Article Text

Abstract
Cancer cells can evade immune surveillance in the body. However, immune checkpoint inhibitors can interrupt this evasion and enhance the antitumor activity of T cells. Other mechanisms for promoting antitumor T-cell function are the targeting of costimulatory molecules expressed on the surface of T cells, such as 4-1BB, OX40, inducible T-cell costimulator and glucocorticoid-induced tumor necrosis factor receptor. In addition, CD40 targets the modulation of the activation of antigen-presenting cells, which ultimately leads to T-cell activation. Agonists of these costimulatory molecules have demonstrated promising results in preclinical and early-phase trials and are now being tested in ongoing clinical trials. In addition, researchers are conducting trials of combinations of such immune modulators with checkpoint blockade, radiotherapy and cytotoxic chemotherapeutic drugs in patients with advanced tumors. This review gives a comprehensive picture of the current knowledge of T-cell agonists based on their use in recent and ongoing clinical trials.
- T-lymphocytes
- review
- receptors
- immunologic
- immunotherapy
- costimulatory and Inhibitory T-cell receptors
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Antitumor immune responses are complex, involving multiple steps and various types of cells, and depend on the interplay of innate and adaptive immune systems. Immunotherapies targeting innate, adaptive immune cells or molecules have demonstrated therapeutic efficacy for a broad range of human malignancies.1–5 Most recently, immunotherapies targeting the adaptive immune system, specifically, T cells, have improved tumor control.4 5 Full T-cell activation requires three signals: T-cell receptor (TCR) signaling, costimulatory signaling and cytokine support.6 TCR signaling occurs through TCR recognition of a neoantigen uniquely expressed on tumor cells. Neoantigens are encoded by the mutated DNA of tumor cells, and their peptide epitopes are distinct from those derived from the normal human genome.7 They are processed and then displayed in major histocompatibility complexes on the surfaces of tumor cells and antigen-presenting cells (APCs).8 These neoantigen peptide-major histocompatibility complexes can be recognized by the TCRs of antigen-specific T cells. Therapies manipulating TCR signaling, such as chimeric antigen receptor T-cell therapy, are already used in the clinic.5
Multiple costimulatory pathways can result in the activation of T cells (figure 1).9 CD80/CD86-CD28 signaling is a major costimulatory signaling cascade contributing to T-cell activation and cytokine release.10 And the T-cell checkpoint inhibitor cytotoxic T-lymphocyte-associated protein 4 competitively binds to CD80/CD86 with a higher affinity and leads to T-cell suppression.11 Inducible T-cell costimulator (ICOS), which interacts with the ICOS ligand, is an inducible costimulatory receptor expressed on activated T cells.12 4-1BB, OX40, glucocorticoid-induced tumor necrosis factor (TNF) receptor (GITR) and other receptors in the TNF superfamily can synergize with TCR signaling to enhance T-cell responses and survival.13

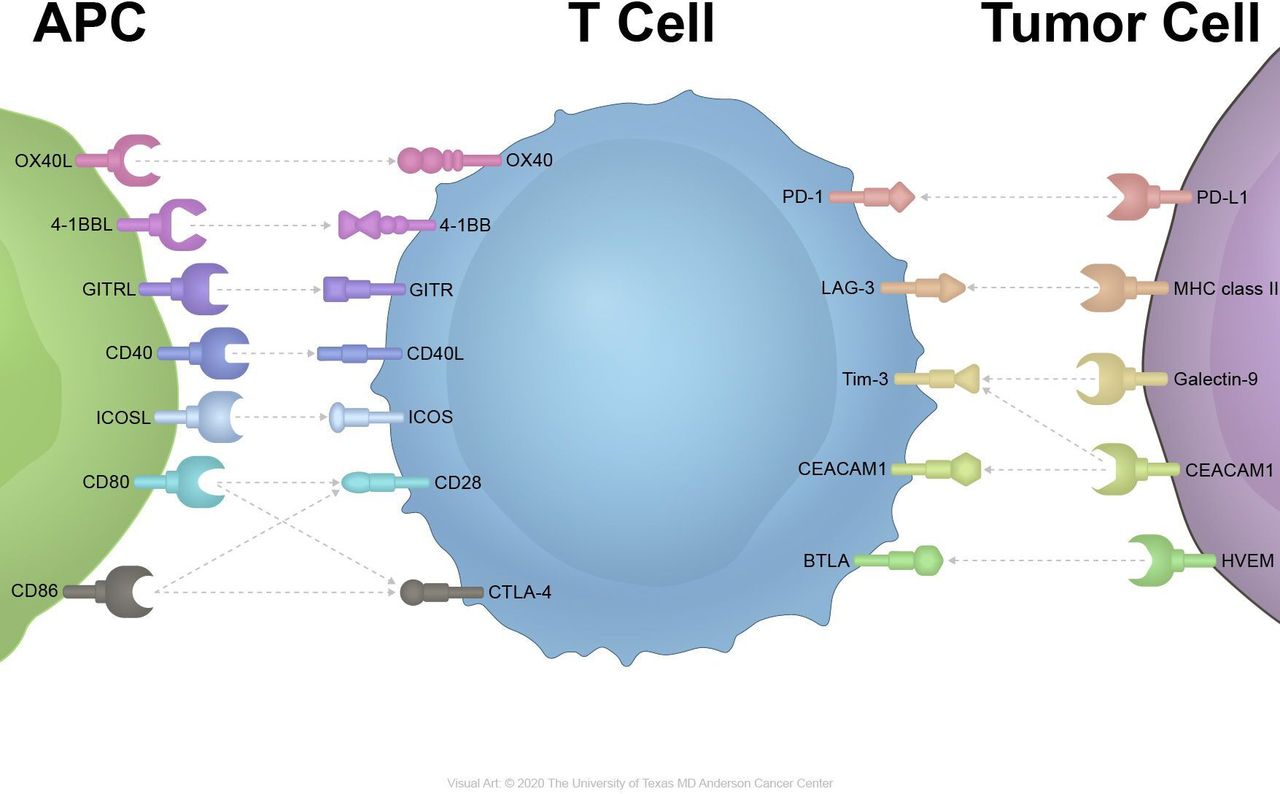{kind=link}
Inhibitory and stimulatory receptors on immune cells and cancer cells. APC, antigen-presenting cells; GITR, glucocorticoid-induced tumor necrosis factor receptor; ICOS, inducible T-cell costimulator.
Despite the success of checkpoint inhibitors in the treatment of cancer, more than 80% of patients do not respond to treatment or eventually experience resistance. Therefore, the focus of efforts to improve T cells’ antitumor responses has shifted to modifying signal through the use of agonistic antibodies targeting these molecules to boost antitumor T-cell responses. Common targets include ICOS, 4-1BB, OX40 and GITR. In addition, costimulatory receptors on APCs such as CD40 provide another means of improving T cells’ antitumor responses because they induce the expression of costimulatory ligands and the secretion of cytokines that drive antitumor activity.14
In this review, we discuss the current use of T-cell agonists in cancer immunotherapy, challenges regarding the timing of agonistic drug delivery and optimal combinations of checkpoint inhibitors, chemotherapy and/or radiotherapy.
OX40
OX40 (CD134), a member of the TNF receptor superfamily 4, is expressed mostly on activated CD4+ and CD8+ T cells and Foxp3+CD4+ regulatory T cells (Tregs). Intratumoral Tregs have particularly high levels of OX40 expression. The expression of OX40 is driven by T-cell activation and is transient, peaking 24–48 hours after T-cell activation and typically lasting 3–4 days.15 In contrast, the ligand of OX40 (CD252) is expressed on activated APCs, specifically dendritic cells (DCs), B cells and macrophages.15 16 OX40 is expressed frequently in breast cancer, melanoma, head and neck cancer, colon cancer, and B cell lymphoma cells.17–19
The signals from the binding of OX40 and its ligand promote effector T-cell expansion and survival by enhancing the expression of cyclin A, cyclin-dependent kinases, Bcl-2 antiapoptotic molecules, multiple cytokines and related receptors like interleukin (IL)-2.20 In addition, OX40 signaling promotes the generation of memory T cells and inhibits the function of Tregs.16 Several in vivo studies have demonstrated that OX40 antagonizes Foxp3+ induction in naïve CD4 T cells and inhibits IL-10 expression in inducible Tregs. Moreover, agonistic OX40 antibodies help deplete tumor-infiltrating Tregs that express OX40 via the antibody-dependent cell cytotoxicity that myeloid and natural killer cells induce after interacting with Tregs.21 In murine tumor models, an agonistic OX40 antibody, when combined with a transforming growth factor-β1 antagonist, increased the expression of interferon (IFN)-γ, downregulated IL-4 production and blocked transforming growth factor-β1-mediated Treg induction.22 However, there is evidence that the effects of agonistic OX40 antibodies can be changed under certain situations, such as when IFN-γ and IL-4 are absent. In these circumstances, Tregs could proliferate on OX40 signaling. Thus, OX40 affects the ability of Tregs to suppress immune reactions in both positive and negative directions and on the basis of the cytokine milieu.23
Thirty patients with advanced cancer (most common subtypes: melanoma and gastrointestinal, renal cell and prostate cancers) were included in the first phase 1 trial of an OX40 agonist as an anti-9B12 murine antibody.24 The most common toxic effects were lymphopenia, fatigue, rash and flu-like symptoms with fever and chills. The maximum tolerated dose (MTD) was never reached during the trial. Twelve of 30 patients had tumor shrinkage. The longest interval of stable disease (SD) lasted 470 days in a patient with renal cancer. The proliferation rate for both CD4+ and CD8+ T cells increased substantially in a dose-dependent manner, as determined by flow cytometric analysis. In contrast, the level of expression of CD4+/Foxp3+ did not increase and upregulation of OX40 was greater in tumor interstitial Tregs than in peripheral blood Tregs. However, high levels of the human antibody neutralizing this murine antibody limited the treatment to only one cycle, not multiple cycles (table 1).
Completed clinical trials
The OX40 agonist MEDI0562 was tested in a phase 1 trial of patients with advanced solid tumors. As of April 1 2016, 55 patients had received MEDI0562, with 96% of the patients experiencing adverse events (AEs).25 Sixty-seven per cent of these AEs were treatment-related, but the researchers detected no serious treatment-related AEs (TRAEs) or immune-related AEs, including dose-limiting toxicity (DLT). Paired biopsy analyzes suggested accrual of PD-L1 expression and upregulation of CD8+ T-cell infiltration in two of three evaluable patients. One patient with head and neck squamous cell carcinoma had a partial response (PR) of at least 3.7 months,26 and updated results published in 2018 reported that two patients had had PRs (table 1).25
When given in combination with other immune-modulating agents, OX40 agonists and PD-1 blockade together have led to greater effects than has either agent alone in some murine models.27 In an orthotopically transplanted murine mammary tumor virus polyoma middle T murine model of mammary cancer refractory to PD-1 blockade, OX40 stimulation delayed tumor growth.27 Of note, delayed PD-1 administration increased the durable responses of CD4+ and CD8+ T cells to tumors. A phase 1 trial of the OX40 agonist MOXR0916 given with atezolizumab included 44 patients with advanced solid tumors who were placed in dose-escalation and serial biopsy groups. The majority of the patients had tolerable safety profiles with mostly grade 1 and no grade 4 or 5 toxicity.28 However, only 2 of 51 patients in the extended trial had responses to the drug and the sponsor decided to discontinue the trial (table 1).
Another OX40 agonist, PF-04518600, is currently being tested alone and in combination with other drugs.29 In a phase 1 clinical trial involving 52 patients with melanoma, hepatocellular carcinoma, head and neck squamous cell carcinomaor renal cell carcinoma, DLT was not reported at doses up to 10 mg/kg; thus, safety was confirmed and a dose expansion cohort for the recommended dose for phase 2 was enrolled from the hepatocellular carcinoma group. Thirty-three (63%) of these patients had no cancer progression after treatment with PF-04518600.30 Other trials of OX40 agonists, such as BMS-986178 given with or without nivolumab or ipilimumab (NCT02737475) and GSK-3174998 given alone or in combination with pembrolizumab (NCT02528357), are ongoing and results are pending (table 2).
Selected ongoing and/or recruiting clinical trials in advanced malignant tumor
Combining OX40 agonist-based treatment with radiotherapy controls tumor growth in murine models, and researchers are currently determining the optimal timing of the two treatments.31 32 In one trial, mice injected with CT26 colorectal tumor cells received radiation plus anti-OX40 antibodies via multiple methods. In this trial, giving anti-OX40 prior to radiotherapy was the most effective treatment method and the optimal time for administering anti-OX40 antibodies was 1 day before radiotherapy.33
4-1BB
The costimulatory molecule 4-1BB is found on both CD4+ and CD8+ T cells in mice. In humans, 4-1BB is expressed on activated CD8+ T cells, activated natural killer cells, natural killer T cells, Tregs, DCs and other myeloid-lineage cells. The ligand of 4-1BB is 4-1BBL, which is inducibly expressed on activated APCs, myeloid progenitor and hematopoietic stem cells.34 35 4-1BB expression levels peak about 12 hours after stimulation and decline within 72 hours.36
On conjunction with its ligand CD137L, 4-1BB forms a heterotrimer with the TNF-associated factors TRAF1 and TRAF2, resulting in downstream activation of the nuclear factor kappa-light-chain-enhancer of activated B cells, extracellular signal-regulated kinase, c-Jun N-terminal kinase and mitogen-activated protein kinase signaling pathways.37 Ultimately, this enhances the cytotoxic T-cell effect via upregulation of the antiapoptotic molecules Bcl-xL and Bfl-1 via TNF-α and IFN-γ secretion.35 38 4-1BB signaling also prolongs cytotoxic T-cell survival.
With regard to Tregs, the role of 4-1BB is controversial. 4-1BB activation is reported to inhibit the formation of inducible Tregs via downregulation of Foxp3 and to possibly inhibit Treg suppression via activation of the Akt pathway.39 On the other hand, scientists also reported that a soluble form of 4-1BBL promotes Treg expansion and immune-inhibitory function and that IL-2 upregulates 4-1BB on Tregs.35 40 As for natural killer cells, Fc receptor engagement-upregulated 4-1BB expression enhances antibody-dependent cell-mediated cytotoxicity.41
In addition to being a target for immunotherapy, 4-1BB signaling has shown tremendous promise in the generation of the generation of chimeric antigen receptors.42 43 To increase T cells’ persistence, activity and ability to expand, chimeric antigen receptor T-cell therapy needs a costimulatory domain incorporated with the CD3ξ chain’s cytoplasmic domain. 4-1BB is a candidate costimulatory domain, and CD19 chimeric antigen receptor T-cell therapy with 4-1BB has shown promising results in the treatment of hematological malignancies.44 45 Several studies have combined targeting 4-1BB with chimeric antigen receptor T-cell therapy.46 47
In preclinical studies, 4-1BB agonists demonstrated antitumor efficacy in several solid tumor models.48 49 For example, in a CT26 colorectal cancer mouse model, a 4-1BB agonist demonstrated dose-dependent suppression of tumor proliferation.50 Based on this preclinical study, researchers developed fully human monoclonal antibodies against 4-1BB, such as urelumab (BMS-663513) and utomilumab (PF-05082566).51
Urelumab is a fully human IgG4 with a point mutation of S228P. In phase 1 and 2 trials of urelumab, 115 patients with advanced or metastatic melanoma, renal cell carcinoma or ovarian cancer were randomized to receive different dose levels of urelumab. Overall, fatigue, reversible grades 3–4 transaminitis and grades 3–4 neutropenia were the most common AEs. The optimal dose of urelumab was not identified, but three patients experienced SD.52 Likewise, a biomarker study of urelumab demonstrated increased levels of the activation molecules of peripheral CD8 T cells and IFN-inducible genes in peripheral blood.52 In a phase 2 trial of urelumab, patients with metastatic melanoma were placed in four groups and given treatment; however, the trial was terminated because of a high incidence of grade 4 hepatotoxicity. Eventually, enrolment in the trial was ended following two hepatotoxicity-related deaths in December 2008.53 A phase 1b monotherapy trial of urelumab resumed in 2012. In this study, two deaths occurred at the higher doses given to patients (1 and 5 mg/kg). A dosage of 0.1 mg/kg given every 3 weeks was considered safe, although 16% of patients given this dose experienced fatigue and 13% experienced nausea.53
Utomilumab, another fully human engineered IgG2 antibody against 4-1BB, was studied in a phase 1 monotherapy trial of 55 patients with advanced solid cancers (Meckel cell carcinoma of the skin, soft tissue sarcoma, and colorectal, gastric, pancreatic, lung, hepatobiliary and breast cancers) or lymphoma.54 A 3+3 dose escalation model was used, and the primary endpoint for the first two cycles was DLT. As a result, utomilumab was tolerated by all patients. The most common AEs were fatigue, fever, decreased appetite and dizziness, and none of the patients experienced liver toxicity. The objective response rate was 3.8%, and patients with Merkel cell carcinoma had a higher response rate, including one complete response (CR), than did patients with other cancers. The best overall response, SD, was seen in 24.5% of patients across all dose-level groups.54
A great deal of evidence demonstrates that 4-1BB agonists induce synergism with other immunotherapy or cytotoxic chemotherapy treatments. In a melanoma cell line model in which a 4-1BB agonist was combined with pembrolizumab, the combination increased antitumor activity in melanoma cells. Specifically, it elevated the CD8+/Treg ratio and increased the activity of tumor-specific cytotoxic T lymphocytes.55 In an ovarian cancer cell model, dual targeting of 4-1BB and PD-1 receptors improved survival and increased effector CD8+ T-cell density but decreased the number of Tregs and myeloid-derived suppressor cells.56 A study in which 4-1BB was given in combination with radiotherapy to mice with implanted tumor cells had similar results.57 In these murine models, the combination led to significant growth retardation in breast tumors. In addition, antitumor activity increased in a lung tumor model, although only at the highest evaluated radiation dose.57 Finally, other studies have shown that administering 4-1BB agonist with chemotherapeutic drugs, including 5-fluorouracil, DNA-alkylating platinum-containing derivatives and cyclophosphamide, led to tumor progression and increased survival rates in mouse models with certain types of solid tumors, such as renal cancer and colorectal cancer.58–60
On the basis of preclinical data, Tolcher et al 61 conducted a phase 1b study of utomilumab plus pembrolizumab for the treatment of advanced solid tumors, including non-small cell lung cancer, renal cell cancer, and head and neck squamous cell carcinoma. Twenty-three patients participated in the trial and were given utomilumab (0.45–5.00 mg/kg) and pembrolizumab (2 mg/kg) every 3 weeks. No patients experienced DLT, and TRAEs were manageable and grade 1 or 2. The objective response rate was 26% (6 of 23 patients), and one complete remission occurred in a patient with small cell lung cancer. In five of the six responders, the responses were maintained for more than 6 months.
A phase 1/2 study reported by Massarelli et al 62 in 2016 demonstrated the safety of the combination of urelumab and nivolumab in patients with advanced malignancies. In patients given urelumab alone, TRAEs occurred in only 7% of patients and led to discontinuation of the study in 5% of them. Six patients in the response group had lymphoma. In 104 patients with melanoma, non-small cell lung cancer, head and neck cancer squamous cell carcinoma, or diffuse large B cell lymphoma, only 7% of the patients had TRAEs, and TRAEs led to study discontinuation (the primary end point) in 6% of the patients. The objective response rate was 10.5% (9 of 86 patients). In this study, adding nivolumab to urelumab did not produce substantial additive/synergistic benefits at the evaluated dose levels. Several trials of 4-1BB agonists are currently recruiting patients (table 1).
Glucocorticoid-induced tumor necrosis factor receptor
GITR (CD357), a member of TNF receptor superfamily 18, is highly expressed on Tregs—which contributes to Tregs’ expansion and differentiation63—and at low levels on naïve and memory T cells.64 It peaks after 2–3 days of single administration and declines by day 5. The complementary ligand is GITR ligand, which is expressed at low levels in APCs such as DCs, macrophages and B cells. GITR boosts T-lymphocyte activity after suboptimal TCR stimulation by upregulating IL-2 and IFN-γ.65 66 It also enhances T-cell survival by inhibiting TCR activation-induced apoptosis.67 Preclinical data on GITR monotherapy demonstrated that the agonist anti-GITR antibody DTA-1 was efficacious against cancer cells in B16 melanoma mouse models.68 It stimulates both CD4+ and CD8+ T cells and inhibits intratumoral Tregs by altering their stability.
A phase 1 study of the GITR monoclonal antibody of TRX-518 was performed in 40 patients with metastatic solid tumors such as melanoma and non-small cell lung and colorectal cancers.69 The patients tolerated TRX-518 well and did not experience DLT. The incidence rate for TRAEs was 15%, and the most common events were cough and fatigue (both 28%). Four of 28 patients had immune-related SD, although the efficacy data in the study were limited.69 Also, a phase 1 study of AMG228, another GITR agonist, involved 30 patients with advanced solid tumors, including colorectal, head and neck squamous cell, urothelial and non-small cell lung cancers.70 The most common TRAEs were fatigue, infusion-related reactions, fever and decreased appetite. Three fatal AEs, consisting of pneumonitis, acute hypoxemic respiratory failure and progressive disease were reported, but in the remaining patients, the MTD was not reached. Twenty-seven patients were evaluable by using immune-related response criteria and seven had SD. However, T-cell activation and antitumor activity were not correlated with GITR coverage in tumors or peripheral blood. Another GITR agonist, MEDI1873, was investigated in a phase 1 study.71 Forty patients with advanced tumors (including non-small cell lung, head and neck squamous cell and colorectal cancers) were enrolled, 82.5% of whom experienced drug-related AEs. Three patients experienced DLT, and one presented with non-ST-elevation myocardial infarction at the maximum dose of MEDI1873; however, the MTD was not reached. The best overall response was SD, occurring in 42.5% of the patients, and SD was maintained in three patients for more than 52 weeks. In an associated translational research study, MEDI1873 was involved in GITR expression on CD4+ T cells and increased it in CD4+/Ki67+ T cells at doses greater than 25 mg. Responses in intratumoral T cells were similar to those in extratumoral T cells.71
The combination of a GITR agonist with other treatments was explored as well. In a preclinical study with an ID8 murine ovarian cancer model, the combination of a GITR agonist and a PD-1 inhibitor inhibited peritoneal ID8 tumor growth by increasing the responses of memory immune cells to tumor cells and the frequencies of IFN-γ-producing effector T cells. In addition, it suppressed Tregs and myeloid-derived suppressor cells by shifting an immunosuppressive tumor milieu to an immunostimulatory state.65 72 Furthermore, adding chemotherapy (cisplatin and paclitaxel) to the GITR agonist and PD-1 inhibitor enhanced the synergy of the treatments.72
An active study of the anti-GITR agonist BMS-986156 and nivolumab suggested that the incidence rate for grade 1 TRAEs was 70% in patients receiving combination therapy compared with 59% in those receiving monotherapy; no patients in either group experienced DLT (table 1).73 A comparison study of MK-1248 monotherapy and MK-1248 in combination with pembrolizumab in 37 patients showed that 17 patients (45.9%) had at least one TRAE and that most AEs were manageable and not accompanied by DLT. The incidence of serious AEs did not differ markedly between the two groups: 6 of 20 patients receiving monotherapy and 5 of 17 receiving combination treatment. In terms of efficacy, of the patients in the combination treatment group, one had a CR and two had a PR.74 A phase 1 study of MK4166 (NCT02132754) with and without pembrolizumab demonstrated a similar safety profile but higher response rates in immune checkpoint inhibitor-naïve melanoma patients.75
CD40
CD40 is a member of TNF receptor superfamily 5. It is expressed on DCs, B lymphocytes, monocytes, and vascular and epithelial cells. Its ligand, CD40L (CD154), is expressed transiently on activated T lymphocytes.76 77 CD40 signaling in DCs promotes proinflammatory cytokine release, upregulates costimulatory molecules and facilitates the cross-presentation of antigens.78 CD40 ligation of B cells also increases B cells’ antigen-presentation capacity and promotes immunoglobulin class switching.79 80 CD40 is not a direct T-cell agonist. However, its increased antigen presentation and immunoglobulin class-switching enhances T-cell activation indirectly. CD40 also affects the expression of proapoptotic and antiapoptotic genes on different types of cells. In normal and certain low-grade malignant B cells, CD40 ligation rapidly rescues cells from apoptosis via the nuclear factor-κB pathway, whereas CD40 ligation causes the apoptosis of breast carcinoma cells by upregulating Bax expression.76 Macrophages activated by CD40 can become tumoricidal, facilitate the depletion of tumor stroma, and cause tumor regression independent of T cells.
A preclinical study of CD40 agonists in pancreatic ductal carcinoma suggested that these agonistic antibodies activate macrophages, directly causing tumor regression, and that this process is independent of T cells.81 Based on this scientific rationale, a CD40 agonistic antibody from multimeric versions of CD40L was evaluated in 32 patients with solid tumors or lymphoma.82 Two patients had objective responses. In addition, the researchers conducting the study determined that serum liver transaminase was a scale for defining MTD. In another study, CP-870,893, another fully selective CD40 agonist, was given to 29 patients with advanced tumors (mostly melanomas), in whom manageable CPS was the most common AE. Four patients with melanoma had objective responses at restaging.83 CDX-1140, APX005M and SEA-CD40 are other candidate agonistic CD40 monoclonal antibodies for cancer immunotherapy, but they resulted in minor antitumor responses in early-phase studies (table 1).
Studies of some preclinical tumor models have shown that combination therapies are promising as means of overcoming immune refractoriness. In a pancreatic carcinoma model, T-cell immunity induced by a CD40 agonist overcame resistance to PD-L1 or CTLA-4 blockage.84
Based on the assumption that CP-870,893 has a synergic effect with chemotherapy when given at the single-agent MTD, it has been extensively tested in combination with cytotoxic chemotherapy. In a study of 22 patients with treatment-naïve advanced pancreatic cancer, patients were given gemcitabine and CP-870,893.85 The drug was well tolerated. One patient experienced DLT (a grade 4 cerebrovascular accident) at a dose that was regarded as the MTD. The most common TRAE was cytokine release syndrome. Patients given combination therapy had better prognoses than did those given monotherapy, with four patients having PRs and 11 patients having SD. CP-870, 893 has also shown good results when combined with paclitaxel/cisplatin and administered to cohorts of mostly melanoma patients.86 In other studies, CP-870, 893 combined with cisplatin and pemetrexed in patients with malignant pleural mesothelioma had tolerable toxicity profiles and tumor control effects (overall response rate, 40%, respectively)87 (table 1).
An active phase 1b trial of the anti-CD40 antibody APX005M given to patients with pancreatic cancer in combination with gemcitabine and nab-paclitaxel with or without nivolumab (NCT03214250) showed favorable results.88 In the DLT-evaluable population (24 patients), the overall response rate was 54%. In the subgroup who received nivolumab, the overall response rate was 67%. A phase 2 study of APX005M is now underway, as are other combination trials of APX005M with PD-1, PD-L1 or Flt3 ligand (table 2).
ICOS (CD278)
ICOS is a member of the CD28 superfamily of costimulatory molecules. Like other members of this superfamily, its expression can be induced rapidly by T-cell activation and delivers secondary costimulatory signals inside T cells.89 90 The ligand of ICOS is ICOS ligand, which is mostly expressed on B cells, macrophages and DCs. The expression of ICOS ligand can be induced by IFN-γand TNF-α.91 ICOS ligand-ICOS signaling modestly promotes T-cell proliferation and differentiation and substantially enhances the production of cytokines such as IL-4, IL-5, IL-10, IFN-γ and TNF-α. It also induces Foxp3 transcription and suppresses Tregs.92 93
The efficacy of JTX2011, a cross-reactive humanized ICOS agonist, was tested first in murine cancer models. The antibody showed activity when given as monotherapy and in combination with anti-PD-L1 antibodies, and its level of activity corresponded with the level of ICOS expression on tumor interstitial Tregs.94 This study also demonstrated that non-small cell lung cancer and head and neck squamous cell carcinoma have higher percentages of ICOS-expressing cell infiltrates as demonstrated by an integrated expression analysis of human tumors. Subsequent, active phase 1/2 trials in patients with advanced malignancies have tested JTX2011 when given as monotherapy and in combination with immune checkpoint inhibitors.95 The study demonstrated that JTX2011-based therapy was tolerable, but it did not demonstrate promising results in terms of response. Of the 67 patients given JTX2011 monotherapy, only 1 (1.5%) had a PR. Of the 106 patients who received JTX2011 in combination with nivolumab, 8 (7.5%) had PRs (table 2).
The anti-ICOS monoclonal antibody KY1044 targets depletion of intratumoral Tregs and stimulation of T effectors.96 A phase 1 trial of KY1044 as monotherapy or in combination with atezolizumab is underway (NCT03829501).97 A first-in-human study of the ICOS agonist GSK3359609 was performed with the agonist given as monotherapy and in combination with pembrolizumab.98 The primary basket trial enrolled 98 patients with metastatic or relapsed invasive cancer and demonstrated drug safety in both monotherapy and combination therapy groups without reaching the MTD. In an expansion cohort of patients with head and neck squamous cell carcinoma, 51 patients received either monotherapy or combination therapy with pembrolizumab.99 The overall response rate was 8% (1 of 8 patients) for the monotherapy group and 28% (8 of 29 patients) for the combination therapy group. The combination treatment showed promising antitumor activity and a manageable safety profile. A phase 3 trial of KY1044 and atezolizumab in patients with PD-L1-positive head and neck squamous cell carcinoma is ongoing (NCT04128696) (table 1).
Challenges associated with targeting costimulatory pathways to improve antitumor immunity
Although checkpoint blockade immunotherapy advances cancer treatment, the resulting tumor control outcomes vary among patients. Various factors contribute to resistance to checkpoint blockade immunotherapy, such as changes in tumor mutations, T-cell infiltration, suppressive tumor microenvironments, antigen presentation deficiencies and other non-specific issues.100 Likewise, therapies targeting costimulatory molecule pathways are also complicated by tumor subtypes, expression pattern of the target molecules, and how to combine with other immune check point inhibitor or chemotherapy. In particular, the use of a single agonist drug has shown a lack of response in early-phase trials, and the overall response to these agonists is, contrary to researchers’ expectations, very low (less than 10%).101 However, when combined with other treatment methods, synergism has been observed. Overall, a deeper understanding of the impact of these agents in vivo is needed to help identify logical combination approaches that can be tested in the clinic
Regarding subtypes of cancers, patients treated with urelumab with nivolumab and MK-4166 with pembrolizumab in melanoma patients have had overall response rates of more than 50%. Moreover, patients with pancreatic cancer—a disease known to respond poorly to immunotherapy—who were treated with CD40 with chemotherapy and immune checkpoint inhibitors.85 88 In other studies, PRs have been seen in patients with head and neck squamous cell cancer who were treated with the OX40 agonist MEDI0562 urelumab plus nivolumab, or utomilumab plus pembrolizumab (one case each).25 61 62 CRs and PRs were also seen in patients with renal cell carcinoma who were treated with utomilumab plus pembrolizumab.61 In addition, PRs were seen in patients with non-small cell lung cancer who received 4-1BB agonist with anti PD-L1.61 62 Patients with small cell lung cancer, bladder cancer, and anaplastic thyroid cancer have also responded to combination therapy.25 61
The main function of a T-cell agonist is to enhance T-cell proliferation and survival without driving Treg expansion. However, most costimulatory molecules targeted are expressed for only a brief time following stimulation. Another hurdle to this form of therapy is that repeated T cell stimulation by agonistic approaches can led to exhaustion.102 Therefore, the concept of treatment duration has been examined during long-term follow-up for immune modulator trials. In fact, some studies have shown that T-cell activation decreases after a few cycles of the agonistic agent demonstrating that this may be a critical challenge in this approach. Two major avenues are being explored to address overcome the T-cell exhaustion hypothesis. The first avenue is to examine the impact of the duration of treatment. How many cycles are needed to induce a properly activated T-cell response without resulting in exhaustion? For example, the OX40 agonist as an anti-9B12 murine antibody has an effect within only one cycle, not multiple cycle.24
The second avenue is the sequencing of drug administration in agonist combination studies (ie, agonist followed by antagonist vs antagonist followed by agonist vs concurrent administration). In the orthotopically transplanted murine mammary tumor virus polyoma middle T murine model of mammary cancer refractory to PD-1 blockade, OX40 stimulation followed by PD-1 blockage delayed tumor growth and increased the durable responses of CD4+ and CD8+ T cells to tumors.62
Therefore, the concept of treatment duration has been examined in long-term follow-up in immune modulator trials.103 In combination of OX40 and concurrent anti-PD-L1 blockade led to rapid, intense intratumoral T-cell proliferation in patients with solid cancer but also induced exhaustion of T-cell immunoglobulin and mucin-domain-containing-3+CD8+ cells in the later stages of immune response.27 In contrast, an encouraging finding regarding T-cell agonists is that sequential administration of these agonists with PD-1 or PD-L1 can reverse T-cell exhaustion and maintain its effect, even in mammary tumor virus polyoma middle T mammary cancer models resistant to PD-1 blockage.
Several studies have demonstrated that radiotherapy can be synergistic with immune check point inhibitor treatment.104–107 When polyomavirus middle T mammary cancer murine models with anti-PD-L1 agent resistance were treated with sequential OX40 activation and radiotherapy, the antitumor activity of the OX40 agonist was profoundly enhanced.27 It has also been shown that 4-1BB administered with radiation therapy enhances antitumor activity.57 Interestingly, treatment with both CTLA-4 and OX40 antibody after radiation may be optimal due to the ability of this combination treatment to increase antigen expression after radiation. The advantage of sequential therapy may be related to the exhaustion of Tregs, release of tumor antigens and enhanced proliferation of CD4 and CD8 cells. Thus, questions emerge about the timing of T-cell agonist treatment when it is combined with other treatment methods.
To improve the efficacy of agonist therapies, it is important to understand the expression level of the costimulatory receptor within the tumor microenvironment by examining expression on tumor infiltrating lymphocytes. Because inefficient T-cell infiltration may be due to a reduced influx of tumor-infiltrating APCs, presence of suppressive or immature APC subsets or dysfunctional tumor vasculature.100 108 If the number of tumor-infiltrating APCs is adequate, T-cell infiltration may possibly be increased through the use of immunotherapies that increase chemokine release by APCs or radiotherapies or chemotherapies that can create an immune-permissive tumor microenvironment by inducing inflammatory immune responses. In situations in which the influx of APCs is reduced at a tumor site, radiotherapy or chemotherapy may be given before the administration of T-cell-agonist-based reagents or checkpoint inhibitors to release chemokines such as CCL2 and CCL5 that attract DCs to the tumor.109 110
The timing of drug administration should be calculated based on drug half-lives and the expression peaks for the agonist target. However, this method of calculating administration time needs further refinement.27 Based on the lack of accuracy in pharmacokinetics, agonists might be used in suboptimal settings and lead to failure in early-phase trials. Unfortunately, researchers have yet to determine the optimal timing, number of doses, and methods to use when delivering multiple treatment modalities.
The control of immune-related AEs can be problematic in immunotherapy. Agonists of T cells can boost the original immune effects of cancer, have unexpected off-target effects, or aggravate pre-existing autoimmune disease. More than half of cancer patients given previously approved immune checkpoint inhibitors experience toxic effects, including dermatologic and gastrointestinal effects.111 Most toxic effects are manageable with proper care. However, severe toxicity precludes patients from further drug administration and can even lead to death. Sufficient data to establish complete safety profiles for T-cell agonists are lacking. Therefore, investigators should always take into consideration both expected and unexpected TRAEs. The majority of AEs greater than grade 2 are pneumonitis, transaminitis and hematological toxic effects.28 52 Some agents cause DLT, including non-ST-elevation myocardial infarctions, at doses below the MTD.71
Finally, we do not have any useful biomarkers with which to gage responses to immune agonists. In the case of immune check-point inhibitors, PD-L1 expression and the tumor mutation burden are now used as biomarkers of response to immunotherapy.112 113 However, these markers are insufficient and there are no known biomarkers for responses to T-cell agonists. Hence, one of the reasons that researchers have not seen strong signals of success in agonist trials might be that all patients were enrolled without stratification based on useful biomarkers.
Summary
With the development of immunotherapy, more methods than ever are available to promote the immune system’s resistance to tumor cells. The currently approved immunotherapies enhance the immune system primarily by targeting immune checkpoint molecules related to immune-escape mechanisms. T-cell activation requires not only a TCR signal but also a second costimulatory signal. OX40, 4-1BB, GITR and ICOS are costimulatory molecules essential to complete T-cell activation. Targeting CD40 on B cells and DCs results in a secondary boost in T-cell activation via antigen presentation and cytokine secretion. Agonists of costimulatory molecules may lead to improved T-cell activation and tumor control. In preclinical studies, some agents have demonstrated clinical efficacy in tumor control. A number of these agonistic drugs have proceeded to first-in-human clinical trials. Although some agents have failed to fulfill their intended goals, others have demonstrated promising results in early phase I trials. In particular, the combination of immune checkpoint inhibitors and chemotherapy or radiotherapy has also inhibited tumor progression, and a potential synergistic mechanism of combination therapy may play a role in the positive results seen in both animal and human models. Based on these potential benefits of combination therapy, combination trials have been under development, especially those involving checkpoint inhibitors. Most agents demonstrated their safety in preclinical or early-phase studies without reaching the DLT. Investigators expect these agents to demonstrate clinical benefits in ongoing last-phase trials, and phase 2 and 3 trials of these drugs for various types of solid tumors are ongoing. Moreover, other types of immunotherapy, such as oncolytic virus therapy and chimeric antigen receptor T-cell therapy, can be combined, and neoantigen vaccines can potentiate responses to T-cell agonists. More studies of combination treatments are warranted.
Nevertheless, some challenges must be managed before T-cell agonists can be widely used in cancer immunotherapy. Inadequate T cell infiltration and activation, reduced influx and activity of APCs, and depressive tumor microenvironments impede the antitumor function of costimulatory agonists. In addition, finding ways to incorporate T-cell agonist-based treatments with other types of cancer treatment and to alleviate concerns about possible immune deletion due to constant T-cell stimulation are important issues. The sequence and dose of treatments is related to enhanced responses and determining optimal treatment delivery methods will be critical in planning combination trials. Lastly, an important challenge is finding a way to manage immune-related AEs. More than half of patients receiving immune checkpoint inhibitors experience more than one kind of AE. T-cell agonists enhance the same immune responses as those enhanced by immune checkpoint inhibitors, so they also may cause AEs. Moreover, combination therapy trials involving T-cell agonists may induce serious AEs, as well. The discovery of potential biomarkers is important to the prediction of benefits from immune-agonist treatment.
Immune costimulatory agonists are powerful candidates for future immunotherapy and have provided promising results in early-phase trials. T-cell agonists will show promising ways against cancer.
Acknowledgments
The authors would like to thank Sunita Patterson and Laura L. Russell from the Scientific Publications Team, Research Medical Library at The University of Texas MD Anderson Cancer Center, who helped with scientific editing of this manuscript.
References
Footnotes
Twitter @AnaingMD
YC and YS contributed equally.
Contributors YC and YS drafted the manuscript. CLH and AN were involved in the writing and critical review of the manuscript. JH and GC participated in the critical review of the content of the manuscript. All authors read and provided final approval of the version to be published.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests AN declaresResearch funding from NCI; EMD Serono; MedImmune; Healios Onc. Nutrition; Atterocor/Millendo; Amplimmune; ARMO BioSciences; Karyopharm Therapeutics; Incyte; Novartis; Regeneron; Merck; BMS; Pfizer, CytomX Therapeutics; Neon Therapeutics; Calithera Biosciences; TopAlliance Biosciences; Eli Lilly; Kymab; PsiOxus; Arcus Biosciences;· On advisory board of CytomX Therapeutics, Novartis and Genome & Company· Travel and accommodation expense from ARMO BioSciences. JH declares Research funding: Immune Deficiency Foundation, Jeffery Modell Foundation and chao physician-scientist. Baxalta Advisory board: Takeda, CSL-BehringWe will provide COI disclosure form from all authors if published.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.