Article Text

Abstract
Tumors evade immune-mediated recognition through multiple mechanisms of immune escape. On chronic tumor antigen exposure, T cells become dysfunctional/exhausted and upregulate various checkpoint inhibitory receptors (IRs) that limit T cells’ survival and function. During the last decade, immunotherapies targeting IRs such as programmed cell death receptor 1 (PD-1) and anticytotoxic T lymphocyte-associated antigen 4 (CTLA-4) have provided ample evidence of clinical benefits in many solid tumors. Beyond CTLA-4 and PD-1, multiple other IRs are also targeted with immune checkpoint blockade in the clinic. Specifically, T cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT) is a promising new target for cancer immunotherapy. TIGIT is upregulated by immune cells, including activated T cells, natural killer cells, and regulatory T cells. TIGIT binds to two ligands, CD155 (PVR) and CD112 (PVRL2, nectin-2), that are expressed by tumor cells and antigen-presenting cells in the tumor microenvironment. There is now ample evidence that the TIGIT pathway regulates T cell-mediated and natural killer cell-mediated tumor recognition in vivo and in vitro. Dual PD-1/TIGIT blockade potently increases tumor antigen-specific CD8+ T cell expansion and function in vitro and promotes tumor rejection in mouse tumor models. These findings support development of ongoing clinical trials with dual PD-1/TIGIT blockade in patients with cancer.
- costimulatory and inhibitory T-cell receptors
- immunotherapy
- therapies
- investigational
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Ample evidence supports the role of inhibitory receptors (IRs) in regulating innate and adaptive immunity in chronic viral infections and cancer.1 2 On chronic antigen stimulation, T cells become dysfunctional/exhausted and upregulate many IRs, including programmed cell death receptor 1 (PD-1) and T cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT). At the same time, IR ligands are expressed by tumor cells and antigen-presenting cells (APCs) in the tumor microenvironment (TME). Targeting IRs with monoclonal antibodies (mAbs) has proven beneficial in mouse tumor models and humans, and immune checkpoint blockade (ICB) with anticytotoxic T lymphocyte-associated antigen 4 (CTLA-4), anti-PD-1, or both mAbs are standard treatments for many solid tumors.3–6 Further, multiple lines of evidence support that TIGIT plays a critical role in limiting adaptive and innate immunity against tumors.7–10 Here, we review results supporting the role of TIGIT in cancer immunology and the potency of TIGIT-based cancer immunotherapy.
TIGIT axis and ligands
TIGIT (also called WUCAM, Vstm3, VSIG9) is a receptor of the Ig superfamily, which plays a critical role in limiting adaptive and innate immunity.5–8 TIGIT participates in a complex regulatory network involving multiple IRs (eg, CD96/TACTILE, CD112R/PVRIG), one competing costimulatory receptor (DNAM-1/CD226), and multiple ligands (eg, CD155 (PVR/NECL-5), CD112 (Nectin-2/PVRL2)8 9 11–13, figure 1). Hence, there is some similarity with the CD28/CTLA-4/CD80/CD86 pathway, for which inhibitory and costimulatory receptors compete for binding to the same ligands. In sharp contrast with CTLA-4 −/− mice, TIGIT−/− mice do not develop autoimmunity.10 However, as compared with wild-type mice, Tigit−/− mice develop more severe experimental autoimmune encephalitis when immunized with myelin oligodendrocyte glycoprotein.10 Such an observation supports the role of TIGIT as a negative regulator of T cell functions.
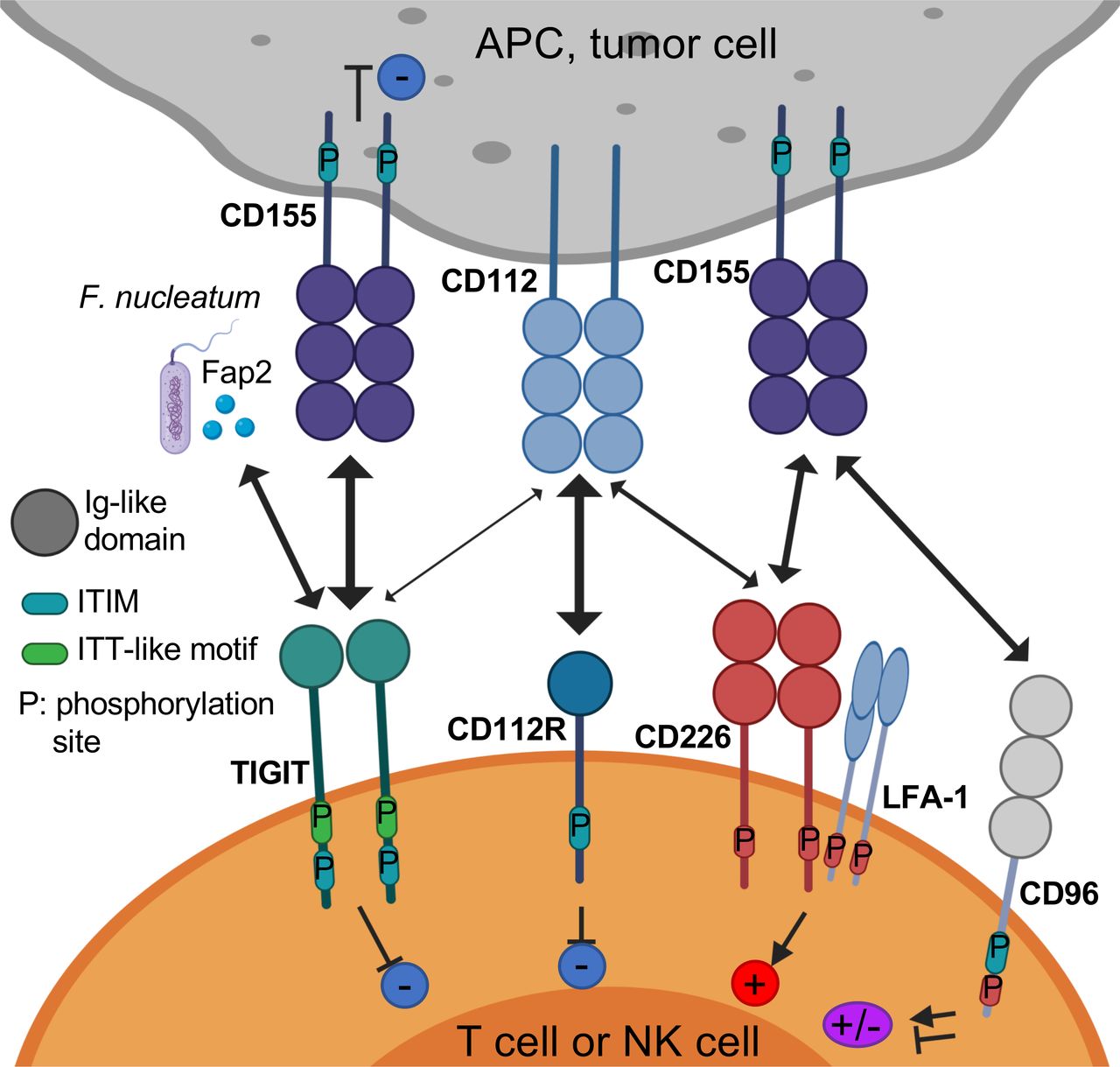
The TIGIT/CD226/CD96/CD112R axis. TIGIT, CD226, CD96, and CD112R are expressed on activated T cells and NK cells. TIGIT ligands CD115 and CD112 are expressed on APCs or tumor cells. TIGIT binds CD155 and CD112 as well as Fap2, a gut bacterium-derived protein. TIGIT, CD96, CD112R, and CD155 contain ITIM motifs in their cytoplasmic tail that trigger inhibitory signals. TIGIT also contains an ITT-like motif. CD226 associates with LFA-1 and binds CD155 to deliver a positive signal. CD96 binds CD155, and whether this triggers inhibitory or activating signals in human T cells remain to be determined. CD112R binds CD112 to deliver an inhibitory signal through its ITIM. APCs, antigen-presenting cells; ITIM, immunoreceptor tyrosine-based inhibitory motif; ITT, Ig tail-tyrosine; NK cells, natural killer cells; TIGIT, T cell immunoreceptor with immunoglobulin and ITIM domain.
TIGIT is expressed by activated CD8+ T and CD4+ T cells, natural killer (NK) cells, regulatory T cells (Tregs), and follicular T helper cells in humans.7 8 14 15 In sharp contrast with DNAM-1/CD226, TIGIT is weakly expressed by naive T cells. In cancer, TIGIT is coexpressed with PD-1 on tumor antigen-specific CD8+ T cells and CD8+ tumor-infiltrating lymphocytes (TILs) in mice and humans.16 17 It is also coexpressed with other IRs, such as T cell immunoglobulin and mucin domain-containing molecule-3 (TIM-3) and lymphocyte activation gene 3 (LAG-3), on exhausted CD8+ T cell subsets in tumors.16 17 Further, TIGIT is highly expressed by Tregs in peripheral blood mononuclear cells of healthy donors and patients with cancer and further upregulated in the TME.18 19
Increased TIGIT expression is associated with hypomethylation and FOXP3 binding at the TIGIT locus in Tregs, and delineates Tregs from activated effector CD4+ T cells.20 In contrast to mouse splenic NK cells, circulating human NK cells exhibit high TIGIT expression, which regulates their tumor killing activity.21 As compared with TIGIT− NK cells, TIGIT+ NK cells exhibit higher cytotoxic capacity and maturation but paradoxically lower cytotoxicity against CD155+ major histocompatibility complex (MHC) class I-deficient melanoma cells.
In sharp contrast with CD8+ T cells, NK cells present at low frequencies in metastatic tumors are dysfunctional, and downregulate both TIGIT and CD226 expression.22 Membrane-bound CD155 triggers CD226 internalization and degradation, resulting in decreased NK cell-mediated tumor reactivity.22 TIGIT binds two ligands, CD155 and CD112 (figure 1 and table 1), that are expressed on monocytes, dendritic cells (DCs), and many non-hematopoietic cells including tumor cells of different histological types.9 16 23–25 TIGIT binds CD155 with higher affinity than competing receptors CD226 and CD968 9 (table 1). While TIGIT weakly binds CD112, CD112R binds CD112 with higher affinity than CD226.13 Interestingly, CD155 expression increases on reactive oxygen species-dependent activation of the DNA damage response, which regulates interactions of NK cells with T cells and with myeloid-derived suppressive cells (MDSCs).26 27 In addition, the Fap2 protein from Fusobacterium nucleatum, an anaerobic Gram− commensal bacteria associated with colorectal carcinoma, binds directly to TIGIT but not CD226 to inhibit NK-cell and T cell mediated tumor reactivity.28 These findings suggest that the gut microbiome regulates innate immune responses in a TIGIT-mediated fashion.
Ligand binding affinities for TIGIT, CD226, and CD112R
TIGIT structure and signaling
TIGIT is composed of an extracellular immunoglobulin (Ig) variable domain, a type 1 transmembrane domain, and a cytoplasmic tail with two inhibitory motifs conserved in mouse and human: an immunoreceptor tyrosine-based inhibitory motif (ITIM) and an Ig tail-tyrosine (ITT)-like motif.7–10 The crystal structure of TIGIT bound to CD155 reveals that two TIGIT/CD155 dimers assemble into a heterotetramer with a core TIGIT/TIGIT cis-homodimer, with each TIGIT molecule binding to one CD155 molecule.29 This cis–trans receptor clustering mediates cell adhesion and signaling.
In mice, phosphorylation of either the ITIM (Y227) or ITT-like motif residue (Y233) can trigger TIGIT inhibitory signal. However, in human NK cell line YTS, TIGIT/CD155 engagement initiates major inhibitory signaling through an ITT-like motif, while the ITIM motif mediates a minor inhibitory signal.8 30 On TIGIT/CD155 ligation, the ITT-like motif is phosphorylated at Tyr225 and binds to cytosolic adapter Grb230 and β-arrestin 231 to recruit SH2-containing inositol phosphatase-1 (SHIP-1). SHIP-1 impedes phosphoinositide 3 kinase and mitogen-activated protein kinase signaling.30 SHIP-1 also impairs TRAF6 and NF-κB activation,31 leading to inhibition of interferon (IFN)-γ production by NK cells.
Mechanisms of inhibition
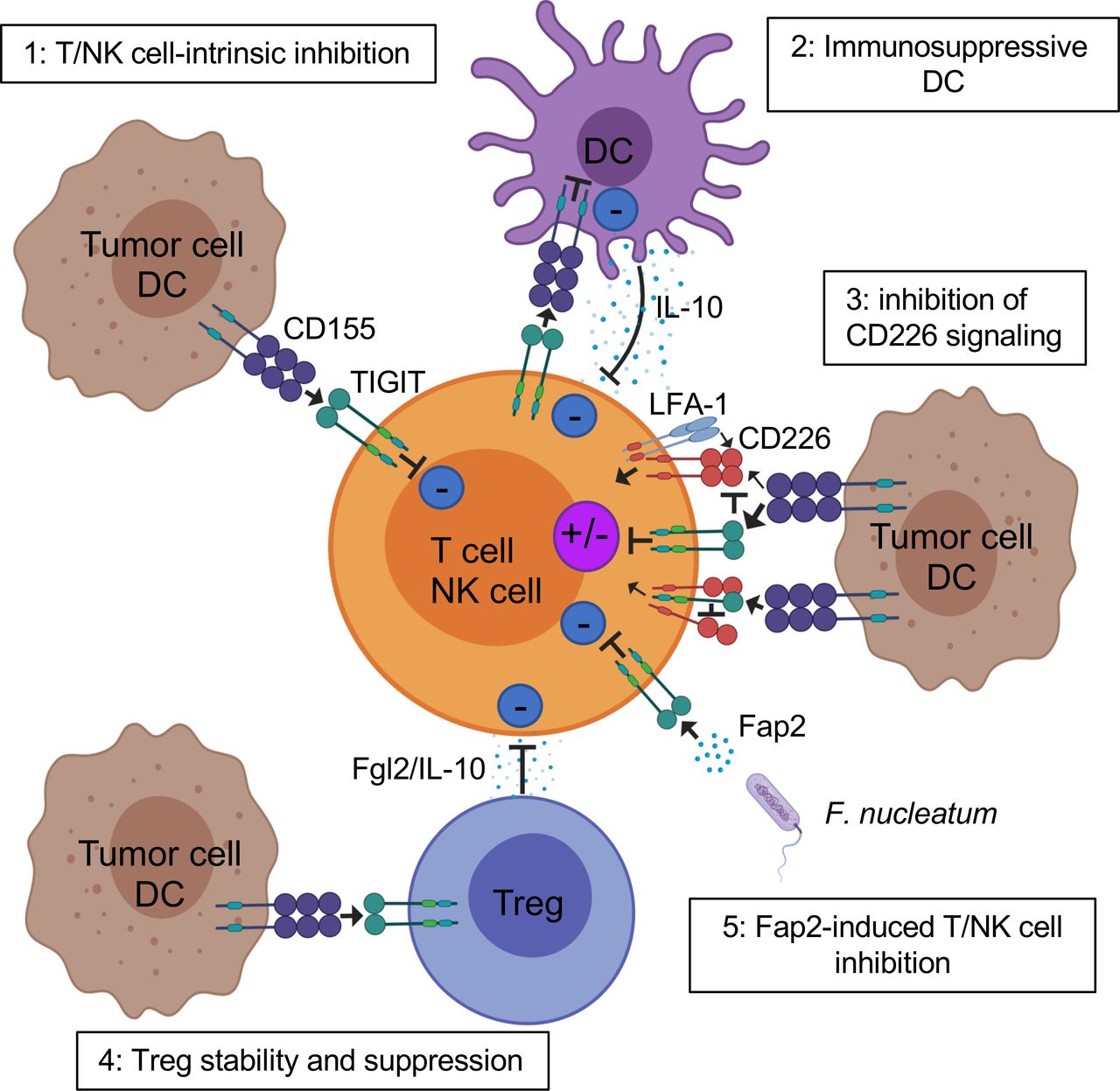
TIGIT potently inhibits innate and adaptive immunity through multiple mechanisms (figure 2). First, in mouse models, TIGIT indirectly impedes T cell function by binding to CD155 on DCs.9 TIGIT engagement on DCs induces CD155 phosphorylation and triggers a signaling cascade promoting tolerogenic DCs with decreased production of interleukin (IL)-12 and increased production of IL-10.9
{kind=link}
{kind=link}
Mechanisms of TIGIT inhibition of T cells in the TME. TIGIT displays multiple inhibitory mechanisms in T cells. 1: TIGIT binds CD155 and triggers direct inhibitory signals in T cells. 2: TIGIT binds CD155 on APCs to trigger IL-10 production and decrease IL-12 production, which indirectly inhibits T cells. 3: TIGIT binds CD155 with higher affinity than CD226 or disrupts CD226 homodimerization to impede CD226-mediated T cell activation. 4: TIGIT signaling in Tregs enhances their immunosuppressive functions. 5: Fap2 protein from the gut bacteria Fusobacterium nucleatum binds TIGIT to trigger inhibitory signals. APCs, antigen-presenting cells; IL, interleukin; ITIM, immunoreceptor tyrosine-based inhibitory motif; TIGIT, T cell immunoreceptor with immunoglobulin and ITIM domain; Tregs, regulatory T cells.
Second, TIGIT exhibits direct immune cell-intrinsic inhibitory effects. Agonistic anti-TIGIT antibodies inhibit T cell proliferation and function by attenuating T cell receptor (TCR)-driven activation signals.10 14 24 In mice and humans, TIGIT inhibits NK cell degranulation, cytokine production, and NK cell-mediated cytotoxicity of CD155-expressing tumor cells.8 30–32 Further, interaction of TIGIT+ NK cells with MDSCs expressing CD155 decreases phosphorylation of ZAP70/Syk and ERK1/2, reducing the cytolytic capacity of NK cells.27
Third, multiple lines of evidence show that TIGIT impedes CD155-mediated CD226 activation. CD226 is a costimulatory receptor widely expressed by immune cells, including T cells, NK cells, monocytes, and platelets.33 34 CD226 associates with LFA-1 to promote cell contact and triggers TCR signaling.35 This receptor also fosters production of proinflammatory cytokines by CD4+ T cells on binding to CD155.36 CD226 is directly involved in tumor recognition by T cells and NK cells in mice and humans,33 37 and CD226-deficient mouse CD8+ T cells and NK cells display immunological synapse defects impairing antitumor immunity.38 39 TIGIT binds CD155 with higher affinity than CD226, thus limiting CD226-mediated activation.8–10 TIGIT also directly binds CD226 in cis, disrupting its homodimerization and binding capacity to CD155.17
Fourth, the balance of TIGIT/CD226 expression regulates the effector function of T cells and NK cells. Abrogation of TIGIT expression with shRNA in TCR-activated CD4+ T cells increases T-bet expression and IFN-γ production, which are abolished on CD226 or CD155 blockade. In contrast, CD226 knockdown decreases T-bet expression and IFN-γ production.24 Further, CD226 blockade abrogates the effects of dual PD-1 and TIGIT blockade on proliferation and cytokine production of tumor antigen-specific CD8+ T cells in melanoma.16 Similarly, in CT26 tumor-bearing mice, the antitumor effects of dual programmed cell death-ligand 1 (PD-L1)/TIGIT blockade occur in a CD226-dependent fashion and are abolished on CD226 blockade.17 Interestingly, blocking anti-PD-1 and agonistic anti-GITR mAbs increases overall survival of MC38 tumor-bearing mice. In this model, PD-1 inhibition rescues CD8+ T cell dysfunction by inhibiting SHP2-mediated CD226 dephosphorylation, while anti-GITR mAbs decrease TIGIT expression.40 These important findings support that beyond PD-1 and TIGIT blockade, other ICBs enhance T cell-mediated tumor rejection by favorably tipping the balance between CD226 and TIGIT in CD8+ T cells.
Fifth, TIGIT acts in Tregs to augment immunosuppressive function and stability. TIGIT is highly expressed by a subset of natural Tregs in mice18 and the majority of Tregs in humans,18 19 41 and TIGIT upregulation in Tregs is associated with hypomethylation and Foxp3 binding at the TIGIT locus.20 TIGIT+ Tregs are more suppressive than TIGIT− Tregs in healthy donors and patients with melanoma.19 41 Further, TIGIT+ Tregs in the periphery and at tumor sites upregulate many Treg gene signature markers as compared with TIGIT− Tregs,18 including Foxp3, Helios, neuropilin-1, CTLA-4, PD-1, and LAG-3.18 19 TIGIT+ Tregs also suppress proinflammatory Th1 and Th17 but not Th2-type T cell responses.18 42 On TIGIT ligation, TIGIT+ Tregs produce IL-10 and fibrinogen-like protein 2, which mediate T cell suppression.18
Interestingly, human Foxp3+ Tregs exhibit lower CD226 expression than Foxp3− CD4+ T cells.19 41 CD226 is also downregulated by Tregs in metastatic melanoma as compared with the periphery, resulting in an increased TIGIT/CD226 ratio.19 TIGIT and CD226 oppose each other to augment or disrupt, respectively, Treg suppression and stability.19 A high TIGIT/CD226 ratio in Tregs appears to correlate with increased Treg frequencies in tumors and poor clinical outcome on ICB. Additional studies are needed to determine whether the TIGIT/CD226 ratio in Tregs may represent a biomarker of clinical response to ICB in patients with solid tumors.
Mice-bearing tumors with CD155 loss on host cells or tumor cells exhibit reduced tumor growth and enhanced effector functions of CD8+ and NK cells.43 While CD155 loss on host cells appears to act in a CD226-dependent fashion, CD155 loss in tumor cells promotes tumor growth and metastasis through tumor-intrinsic mechanisms. Further, CD155 deletion on both host and tumor cells results in greater tumor inhibition and increased effects of ICB. In addition, tumor CD155 expression is associated with increased tumor-infiltrating PD-1+ CD8+ T cells and resistance to anti-PD-1 immunotherapy in metastatic melanoma.44 Collectively, these findings suggest that targeting the CD155 pathway with combinatorial ICB (TIGIT and CD96) may improve response to PD-1 blockade.
TIGIT in cancer immunotherapy
Dual PD-1 and TIGIT blockade is a promising combinatorial immunotherapy of cancer. While each single blockade does not significantly impede growth of CT26 tumors in mice, dual TIGIT and PD-1/PD-L1 blockade synergizes to augment proliferation and function of antitumor CD8+ T cells, resulting in protective memory T cells, complete tumor rejection, and prolonged overall survival.17 45 46 These effects are abrogated on CD8+ T cell depletion, supporting a critical role of CD8+ T cell-mediated tumor reactivity.
Dual PD-1/TIGIT blockade also enhances proliferation and function of tumor antigen-specific CD8+ T cells and TILs isolated from patients with melanoma as compared with single blockade.16 47 Interestingly, dual PD-L1/TIGIT blockade (atezolizumab/tiragolumab) appears to provide superior clinical benefits as compared with PD-L1 blockade alone as a first-line therapy for patients with PD-L1-positive non-small cell lung cancers, despite similar toxicity profiles.48 However, these observations need to be confirmed in large randomized clinical trials.
The effects of dual PD-1/TIGIT blockade in mouse tumor models and in vitro are abrogated on CD226 blockade, suggesting that TIGIT blockade acts primarily by tipping CD155-mediated signaling towards CD226 activation.16 17 In addition, PD-1 induces SHP2-mediated CD226 dephosphorylation, supporting the need for dual PD-1/TIGIT blockade to promote CD226 signaling.40 Along this line, CD8+ TILs downregulate CD226 expression in multiple solid tumors, including melanoma, which may represent a significant obstacle limiting the effects of dual PD-1/TIGIT blockade in patients with cancer.16 49 Membrane-bound CD155 plays a critical role in mediating CD226 downregulation by immune cells in the TME via CD226 internalization and degradation, supporting the role of CD155-mediated immune dysfunction.22
In experimental models, TIGIT blockade or TIGIT deletion promotes NK cell-mediated antitumor reactivity in vitro and in vivo.8 27 30 32 50 Strikingly, one recent study of B16 melanoma and CT26 lung metastasis mouse models suggests that TIGIT blockade alone or in combination with PD-1 blockade acts primarily on NK cells to augment CD8+ T cell-mediated antitumor responses and impede tumor growth. In these experimental models, NK cell-specific TIGIT deficiency and NK cell depletion compromised the effects of TIGIT blockade.45 These findings are at odds with many observations supporting that TIGIT blockade alone fails to significantly augment CD8+ T cell immunity and promote tumor rejection in wild-type mice transplanted with solid tumors17 51 and in expanding tumor antigen-specific CD8+ T cell responses16 as compared with combined dual PD-1/TIGIT blockade. The mechanisms supporting potential helper effects of NK cells on CD8+ TILs as well as the relevance of these findings to patients with cancer remain elusive. Whether and how NK cells contribute to environmental cues guiding CD8+ T cell priming, maturation, and memory differentiation needs to be thoroughly determined. Interestingly, IL-15 together with TIGIT blockade increases NK cell-mediated melanoma cytotoxicity in vitro and decreases tumor metastasis in mouse melanoma models.22 These findings support development of novel combinatorial immunotherapy with IL-15 and TIGIT blockade to promote NK cell-mediated destruction of MHC class I-deficient melanoma, which is refractory to CD8+ T cell-mediated immunity.
Besides PD-1 blockade, other ICBs combined with TIGIT blockade also enhance antitumor immune responses. TIGIT blockade has been tested together with ICB targeting IRs outside of the TIGIT network. For example, TIGIT and TIM-3 synergize to suppress antitumor immune responses in mice.42 Adoptive transfer of mixed T cell subsets in subcutaneous B16F10-bearing Rag−/− mice, including CD8+ T cells, CD4+ T cells, and Tregs from wild-type and Tigit−/− mice, show that TIGIT depletion in Tregs but not CD8+ T cells decreases tumor growth. These data suggest that TIGIT can act primarily in Tregs to impede antitumor CD8+ T cell responses and promote tumor growth. TIGIT+ Treg-infiltrating tumors upregulate TIM-3, and blocking TIM-3 in Tigit−/− mice further decreases tumor size and increases overall survival.42 Because TIGIT competes with the IRs CD96 and CD112R for binding to its ligands, multiple studies have investigated the immunological and clinical effects of combinatorial therapies targeting PD-1 together with TIGIT and/or other IRs within the TIGIT network, including CD96 and CD112R. TIGIT synergizes with CD96 to inhibit antitumor responses—in tumor-bearing mouse models with lung metastasis, antitumoral effects of CD96 blockade are higher in Tigit−/− mice.52 CD96 blockade appears more effective in combination with anti-CTLA-4 or anti-PD-1, and its effects depend on NKs, CD226 signaling, and IFN-γ production. Further, TIGIT blockade alone or in combination with PD-1 blockade adds to CD96 blockade to significantly reduce B16 melanoma growth in wild-type and Cd155−/− mouse models.43 Notably, the role of CD96 as an IR remains controversial because there is also evidence that it can act as a costimulatory receptor in CD8+ T cells.53 Multiple experimental studies in mice and in vitro suggest that CD112R blockade combined with TIGIT blockade increases antitumor immune responses. CD112R blockade synergizes with TIGIT blockade to enhance human NK cell-triggered antibody-dependent cellular cytotoxicity (ADCC) against breast tumor cell lines in vitro.50 Dual CD112R/PD-L1 blockade also confers improved outcomes as compared with single blockade in mice with MC38 tumors.54 Further, CD112R blockade alone or combined with either TIGIT blockade or PD-1 blockade or both increases cytokine production by TILs from ovarian, endometrial, and lung tumors in the presence of allogeneic melanoma cells expressing surface-bound anti-CD3 antibody.55 However, the relevance of these findings to human cancer and autologous CD8+ T cell responses against well-defined tumor antigens remains to be demonstrated.
Multiple studies have suggested that anti-CTLA-4 mAbs act through ADCC-mediated Treg depletion.56–59 Because Tregs highly express TIGIT in the TME, one wonders whether anti-TIGIT mAbs with Fc-binding capability induce Treg depletion. Interestingly, in mouse tumor models, ICB with Fc variants of anti-TIGIT mAbs shows that selective FcγR coengagement on APCs enhances antigen-specific T cell responses and tumor reactivity without evidence of Treg depletion.60 Whether the antitumor effects of anti-TIGIT antibodies in patients with cancer are Fc-dependent remains to be determined. The answer to this critical question may be provided by multiple phase I and II clinical trials (table 2) that are testing Fc-engineered anti-TIGIT mAbs: IgG1 (MTIG7192/Genentech, MK-7684/Merck, and OP-313M32/Oncomed), inert-Fc IgG1 (BMS-986207/Bristol-Myers Squibb; AB-154/Arcus), and IgG4 (ASP8374/Potenza/Astellas).
Clinical trials targeting TIGIT, CD112R, and CD226
Concluding remarks, challenges, and critical questions
TIGIT is a promising target in cancer immunotherapy, particularly in combination with PD-1 blockade. Moving forward with ongoing TIGIT-based clinical trials in patients with cancer, however, we need to address many key questions and challenges. First, what mechanisms drive the effects of TIGIT blockade in patients with cancer? Are these effects primarily mediated by its direct activity in CD8+ T cells, Tregs, or both? Can TIGIT blockade reprogram APCs in the TME to increase T cell priming/activation? Can these effects be context-dependent and vary according to the disease stage? Can TIGIT blockade mediate NK cell-mediated tumor reactivity against MHC class I-deficient human tumors in vivo, and will this be an opportunity to provide clinical benefits to a subset of PD-1-refractory patients with cancer? And, in addition to dual PD-1/TIGIT blockade, is there any potential synergy/additive effect of CD112R or CD96 blockade as suggested by mouse tumor models and in vitro studies? In this regard, one has to keep in mind that the role of CD96 as an IR remains controversial.53 Further, evidence that CD112R blockade can potently enhance autologous human tumor antigen-specific CD8+ T cells is still missing. The answer to this important question may come from the phase I clinical trial evaluating the effects of one CD112R inhibitor alone or in combination with anti-PD-1 mAbs in patients with advanced solid tumors (NCT03667716, table 2).
In addition, CD226 plays a critical role as a master regulator of dual PD-1/TIGIT blockade. Its downregulation by CD8+ T cells and NK cells in the TME may represent a major obstacle for the success of dual PD-1/TIGIT blockade in the clinic. Therefore, it appears essential to design novel strategies to augment CD226 expression and signaling or prevent its downregulation in the TME. It is noteworthy that one ongoing clinical trial is testing agonistic anti-CD226 in multiple cancers (NCT04099277, table 2). Because of the role of CD226 in mediating platelet adhesion and activation, however, potential hematological adverse events will need to be monitored carefully.34 Finally, the many ongoing clinical trials using different Fc-engineered anti-TIGIT mAbs will likely help determine the role of FcγR coengagement in promoting the effects of TIGIT blockade in patients with cancer.
References
Footnotes
Contributors J-MC and HMZ wrote the manuscript.
Funding This work was supported by NIH/NCI grants R01CA228181 and R01CA222203 (to HMZ), a research grant by Bristol-Myers Squibb (to HMZ), a cancer vaccine collaborative clinical strategy team grant (to HMZ), NCI grant P50CA121973 (to JMK), and the James W and Frances G McGlothlin Chair in Melanoma Immunotherapy (to HMZ).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Commissioned; externally peer reviewed.