive at the 3-year landmark (especially in cases in which treatment had been stopped) will likely experience prolonged cancer remission. Quality-of-life and mixture-cure modeling data, as well as metrics such as treatment-free survival, are helping to define the value of this long-term survival. In this review, we describe the current treatment landscape for melanoma and discuss the long-term survival data with immunotherapies and targeted therapies, discussing how to best evaluate the value of long-term survival. We propose that some patients might be considered functionally cured if they have responded to treatment and remained treatment-free for at least 2 years without disease progression. Finally, we consider that, while there have been major advances in the treatment of melanoma in the past decade, there remains a need to improve outcomes for the patients with melanoma who do not experience durable survival.
- immunomodulation
- immunotherapy
- CTLA-4 antigen
- review
- programmed cell death 1 receptor
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Despite intensive efforts, the goal of curing cancer has proven to be elusive for most advanced solid tumors, including melanoma. To achieve this goal, an in-depth understanding of melanoma biology, tumor development, and the body’s innate defense against tumor progression is required. Advances in our understanding of melanoma have facilitated the development and subsequent approval of various novel cancer therapies with the ability to substantially improve outcomes.
Melanoma is the paradigm of how the treatment landscape can be revolutionized over a decade. Since 2011, a series of novel immuno-oncology (I-O) agents, targeted therapies, and combination regimens have been approved.1 Robust data from phase III trials2–7 and subsequent real-world evidence8–10 have unequivocally demonstrated overall survival (OS) and progression-free survival (PFS) benefits with these agents. As the follow-up of pivotal trials continues, a substantial body of long-term data is emerging that supports the durability of survival with some agents.11–17 These data are altering practice in ways almost inconceivable a decade ago, raising new clinical questions and challenges.
In this review, we consider the current treatment landscape for metastatic melanoma, including the following questions: Are patients with sustained benefit actually cured? Can treatment and/or follow-up of these patients be safely stopped, allowing a return to their lives before diagnosis? How is the value of long-term survival best assessed, and can these metrics guide treatment choice? Does the demonstration of durable outcomes for patients treated in the metastatic setting impact the role of local or regional therapies? How can outcomes be improved for patients with melanoma who do not experience durable survival? Because melanoma was the first tumor type for which I-O agents were approved, it has served as a guiding example for other tumors; therefore, these discussions may prove to be informative for the treatment of different tumor types in the future.
Discussion
The evolution of melanoma treatment
Supported by innovative research tools and techniques, our understanding of tumor biology has improved, particularly in regard to the mechanisms tumors use to escape detection by the immune system. Fundamental to greater understanding has been the development of the immunoediting theory, which describes the interaction between the tumor and the immune system as a three-step process (referred to as the three Es): (1) elimination of tumors at an early stage (also known as the immune surveillance hypothesis); (2) equilibrium (when the immune system controls the tumor); and (3) escape (when tumor cells are fully immunoedited and grow without immune control). The three Es are now accepted as a template for understanding the interactions of cancer cells with the host immune system.18 This concept has important therapeutic consequences because it implies that fully developed tumors are inherently immune-resistant. This has shifted our therapeutic strategies away from stimulating the immune system by vaccination, interleukin (IL)-2, or other strategies and toward counteracting immune escape mechanisms. The first agents were designed to block the inhibitory immune checkpoint receptors cytotoxic T-lymphocyte associated antigen-4 (CTLA-4) and programmed death 1 (PD-1). CTLA-4 is expressed on T cells where it acts to regulate the magnitude of the early, priming stages of T-cell activation. Once the T-cell receptor binds to an antigen, cluster of differentiation (CD)28, a co-stimulatory receptor, amplifies the signal to activate the T cells. CD28 and CTLA-4 share ligands (CD80 (B7.1) and CD86 (B7.2)), for which CTLA-4 has a much greater affinity than CD28. Therefore, expression of CTLA-4 on the T-cell surface outcompetes CD28 for ligand binding, delivering inhibitory signals to the T cell and reducing activation.19 Unlike CTLA-4, the primary role of PD-1 is to limit the activity of T cells during the effector phase of the immune response in the peripheral tissue, limiting autoimmunity. The engagement of PD-1 by its ligands, programmed death-ligand 1 (PD-L1) and PD-L2, results in an inhibitory signal that reduces T-cell proliferation, cytotoxicity, and cytokine production.19 The PD-1 and CTLA-4 pathways are distinct, acting at different times and locations to regulate T-cell activation.20 In tumors, inhibiting these pathways has different effects: blocking CTLA-4 induces de novo antitumor T-cell responses, while PD-1 inhibition restores antitumor T-cell function.19–23 These distinct but potentially complementary effects led to the clinical evaluation of immune checkpoint inhibitor-based combinations.
While immune checkpoint inhibitors are now a mainstay of treatment for patients with metastatic melanoma, other therapeutic approaches designed to modulate a patient’s immune system have also been developed. Adoptive cell transfer involves the infusion of autologous tumor-infiltrating lymphocytes that have been expanded in vitro with IL-2.24 Development of this approach began in the 1980s and responses of long duration were first achieved with the use of lymphoid depletion prior to adoptive cell transfer.25 26 Subsequently, various regimens and production processes have subsequently been evaluated in larger clinical trials. Findings from a recent meta-analysis showed that, especially when combined with high-dose IL-2, adoptive cell transfer achieves durable clinical benefit in a substantial minority of patients; however, the complex production process means that this approach is currently used in a small, but expanding number of treatment centers worldwide.24 Vaccination is a strategy to expand tumor-directed lymphocytes in vivo. Melanoma vaccines have a long history, with numerous approaches having been investigated: the majority of studies have evaluated melanoma vaccines used alone and have not demonstrated clinical efficacy.27–30 In 2015, talimogene laherparepvec (TVEC), an oncolytic virotherapy, was approved by the US Food and Drug Administration (FDA) for the local treatment of unresectable lesions in patients with melanoma recurrence after initial surgery (figure 1).31 32 It has been speculated that the presence of this genetically modified virus in the melanoma cells results in an individualized vaccine. Approval was based on the results of a randomized phase III trial showing significantly improved durable response rates with TVEC versus subcutaneous granulocyte-macrophage colony-stimulating factor in patients with unresectable stage IIIB to stage IV melanoma.33 The final analysis from this trial showed a 33% OS rate at 5 years.34 A number of other vaccines are under evaluation for the treatment of melanoma and may play a role in combination therapy with immune checkpoint inhibitors and/or targeted therapies in the future.35

US Food and Drug Administration approval of melanoma therapies.31 32 Agents shown are approved for metastatic melanoma unless stated and italicized. aFor patients whose tumors express BRAF V600E or V600K. bFor patients whose tumors express BRAF V600E.
At the same time while research into immune checkpoint receptors was being pursued, various tumorigenic mutations were discovered. In particular, BRAF-activating mutations were identified in more than 50% of melanomas and found to play a critical role in driving tumor growth and development. This led to the development of vemurafenib, dabrafenib, and encorafenib which target oncogenic activating mutations in the MAP kinase (MAPK) pathway, and trametinib, cobimetinib, and binimetinib, which target MEK.31 36 Clinical experience has demonstrated that the majority of patients treated with a BRAF inhibitor alone developed resistant disease, primarily involving reactivation of the MAPK pathway.3 Some patients also developed secondary malignancies, such as cutaneous squamous-cell carcinoma, attributed to paradoxical BRAF-inhibitor-induced activation of the MAPK pathway in RAS-mutant tumors. Further research has shown that combined BRAF and MEK inhibition delayed the emergence of resistance and reduced the incidence of cutaneous hyperproliferative lesions compared with single-agent BRAF inhibition, which led to the clinical evaluation of combination regimens that demonstrated improved therapeutic efficacy compared with monotherapy.3
In 2011, the FDA approved the first immune checkpoint inhibitor, ipilimumab, and the first targeted therapy, vemurafenib, for melanoma.31 Subsequently, several other agents have been approved, based on the demonstration of significant OS benefit in randomized trials.1 31 These agents were initially approved for use as monotherapy (figure 1);31 32 however, strong scientific and clinical rationale for combination therapy resulted in the subsequent approval of checkpoint-inhibitor-based and targeted therapy-based combination regimens. More recently, several adjuvant treatments for patients with fully resected stage III or stage IV disease have been approved (figure 1).31 37–40
Metastatic melanoma treatment today
The newer therapies for metastatic melanoma have undoubtedly improved outcomes for many patients, but they have also created new treatment selection challenges for both patients and oncologists. This is particularly true for patients with BRAF-mutant melanoma, for which both I-O agents and targeted therapies are approved.
For patients with BRAF-mutant melanoma, robust data have demonstrated a significant benefit for combination targeted therapy over BRAF-inhibitor monotherapy; so, when a targeted therapy is chosen, combination BRAF/MEK inhibitor treatment is preferred for most patients.3 4 6 7 41 When considering treatment with an I-O agent, note that data from randomized trials has clearly shown that a PD-1 inhibitor is superior to ipilimumab (a CTLA-4 inhibitor) in the first-line setting in terms of both efficacy and safety, regardless of tumor BRAF-mutation status.2 5 41 42 In the absence of head-to-head studies, the choice between the approved PD-1 inhibitors (nivolumab or pembrolizumab) is often based on factors such as treatment schedule, institutional formulary options, and the preference of the treating oncologist. However, as long-term data become available, they may also inform decision-making (table 1).5 11–17 25 43–46
Long-term survival in clinical trials with I-O agents or targeted therapies in patients with advanced/metastatic melanoma
When deciding whether to prescribe nivolumab plus ipilimumab combination therapy or PD-1–inhibitor monotherapy, consider that descriptive data from the CheckMate 067 trial showed superior median OS with the combination versus nivolumab alone (>60 months (median not reached) versus 36.9 months; HR 0.83); this difference was maintained with up to 5 years of follow-up.12 The HR for benefit with the combination over nivolumab was particularly robust in patients with BRAF-mutant disease (0.70; 5-year OS rate 60% vs 46%), elevated (ie, two times the upper limit of normal) lactate dehydrogenase (LDH; 0.73; 5-year OS rate 28% vs 14%), or M1c stage per American Joint Committee on Cancer, seventh edition (0.82; 5-year OS rate 43% vs 35%), respectively. The frequency and severity of treatment-related adverse events (AEs), however, was higher with the combination than with nivolumab alone (any grade 96% vs 87%; grade 3/4 59% vs 23%).12 Consequently, the treatment decision is complex and multifactorial, involving careful consideration and discussion about the risks and potential benefits of the treatment options and, in our experience, with patient preference often being an important factor.
The choice between I-O and targeted therapy for treatment-naive patients with BRAF-mutant disease is difficult because of the lack of data from head-to-head trials. Most clinicians rely on practical experience, real-world and trial data, and expert-opinion publications. However, health economic/quality-of-life (QoL) data and outcomes from other analyzes, such as data modeling, may help to inform decision-making. For example, in a recent matching-adjusted indirect comparison, clinical outcomes with nivolumab plus ipilimumab were compared with dabrafenib plus trametinib or vemurafenib plus cobimetinib combination treatments in patients with BRAF-mutant advanced melanoma.47 After adjusting for differences in baseline characteristics, PFS and response outcomes were similar among the treatments; but, beyond 12 months of treatment, nivolumab plus ipilimumab improved OS compared with targeted therapy. This suggests that the survival benefit with combination immunotherapy emerges later and may be more durable than with targeted therapies. The incidences of AEs (any-grade, grade 3/4) were lowest with dabrafenib plus trametinib (89.5%, 31.6%) and similar between vemurafenib plus cobimetinib (97.6%, 59.5%) and nivolumab plus ipilimumab (94.6%, 54.1%), although the nature of the toxicities is very different between immune checkpoint inhibitors and targeted therapies.48
Several randomized trials have been ongoing to determine the most effective sequence and administration of I-O and targeted therapies for all comers and biomarker-defined subsets of treatment-naive patients with BRAF-mutant melanoma.49–51 The randomized phase II SECOMBIT trial (NCT02631447)49 was designed to determine the best sequential approach with ipilimumab plus nivolumab and encorafenib plus binimetinib. In the phase III DREAMseq trial (NCT02224781),50 patients will be receiving ipilimumab and nivolumab followed by dabrafenib and trametinib or the reverse sequence at the time of documented disease progression. The phase II EBIN trial (NCT03235245)51 was designed to compare encorafenib plus binimetinib followed by nivolumab plus ipilimumab versus nivolumab plus ipilimumab. The phase II ImmunoCobiVem trial (NCT02902029)52 was designed to evaluate cobimetinib plus vemurafenib followed by a programmed death ligand 1 (PD-L1) inhibitor.
Long-term survival outcomes
As patients are followed for 5 years and beyond in the pivotal trials with I-O agents and targeted therapies, long-term data are becoming available and provide new metrics that can contribute to treatment decisions. Available long-term data are summarized in table 1 and figure 2.5 11–17 25 43–46 Because these studies differed in their patient populations and other parameters, it is not appropriate to compare the trials directly. Additionally, different trial start dates may have impacted the patient populations enrolled and the subsequent therapies received. However, when assessing IL-2 or ipilimumab treatment, the improvement in clinical outcomes in patients treated with PD-1-inhibitor monotherapy, targeted therapy, or nivolumab plus ipilimumab becomes clear.
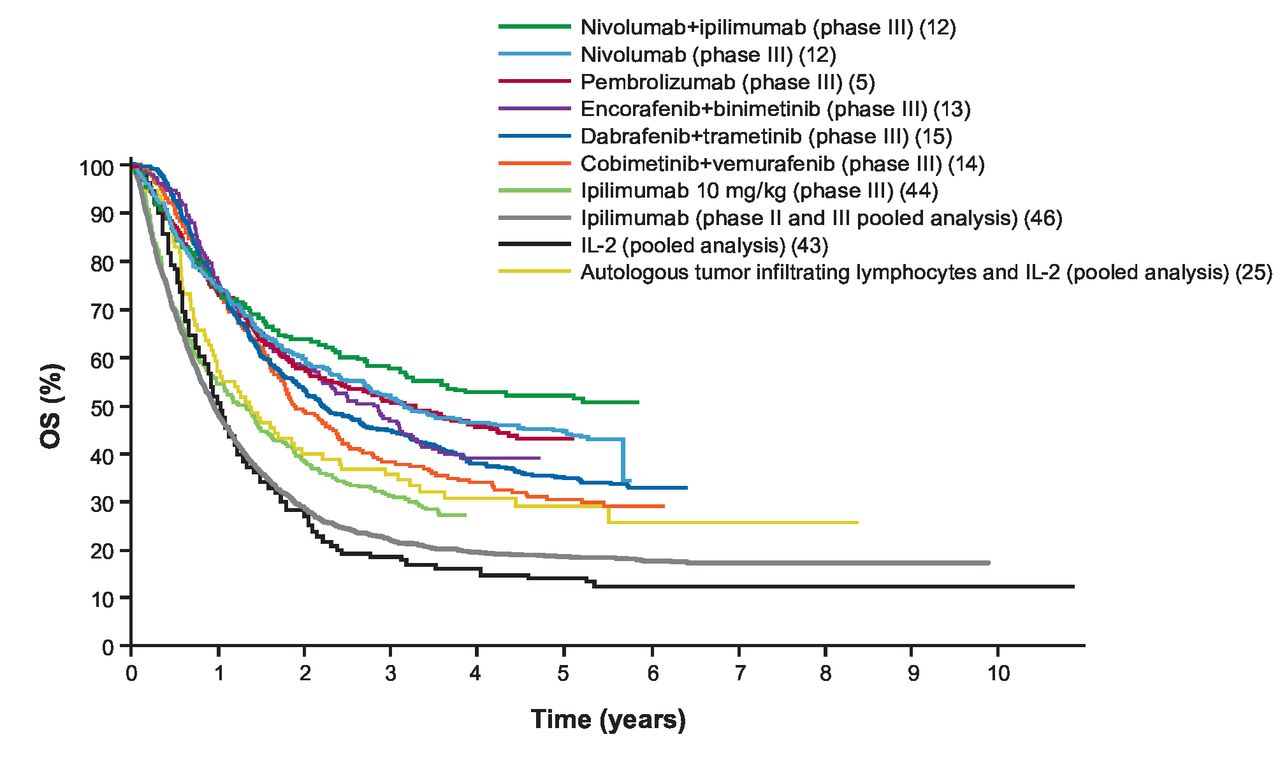
Long-term OS in clinical trials with immuno-oncology agents and targeted therapies in patients with advanced melanoma. The data presented represent first-line treatment options, with the exception of those for IL-2 and pooled ipilimumab. The data were compiled from the references indicated. IL-2, interleukin 2; OS, overall survival.
Across studies, the best survival outcomes have generally been observed in patients with favorable prognostic factors, particularly a normal or low LDH level and/or a low volume of disease. In a pooled survival analysis of dabrafenib and trametinib (in patients with BRAF-mutant tumors), the 5-year survival rate was 43% in patients with LDH at or below the upper limit of normal versus 34% in the general population.15 The highest OS rate, 55%, was found in patients with low/normal LDH and fewer than three sites of disease. In CheckMate 067, there was a similar trend for better survival in patients with favorable prognostic factors: the highest 5-year survival rates with nivolumab plus ipilimumab were observed in patients with normal LDH (60%) or normal LDH with fewer than three sites of disease (64%), compared with 52% in all patients (the 5-year survival rate was 60% for patients with BRAF-mutant tumors).12
When long-term data with immune checkpoint inhibitors became available in a pooled analysis of ipilimumab trials in which some patients were followed for more than 10 years,46 attention was drawn to the shape of the OS curves, specifically the plateau or flattening of the tail of the curve. As shown in figure 2, the immune checkpoint inhibitor and IL-2 curves were similar, plateauing at 3 or 4 years. The OS curves for targeted therapies also appeared to plateau. However, the contributions of patients receiving a subsequent therapy were unclear. Based on current data, the plateaus for the I-O and the targeted agents are expected to be sustained with longer follow-up. This leads to the question of whether the patients represented in the tails of these curves can be considered to be functionally cured.
OS curves are known to be impacted by subsequent therapies. Many patients treated with BRAF plus MEK inhibitor combinations who are still alive may have received, or are still receiving, subsequent therapy with immune checkpoint inhibitors; patients in immune checkpoint inhibitor trials may have received subsequent BRAF plus MEK inhibitor combinations. In the pooled analysis of the COMBI-d and COMBI-v trials, of the 161 patients who were alive at 5 years, 72 (45%) had received further treatment, the most common being immunotherapy (52 patients, 78%).15 In the CheckMate 067 trial, of the 281 patients in the nivolumab-containing arms who were alive at 5 years, 58 (21%) had received subsequent therapy.12 Data were similar for patients with BRAF-mutant melanoma: of the 97 patients who were alive at 5 years, 25 (26%) had received subsequent therapy.53 To fully evaluate the contribution of an agent to OS, it may be helpful to consider the tail of the PFS curve. PFS offers an alternative to OS that is available in a timely manner and is not confounded by subsequent therapies or causes of mortality unrelated to cancer.54 55 In CheckMate 067 and the COMBI-d and COMBI-v pooled analyzes, the PFS curves appeared to flatten after approximately 3 years. In CheckMate 067, PFS rates for the overall population at 3, 4, and 5 years were 39%, 37%, and 36% with nivolumab plus ipilimumab, and 32%, 31%, and 29% with nivolumab alone.12 56 57 PFS rates for patients with BRAF-mutant melanoma at 3, 4, and 5 years were 40%, 39%, and 38% with nivolumab plus ipilimumab, and 22%, 23%, and 22% with nivolumab alone. In COMBI-d and COMBI-v, PFS rates were 24% at 3 years, 21% at 4 years, and 19% at 5 years.15 In the KEYNOTE-006 trial, PFS rates for patients who received either dose of pembrolizumab as a first-line therapy declined from 37% at 2 years, to 33% at 3 years, and 27% at 4 years; the lack of data beyond 4 years makes it unclear whether the curve continued to flatten.5
Defining cure in advanced cancer
Defining cure has not been necessary for most advanced cancers. In 1985, Schipper and colleagues proposed the term ‘functional cure,’ acknowledging that, given the biology of cancer, eradicating all cancer cells or proving that they had been eradicated may not be possible, and seeking to gain long-term control may be more realistic.58 Based on current long-term data, patients could be regarded as functionally cured after 3 or 4 years when the OS curve plateaus; however, at that time, depending on the agent, some patients may still be receiving treatment. For example, in CheckMate 067, stopping nivolumab after a specific time period had not been mandated and, at 5 years, 13% of the patients in the nivolumab-containing arms who were alive were still receiving study treatment. However, the median duration of treatment was short in both arms (3.6 months (range, 0.0 to 57.0) with nivolumab plus ipilimumab combination therapy and 7.6 months (range, 0.0 to 62.9) with nivolumab monotherapy).59 In contrast, in the COMBI-d and COMBI-v pooled analysis, at 5 years, 43% of the patients who were alive were still on study treatment.15 Furthermore, one-half of the patients with complete response (CR) on these medications were shown to relapse at median of 6.6 months after treatment was stopped.60 Findings from a retrospective analysis of the long-term outcomes of 396 patients treated with a PD-1 inhibitor at a single institution showed that most patients discontinued treatment at the time of CR. Most CRs were durable, but the probability of treatment failure 3 years after best overall response was 27%.61
The appropriate time to stop treatment has been the subject of much debate, particularly because different agents were developed with different stopping rules. For example, in the pembrolizumab KEYNOTE-006 trial, patients received up to 2 years of treatment5; whereas, in the CheckMate 067 trial, patients continued until progression, unacceptable toxicity, or withdrawal of consent.57 If a maximum duration of therapy has not been mandated, the decision to stop may be supported by a biopsy showing either the absence of viable tumor or negative 18F-2-fluoro-2-deoxy-d-glucose-positron emission tomography (FDG-PET) imaging. An FDG-PET scan, which provides functional information alongside standard anatomical imaging, may help to determine whether residual lesions in patients with a partial response (PR) or stable disease (SD) contain any viable tumor, thereby identifying patients achieving a complete metabolic response.62 Results of a study designed to determine whether FDG-PET imaging is a better predictor of long-term outcomes than a standard CT scan showed that, after 1 year of immune checkpoint inhibitor treatment, 28% of patients had achieved CR on CT according to Response Evaluation Criteria in Solid Tumors V.1.1.62 In comparison, 68% of patients with a PR on CT had a complete metabolic response according to FDG-PET imaging. Almost all of the patients with a complete metabolic response at 1 year had an ongoing response to therapy thereafter. Acknowledging that longer follow-up is needed, these findings suggest that FDG-PET imaging could help to predict long-term survival and guide the decision to stop therapy. Other studies are planned to investigate the ideal duration of therapy for a specific subset of patients. The subject of how long to treat patients requires data, discussion, and development of best-practice guidelines, along with the identification of surrogate markers that indicate when stopping treatment is safe.
A further consideration is whether a documented radiologic CR is necessary for a patient to be considered functionally cured. Given the concept of a functional cure and the mechanism of action of I-O agents in particular, the long-term absence of progression rather than the documentation of a CR may be a more appropriate metric. Long-term data suggest that achieving a CR is not necessary for a sustained survival benefit, although patients who achieve a CR have more durable benefit over time.15 53 63 In the COMBI-d and COMBI-v pooled analysis, the 5-year OS rates were 71% in patients with a CR, 32% in patients with a PR, and 16% in patients with SD.15 In comparison, the 5-year OS rates in CheckMate 067 were 90%, 65%, and 24% with nivolumab plus ipilimumab combination therapy and 93%, 63%, and 40% with nivolumab monotherapy.53 Data obtained with pembrolizumab in a pooled analysis of KEYNOTE-001 and KEYNOTE-006 showed a similar trend, with 4-year OS rates of 98%, 75%, and 66% in patients with a CR, PR, or SD at week 24, respectively.64
After taking into consideration both the data above and our clinical experience, we propose that patients diagnosed with advanced melanoma may be considered functionally cured if they have responded to treatment and have been off treatment for at least 2 years without disease progression. In our opinion, a patient still receiving treatment cannot be considered functionally cured. Furthermore, while a patient is still on treatment, it cannot be determined whether ongoing treatment is necessary to control their disease.
Evaluating the value of long-term survival
As these data for melanoma treatments continue to mature, landmark survival rates at 5 and 10 years will become increasingly relevant. It is also becoming important to understand what long-term survival means to patients, physicians, payers, and other healthcare professionals. In this regard, new analytical approaches may help to describe long-term survival and inform decision-making. Treatment-free survival (TFS) and treatment-free interval (TFI) are metrics that may be considered alongside traditional efficacy measures. TFS characterizes the proportion of time without both treatment or toxicity on a particular regimen; it can be further separated into time with, and time without treatment-related toxicity of a specified grade.65 TFI captures the time interval from the last dose of study drug to subsequent systemic therapy.12 For example, data suggest that TFI positively impacts QoL and may improve the cost-effectiveness of immune checkpoint inhibitors.66 67 TFI data may help to determine the optimal duration of treatment. In CheckMate 067, the median TFI for patients receiving nivolumab plus ipilimumab combination therapy was 18.1 months compared with 1.8 months with nivolumab monotherapy.12 These data indicate that a short duration of highly active combination therapy may be sufficient for many patients to achieve durable tumor control without requiring ongoing treatment. Additionally, survival at 5 years was similar in patients who discontinued combination therapy due to AEs during the induction phase and in the overall population.12 Emerging data from studies in the neoadjuvant setting also suggest that a short course of active therapy may be sufficient to achieve durable disease control.68 69
The quality of prolonged survival is a potentially important relative determinant of value. The 5-year results from CheckMate 067 showed that QoL was maintained during the TFI after discontinuation of treatment with nivolumab plus ipilimumab combination therapy or nivolumab monotherapy.12 Another analysis (Q-TWiST) provided evidence that longer quality-adjusted time without symptoms or toxicity is attainable for patients treated with nivolumab plus ipilimumab than for those treated with ipilimumab alone.70 QoL data for patients with metastatic melanoma are available from key studies with pembrolizumab71 and dabrafenib plus trametinib,72 73 but the analysis period to date is limited to 2 years after treatment initiation.
For cancer therapies yielding long-term survival and hence functional cures in a proportion of patients, traditional statistical analyzes may not accurately estimate OS due to heterogeneity in patient outcomes. Mixture-cure models can be used to capture survival heterogeneity among patients by assuming that a certain proportion of patients are ‘cured’; the mortality risk among these patients is considered to be the same as that of the general population. Mixture-cure models have been used in various cancer settings, including melanoma in the adjuvant setting, to complement standard survival analyzes. By assuming that a proportion of patients in a study will never experience an event of interest, these models allow a more accurate quantification of the proportion of patients who gain a long-term survival benefit (ie, are cured).74 75 Determining the ‘cure fraction’ with agents in the metastatic setting may provide another valuable comparative metric.76 77
Opportunities to improve survival in more patients with metastatic melanoma
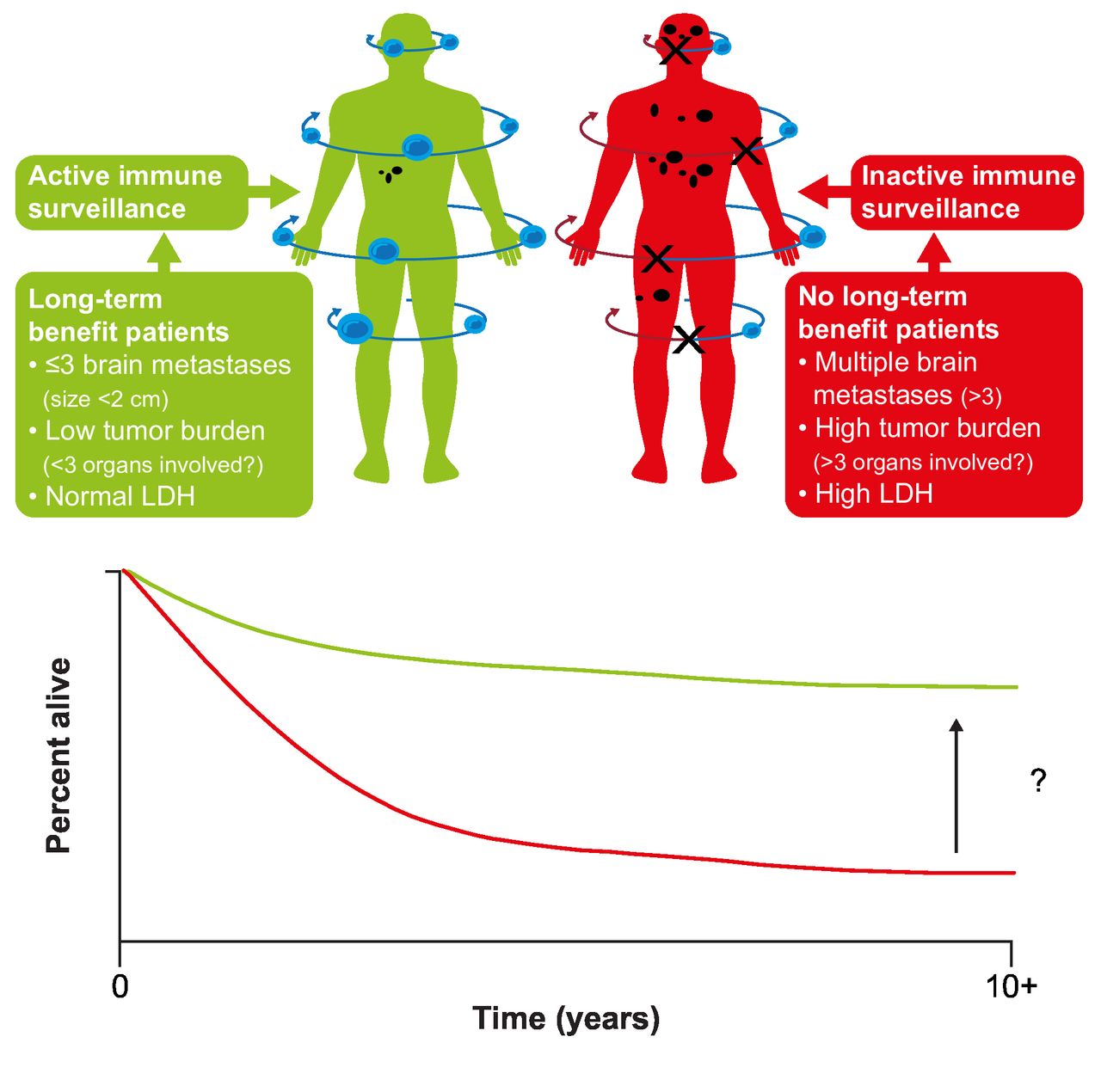
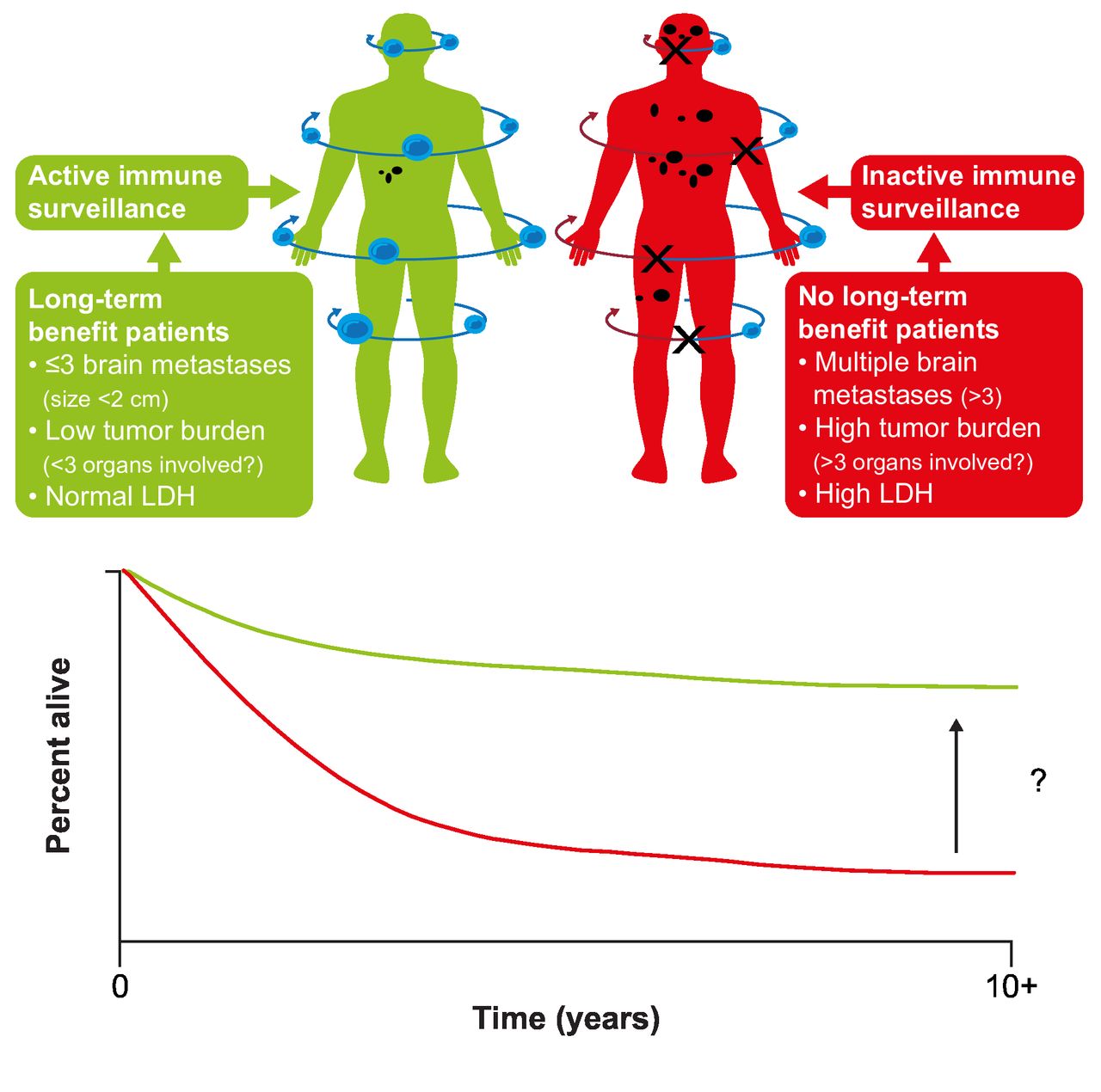
While some patients achieve long-term survival, improving outcomes for those who do not remains an important goal (figure 3).78 One approach is to identify biomarkers or characteristics that identify the patients most likely to achieve long-term survival. Beyond established prognostic factors such as baseline LDH and disease burden/sites of disease, analyzes of data from key phase III studies have failed to identify reliable predictors of poor survival.12 15 79 Analysis of long-term data from a phase I study of nivolumab in patients with advanced solid tumors, including melanoma, suggested that higher absolute lymphocyte count or a higher number of circulating regulatory T cells at baseline was related to survival at 5 years, warranting further evaluation.17 Identifying predictive biomarkers remains a focus of research; potential biomarkers under evaluation include tumor mutational burden and neoantigen expression, T-cell receptor sequencing, immune-related gene expression, and tumor T-cell infiltration.79 Other opportunities to improve long-term survival include identifying effective adjuvant treatments for patients with early-stage disease, improving the efficacy of adjuvant treatment in high-risk patients (perhaps through treatment in the neoadjuvant setting or with novel combinations), and the development of treatment regimens designed to overcome the innate and acquired mechanisms of tumor resistance.
{kind=link}
{kind=link}
{kind=link}
Improving long-term survival in patients with metastatic melanoma. LDH, lactate dehydrogenase. (Figure from Ascierto and Dummer. Immunological effects of BRAF+MEK inhibition.78 Adapted with permission from Taylor & Francis Ltd, http://www.tandfonline.com).
When considering new treatment combinations as a way to improve survival, one approach under evaluation is the use of I-O plus targeted therapy in patients with BRAF-mutant melanoma, based on reports indicating that BRAF and MEK inhibitors may impact tumor antigen expression, T-cell infiltration, and immune-cell populations, making the tumor microenvironment more amenable to I-O therapy.80 Several studies are in progress, and phase III results are becoming available. The phase III IMspire 170 trial (NCT03273153) was designed to evaluate atezolizumab (PD-L1 inhibitor) plus cobimetinib (MEK inhibitor) versus pembrolizumab in untreated patients with BRAF wild-type tumors: unfortunately, it failed to meet its primary endpoint and PFS was not improved.81 Recently, positive results were presented from a phase III triplet trial, IMspire150 (NCT02908672), in which cobimetinib plus vemurafenib was evaluated with or without atezolizumab in untreated patients with BRAF-mutant melanoma. The triplet combination significantly improved PFS compared with cobimetinib plus vemurafenib (15.1 vs 10.6 months), although objective response rates (ORRs) were similar; median OS data, although not mature, suggested a benefit with the triplet regimen. The safety profile of the triplet combination was tolerable and manageable.82 In the phase II KEYNOTE-022 triplet study of dabrafenib plus trametinib with or without pembrolizumab, the difference in median PFS between the arms (16.9 vs 10.7 months) did not reach statistical significance. At 24 months, the PFS rate was numerically higher in the triplet compared with the doublet arm (41% vs 16%), the overall response rate was lower (ORR; 63% vs 72%), the duration of response was longer (55% vs 16% at 24 months), and there was a higher incidence of grade ≥3 treatment-related AEs (58% vs 25%), respectively. Median OS was not reached with the triplet compared with 26.3 months with doublet and the 2-year OS rate was higher with the triplet (63% vs 52%). These data suggest a potential benefit with the triplet combination that will need to be confirmed with longer follow-up.83 84 In COMBI-i (NCT02967692), a phase III study of dabrafenib plus trametinib with or without spartalizumab (PD-1 inhibitor), results from the non-randomized part of the trial included an ORR of 78% and grade ≥3 treatment-related AEs in 72% of patients treated with the triplet therapy.85 While initial ORRs from some trials are high, no information has been reported regarding the durability of these responses and whether they will translate into an OS benefit relative to BRAF plus MEK inhibition or I-O agents alone, or with these approaches used in sequence. The generally high incidences of grade ≥3 AEs with triplet regimens is also a concern. In the future, a more appropriate control arm for such trials may be nivolumab plus ipilimumab, which represents an alternative and possibly more powerful standard of care than the comparators used so far.
IMPemBra (NCT02625337) was a phase II study designed to evaluate several dabrafenib plus trametinib dosing schedules in combination with pembrolizumab.86 Early results from this small trial suggest that intermittent, short-term administration of dabrafenib plus trametinib in combination with pembrolizumab may provide the efficacy benefits expected with triplet therapy while minimizing both toxicity and the development of resistance.87 Results have recently been presented from the single-arm, phase II TRIDeNT study (NCT02910700), which includes treatment with a combination of nivolumab, dabrafenib, and trametinib in patients with BRAF-mutant melanoma.88 Unlike other comparable trials, patients with disease that was refractory to previous PD-1 inhibitor therapy or those with brain metastases are eligible for inclusion. The incidence of grade 3 or 4 treatment-related AEs (65% of patients) was similar to those of other triplet trials. Efficacy data showed an ORR of 100% in PD-1 inhibitor-naive patients (including two CRs) and an ORR of 83% in patients with refractory disease (including one CR); ORRs were similar in patients with and without brain metastases (88%, including two CRs, in patients with brain metastases; 93%, including one CR, in those without). These data suggest that triplet therapy may have particular value, and merits further evaluation in patients with PD-1 inhibitor-refractory disease and/or those with brain metastases.
With the efficacy of triplet regimens still to be determined, a rational approach may be to use what is thought to be the most effective treatment first. Ongoing trials such as SECOMBIT, DREAMseq, and EBIN should ultimately help to determine which sequence of I-O and targeted agents is most active for particular subsets of patients and whether using a PD-1 inhibitor with intermittent or short-term targeted therapy is effective.
Other agents designed to target different components of the immune system or oncogenic processes/pathways are under evaluation, either alone or in combination with a PD-1 inhibitor. In the phase III LEAP-3 study, pembrolizumab is being evaluated in combination with lenvatinib, a multireceptor tyrosine kinase inhibitor, in previously untreated patients with advanced melanoma (NCT03820986).89 Another approach is to combine an I-O agent with a vaccine. A phase III trial (KEYNOTE-034/MASTERKEY265/NCT02263508)90 designed to evaluate TVEC plus pembrolizumab was started after results from a phase Ib study showed that the vaccine followed by combination therapy gave a response rate of 62% in patients with advanced melanoma, with no dose-limiting toxicities reported.91
Agents are under evaluation that target other immune checkpoint receptors, such as lymphocyte-activation gene-3 (LAG-3; eg, relatlimab)92 and glucocorticoid-induced tumor necrosis factor receptor (GITR).93 A phase II/III study of relatlimab plus nivolumab versus nivolumab in patients with untreated metastatic unresectable melanoma was initiated (NCT03470922)94 after phase I/IIa results showed responses with the combination in a heavily pretreated population of patients with melanoma and progression on prior anti-PD-1/anti-PD-L1 therapy. Data from this initial trial also showed that the combination had a safety profile similar to that of nivolumab monotherapy.92 Bempegaldesleukin is a pegylated IL-2 pathway agonist. In the phase I/II PIVOT-02 study, an ORR of 53% (34% CRs) was reported with first-line bempegaldesleukin plus nivolumab treatment of patients with metastatic melanoma; responses were durable and were observed regardless of PD-L1 expression and the combination was well tolerated.95 Bempegaldesleukin plus nivolumab is now under evaluation in a randomized phase III study in patients with previously untreated, unresectable, or metastatic melanoma (NCT03635983).96 Another approach involves using toll-like receptor-9 agonists to stimulate innate and adaptive antitumor immune responses. These agents, including tilsotolimod and CMP-001, are being studied in melanoma.97 98 A randomized, phase II trial with intradermal tilsotolimod in pT3-4 cN0M0 melanoma (INTRIM) is in progress (NCT04126876).99 Data from phase I studies showed acceptable tolerability and evidence of antitumor activity alone (intratumoral tilsotolimod) and in combination with pembrolizumab (CMP-001).97 98 Other agents are designed to target the tumor, although often with the goal of mediating immunomodulatory effects. These include entinostat, a histone deacetylase inhibitor that leads to the downregulation of immunosuppressive cell types in the tumor microenvironment.100 BMS-986179 is an anti-CD73 antibody that prevents the conversion of AMP to adenosine, with the goal of stopping adenosine-associated immunosuppression and tumor progression.101 Another approach involves inhibiting heat shock protein 90 (HSP90), for example, with XL888; preclinical data suggest that inhibiting HSP90 may help to overcome resistance to BRAF plus MEK inhibitor combinations.102
Conclusion
The potential for long-term survival in patients with metastatic melanoma is an important decision-making factor and, accordingly, associated outcomes need to be assessed appropriately and accurately. Metrics that encompass economic or QoL parameters may be particularly helpful in describing the long-term value of agents used to treat these patients. The current data indicate that several treatment approaches offer the potential for long-term survival. Combined CTLA-4 and PD-1 inhibitor immunotherapy results in the highest OS plateau (up to 52% of patients), and in our opinion, may achieve long-term survival (functional cure) in the greatest number of patients. We propose that patients may be considered functionally cured if they have responded to treatment and have been off treatment for at least 2 years without disease progression. There remains a need to improve outcomes for those patients who do not experience long-term OS.
Acknowledgments
Professional medical writing and editorial assistance were provided by Rebecca Turner and Michele Salernitano at Ashfield Healthcare Communications, funded by Bristol Myers Squibb.
References
Footnotes
Contributors OM, MBA, HBK, RD, and PAA all contributed to the conception, content, writing and revisions, and have read and approved the submitted manuscript.
Funding This work was supported by Bristol Myers Squibb.
Competing interests OM: Consulting or advisory role: Amgen, Bristol Myers Squibb, Merck Sharp & Dohme, Novartis, and Roche. Travel, accommodations, and expenses: Bristol Myers Squibb and Merck Sharp & Dohme. Expert testimony: Bristol Myers Squibb. Research funding: Bristol Myers Squibb, Merck Sharp & Dohme, and NeraCare GmbH. MBA: Advisory board: Arrowhead, Bristol Myers Squibb, Exelixis, Fathom, Leads, Merck, Novartis, PACT, Pfizer, Pneuma, Pyxis Oncology, and Werewolf. Consultant: Array, Aveo, Boehringer Ingelheim, Bristol Myers Squibb, Cota, Eisai, Galactone (personal fees), Genentech/Roche, ImmunoCore, Iovance, Merck, Newlink, Novartis, Pfizer, and Surface. Research support to institution: Bristol Myers Squibb, Merck, and Pfizer. Clinical trial involvement: Aveo (Tivo 3), Bristol Myers Squibb (CM-214, CM-004, CM-067, CM-204, CM-218, CM-238, CM-905, X4P-X4 RCCA, X4-RCCB), Genentech/Roche (ImMotion 150, ImMotion 151), Merck (KN- 426, KN-427, KN-029, KN-564), and Pfizer (029 Trial, Javelin 101). Speaker’s bureau: None. Stock options: Pyxis Oncology and Werewolf. HBK: Employment: Bristol Myers Squibb. Stock and other ownership interests: Bristol Myers Squibb. RD: Consulting or advisory role: Amgen, Bristol Myers Squibb, Merck Sharp & Dohme, Novartis, Pierre Fabre, Roche, Sanofi, Sun Pharma, and Takeda. Honoraria: Amgen, Bristol Myers Squibb, Merck Sharp & Dohme, Novartis, Pierre Fabre, Roche, Sanofi, Sun Pharma, and Takeda. Research funding: Amgen, Bristol Myers Squibb, Merck Sharp & Dohme, Novartis, and Roche. PAA: Consulting or advisory role: 4SC, Alkermes, Amgen, Array BioPharma, AstraZeneca, Bristol Myers Squibb, Genmab, Idera, Immunocore, Incyte, Italfarmaco, MedImmune, Merck Serono, Merck Sharp & Dohme, Nektar, NewLink Genetics, Novartis, Pierre Fabre, Roche/Genentech, Sandoz, Sanofi, Sun Pharma, Syndax, and Ultimovacs. Travel, accommodations, and expenses: Merck Sharp & Dohme. Stock and other ownership interests: PrimeVax. Research funding: Array BioPharma, Bristol Myers Squibb, and Roche/Genentech.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer-reviewed.