Article Text

Abstract
Background Tumor relapse constitutes a major challenge for anti-tumoral treatments, including immunotherapies. Indeed, most cancer-related deaths occur during the tumor relapse phase.
Methods We designed a mouse model of tumor relapse in which mice transplanted with E7+ TC1 tumor cells received a single therapeutic vaccination of STxB-E7+IFNα. Unlike the complete regression observed after two vaccinations, such a treatment induced a transient shrinkage of the tumor mass, followed by a rapid tumor outgrowth. To prevent this relapse, we tested the efficacy of a local administration of IFNα together with a systemic therapy with anti-PD1 Ab. The immune response was analyzed during both the tumor regression and relapse phases.
Results We show that, during the regression phase, tumors of mice treated with a single vaccination of STxB-E7 + IFNα harbor fewer activated CD8 T cells and monocytes than tumors doomed to fully regress after two vaccinations. In contrast, the systemic injection of an anti-PD1 Ab combined with the peri-tumoral injection of IFNα in this time frame promotes infiltration of activated CD8 T cells and myeloid cells, which, together, exert a high cytotoxicity in vitro against TC1 cells. Moreover, the IFNα and anti-PD1 Ab combination was found to be more efficient than IFNα or anti-PD1 used alone in preventing tumor relapse and was better able to prolong mice survival.
Conclusions Together, these results indicate that the local increase of IFNα in combination with an anti-PD1 therapy is an effective way to promote efficient and durable innate and adaptive immune responses preventing tumor relapse.
- immunotherapy
- interferon inducers
- lymphocytes
- tumor-infiltrating
- macrophages
- programmed cell death 1 receptor
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- immunotherapy
- interferon inducers
- lymphocytes
- tumor-infiltrating
- macrophages
- programmed cell death 1 receptor
Background
A transient response to therapy followed by recurrence and drug resistance is a common clinical outcome in many types of cancers. Glioblastoma, for example, relapse in nearly all patients, despite aggressive therapy.1 Among patients with ovarian cancer, the rate of recurrence reaches 85%.2 More importantly, tumor relapses are the cause of cancer-related mortality. Thus, the discovery of therapeutic approaches that seek to prevent tumor relapse is essential for improving cancer medicine.
The origin of tumor escape can be intrinsic to the tumor cells, often through the selection of resistant variants. In particular, an initial attack by the immune system frequently selects cancer subclones poorly recognized by the immune system, a phenomenon called “tumor editing”.3 Indeed, some selected tumor variants no longer present tumor-associated antigens recognized by cytotoxic T cells. Others have acquired a resistance to cytotoxic molecules. For example, 40% of non-small cell lung cancers and 80% of breast tumors present HLA alterations, including total or partial HLA class I antigen loss, which leads to their immune escape.4 5
In addition, a chronic inflammatory response frequently develops in the tumor microenvironment, which promotes the expression of inhibitory molecules on immune cells, such as PD1 and CTLA-4. The presence of these molecules at the surface of activated T cells is functionally important for preventing their excessive expansion in various immune responses. However, their induction at the surface of tumor-infiltrating T lymphocytes (TIL) leads to a premature weakening of their response against the tumor.6
Tumor escape may also be due to a tumor microenvironment that protects cancer cells from an immune cell attack. The recruitment of myeloid-derived suppressor cells and T regulatory cells at the tumor site, together with the accumulation of molecules such as IL-10, TGFβ and VEGF,7 contribute to build such an immunosuppressive tumor microenvironment. For instance, TGFβ is often associated with tumor resistance and fibrosis. TGFβ decreases the capacity of myeloid cells to present tumor antigen by decreasing MHCII expression at their surface.8 It also prevents them from producing anti-tumoral cytokines such as type I IFN.9
Therefore, various therapeutic interventions blocking these tumor escape mechanisms have been envisaged to overcome tumor relapse. The targeting of immune checkpoints has evolved as the treatment of choice to preserve and restore the function of anti-tumor T cells. For multiple cancers, anti-PD1 immunotherapy has been successful at enhancing effector CD8+ T-cell responses associated with objective clinical responses. However, a large fraction of patients treated with this approach does not respond or if they do respond, ultimately experience tumor relapse. Thus, multimodal therapies aiming at disrupting different modes of immunosuppression could potentially have synergistic effects and improve the efficacy of current immunotherapies. In line with this, two recent studies have shown that blocking TGFβ increases the therapeutic response of anti-PD-L1 therapy, resulting in tumor regression in an EMT6 breast carcinoma model10 and in the complete elimination of established liver metastases in a colorectal cancer model.11
Another way to achieve this goal would be to directly reinvigorate the different tumor-infiltrating immune cell populations. Indeed, the activation of immune cells triggers negative feedback loops that terminate the immune response. A de novo, acute inflammation may be needed to reboot the immune system against the tumor.12 To this end, type I IFN could directly activate a variety of immune cells, such as dendritic cells, macrophages, monocytes, and both CD4+ and CD8+ T cells.13
Of particular interest is the possibility to activate the tumoricidal activity of macrophages, which are abundant in solid tumors, and which have well-known pro-tumoral and immunosuppressive roles during chronic inflammation. Indeed, recent reports in the literature, including ours, have brought new evidence that myeloid cells, when appropriately activated, can participate in the elimination of tumor cells.14 This has been observed after direct targeting of myeloid cells with an anti-CD40 agonist15 16 as well as after therapies which rely on type I IFN.17–19 In such a context, activated myeloid cells may positively interact with CD8+ T cells and induce tumor regression.18 19
Despite its importance, the number of preclinical models aimed at studying the specific question of tumor relapse is much too low. To model and overcome tumor relapse, we have set up a suboptimal vaccine protocol in which more than 65% of the tumors relapse after an initial phase of regression. We show that in this context, an anti-PD1 combined with a local IFNα therapy prevent tumor recurrences through the development of innate and adaptive immune responses efficiently targeting cancer cells.
Methods
Mice and cell lines
C57BL/6J and FVB/N mice (7 weeks old) were obtained from Janvier Labs or Charles River Laboratories, respectively. Mice were maintained in the specific pathogen-free barrier facility of the Cochin Institute. Animal care was performed by expert technicians in compliance with the Federation of European Laboratory Animal Science association and under the approval of the animal experimentation ethics committee of Paris Descartes. Small and large tumor diameters (d and D) were measured with calipers and the tumor volume was approximated with the volume of a spheroid (d2×D/2).
The TC1 cell line and the TC1-GFP line18 were maintained in DMEM GlutaMAX, with 10% FCS (GE Healthcare), antibiotics (penicillin 50 U/mL, streptomycin 50 µg/mL; GIBCO) and sodium pyruvate (1 mM; GIBCO). Cultured cells were trypsinized, washed three times in serum-free PBS and tumor cell suspensions were injected subcutaneously (105 TC1 cells) in the back of 8-week-old C57BL/6J mice. When the tumors reached a diameter of approximately 6 mm (after 10 days), mice were vaccinated (D0, priming) with a peri-tumoral injection of 15 µg of E7 vaccine (STxB-E7 vaccine)20 and 6×105 U of IFNα4,21 in a total volume of 200 µL. Control mice were injected in parallel with PBS. The next day, the vaccinated mice received the same dose of IFNα. This protocol was repeated a week later in some mice (D7, boost). For experiments conducted in the PyMT-tumor model, FVB/N mice were transplanted in the mammary gland with freshly dissociated PyMT tumor cell suspensions as previously reported.19 After 2 weeks, mice with PyMT tumors (6 mm in a diameter) received one intraperitoneal injection (20 mg/kg) of DMXAA (Sigma). For antibody treatment, mice were injected intraperitoneally with 200 µg of anti-PD1 (#BE0146: clone RMP1-14) purchased from Bioxcell. IFNα (6×105 U) was injected peri-tumorally in 100 µL. Anti-PD1 and IFNα were injected at days 14, 16 and 18 after the priming in TC1-bearing mice or at days 8, 10 and 14 after DMXAA injection.
Preparation of tumor cell suspensions and multicolor flow cytometry
Fresh TC1 tumors were dissociated mechanically and incubated for 30 min at 37°C with DNase I (100 µg/mL; Roche) and collagenase (1 mg/mL; Roche). The cell suspension was filtered on a 40-µm filter then rinsed two times with PBS 2% FCS and 1 mM EDTA. Cells (4×106) were labeled in 96-well round-bottom plates with fluorescent blue reactive dye (Invitrogen) for 20 min at room temperature. Fc receptors were blocked with anti-anti-CD16/CD32 Abs (5 µg/mL; BD Pharmingen). Cells were then labeled with fluorescent Abs (table 1) for 15 min at 4°C with agitation. For the detection of E7-specific CD8 T cells, tumor cell suspensions were stained with Db/E7-Dextramers (Immudex) for 20 min at 4°C, before fluorescent Abs were added for an additional 15 min. After washing in PBS, cells were fixed in 1% PFA, stored at 4°C and acquired the next day with LSR II flow cytometer (BD Bioscience). Data were analyzed with FlowJo V.10.0.7 software.
List of antibodies used in flow cytometry
Cytotoxic assay
Freshly dissociated tumor cell suspensions were stained with anti-F4/80 UltraPureMicroBeads (Miltenyi #130-110-443) and passed through an Automacs (MiltenyiBiotec) following manufacturer’s instructions to purify F4/80+ myeloid cells. The negative fraction was subsequently stained with anti-CD8a MicroBeads (Ly2; Miltenyi #130-117-044) and then CD8+ tumor-infiltrating T cells were purified with the Automacs. Purified CD8+ and F4/80+ cell suspensions were >90% pure. Then, purified myeloid and CD8+ T cells (2×105) were added to a monolayer of TC1-GFP cells (2×104), and co-cultured for 24 hours at 37°C before acquisition of images with an inverted microscope (TE2000-E; Nikon) equipped with a ×20 objective, and Metamorph imaging software. The fraction of the surface covered by GFP was calculated using ImageJ.
Transcriptomic analysis
Total RNA was extracted from fresh tumors using the RNeasy Mini kit (Qiagen). Gene expression was analyzed with the nanostring technology as described earlier.18 The house keeping genes (ACTB, GAPDH and RPL4) were used to normalize for RNA loading differences. The positive control and housekeeping normalized counts were then logarithmically transformed and used for all subsequent analysis.
Statistics
Data were analyzed with GraphPad Prism5 software to run unpaired Student’s t-test or one-way ANOVA plus Tukey test for multiple comparisons. Values ≤0.05 were considered significant. *p<0.05; **p<0.01; ***p<0.001.
Results
Suboptimal vaccination leads to a partial regression followed by tumor relapse
We have previously reported a complete and persistent regression of TC1 transplanted tumors in mice receiving two peri-tumoral injections (p.t.), 7 days apart, of a vaccination that combined STxB-E7 + IFNα (priming and boost).18 The recruitment and activation of both CD8+ T cells and myeloid cells at the tumor site were required to reach this optimal regression. From this model and in order to tackle the relapse phase usually seen in human cancers, we set up an experimental relapse model in which a suboptimal initial treatment, with only one injection of the STxB-E7 vaccine + IFNα (priming only), induces a regression of the TC1 tumors usually between day 8 and day 14 post-priming but followed by a robust regrow (figure 1).
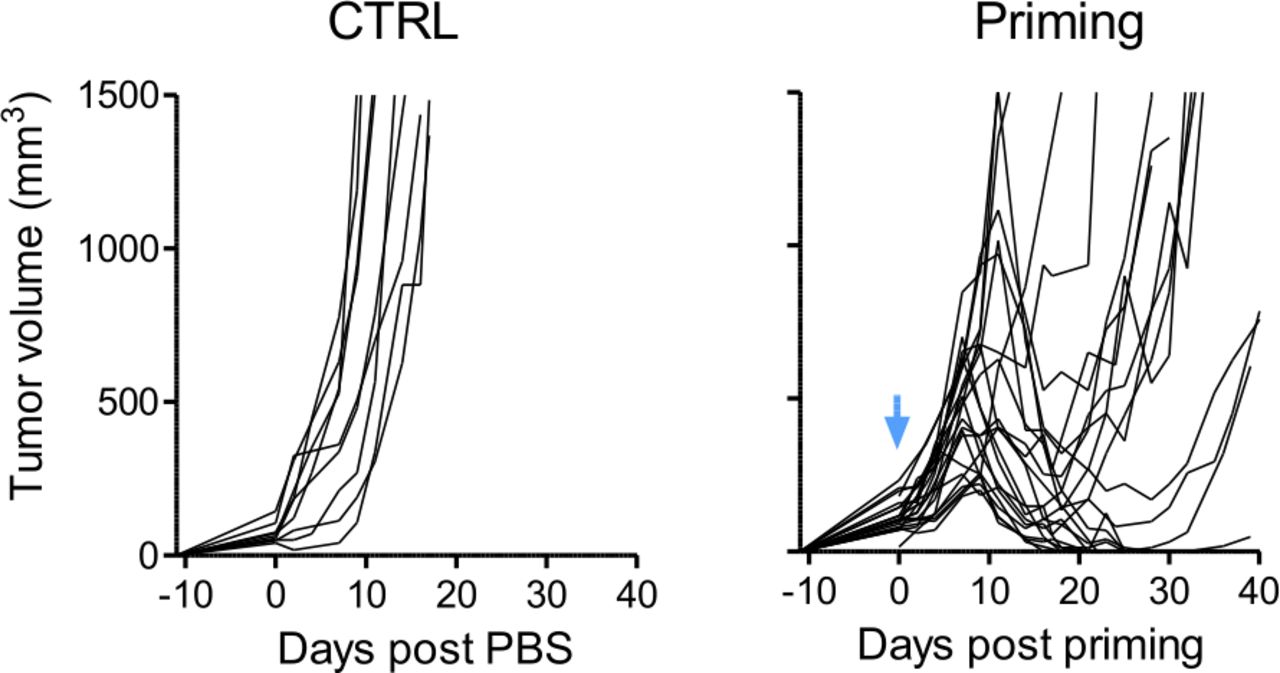
TC1 tumor relapse is obtained after a single injection of E7 vaccine+IFNα. C57BL6/J mice with TC1 tumors received one injection of the E7 vaccine associated with IFNα. Tumor growth curves (individual mice) are shown for both control and treated group. Blue arrow: day of priming (day 0). n=22 or n=9 mice for treated or control tumors, respectively, from 5 independent experiments.
Immune infiltrate influences the tumor fate
We first examined which parameters were most critical in the induction of a complete tumor regression without any relapse. To this end, we first compared the evolution of tumor size after one prime (P), that is, conditions in which a regrow of the tumor is usually observed, with that observed after a priming and boost (PAB) treatment. The two successive PAB vaccinations induced a strong tumor burden reduction, with a 14-fold decrease in tumor size at day 14 after the priming, whereas the suboptimal P protocol induced only a 2-fold reduction in tumor volume at this time point (figure 2A). No such difference was visible at earlier time points, for example, day 10, suggesting that this could be a tipping point in tumor evolution.

Difference in terms of immune infiltrate between PAB and P tumors during tumor regression. (A) Comparison of the tumor volume evolution between control, Primed and Boost (PAB), or Primed only (P)–treated mice during the phase of tumor regression (day 8 to day 14 post first injection of E7 vaccine+IFNα). n=10–17 mice from 3 to 5 independent experiments. B–E: measurements were performed at day 10. (B) The proportion of CD45+ cells in tumors for control (empty circles) or PAB-treated (black circles) or P-treated (blue circles) mice was determined by flow cytometry after dissociation of the tumors. n=27–40 mice from 5 independent experiments. Results are expressed as mean±SEM. Tukey’s multiple comparison test. *p<0.05; **p<0.01; ***p<0.001. (C) Correlation between the percentage of CD45+ cells among the viable cells of the tumors and the tumor volume. (D) The proportion of CD8 T cells, tumor-associated macrophages (TAM) and monocytes among those CD45+ cells for control (empty circles) or PAB-treated (black circles) or P-treated (blue circles) mice are shown. n=12–37 mice from 5 independent experiments. Results are expressed as mean±SEM. Tukey’s multiple comparison test. *p<0.05; **p<0.01; ***p<0.001. (E) Tumors from control, PAB-treated or P-treated mice were analyzed by flow cytometry for the expression of PD1 on CD8 T cells and MHC class II on TAM and monocytes. n=6–27 mice from 3 to 5 independent experiments.
We thus examined closely several features of the tumor microenvironment at this particular time point in mice having received the P or the PAB treatment. Cytokines and chemokines present in the tumor microenvironment of the two groups were not significantly different. Indeed, compared with untreated mice, both treated groups showed increased levels of cytotoxic and cytostatic cytokines such as TNFα, Grzmb, IFNγ and iNOS (online supplemental figure 1A) and of T-cell chemoattractants (CXCL9, CXCL10, CCL5 and CCL3) (online supplemental figure 1B). Moreover, the same level of pro-tumoral, anti-inflammatory cytokines (VEGFA, TGFβ, IL10) were detected in the P-treated and PAB-treated groups (online supplemental figure 1B).
Supplemental material
Next, we characterized the immune cells infiltrating the tumors at day 10 again (online supplemental figure 2). Tumors from mice treated with only one prime were less infiltrated by immune cells, with 30% of immune cells on average in P-treated tumors versus 45% in PAB-treated ones. The global abundance of this CD45+ immune infiltrate was negatively correlated with the tumor size (figure 2B). All the progressing tumors in the control group (PBS-treated mice) showed less than 25% of immune cells whereas in the majority of PAB-treated tumors, which systematically regressed, more than 25% of CD45+ cells were present. Concerning the P-treated tumors, the proportion of immune cells infiltrating the tumors of different mice was highly variable, just as the evolution of their tumor size.
What about the abundance of CD8+ T cells and myeloid cells in these tumors? CD8+ T cells were two times more abundant in PAB-treated than in P-treated groups (figure 2D). The proportion of macrophages (Ly6Cneg Ly6gneg F4/80+ or TAM) was similar in P-treated and PAB-treated groups. However, monocytes (Ly6C+ Ly6gneg F4/80lo) were slightly more abundant in PAB-treated tumors, so that the balance between monocytes and macrophages was skewed in favor of monocytes in PAB mice (figure 2D). Of note, both CD8+ T cells and myeloid cells were activated in both P-treated and PAB-treated tumors, as shown by the expression of PD1 or MHCII in the two cell subsets, respectively (figure 2E).
Taken together, these results suggest that the abundance of the immune cell infiltrate and its internal balance are conspicuous parameters associated with an optimal tumor regression.
Tumor relapse is associated with a decrease in immune cell recruitment and activation
Next, we focused on tumors from P-treated mice. The kinetics and behavior of the immune cell infiltrate was evaluated at different key points of the tumor evolution. We selected day 10 and day 17 post-priming as tipping points for the regressing and the relapsing phases, respectively. This also allowed comparing tumors of similar sizes in the two groups.
The proportion of CD45+ immune cells increased progressively from day 4 and reached a peak at day 10, around the start of the regression phase (30% of immune cells among viable cells) and then decreased rapidly thereafter (around 17% at day 17) (figure 3A and online supplemental figure 3). Intriguingly, while the abundance of total CD8+ T cells at day 17 (17%) was larger than during the regression phase (10%), the frequency of E7-specific CD8+ TIL has dropped from 8% at day 10% to 4% at day 17 (figure 3B). Whereas no difference was observed in the TAM proportions (figure 3B), the proportion of monocytes decreased sharply (about 2-fold) between day 10 and day 17, until reaching the proportion found in tumors before treatment. In addition, while the expression of PD1 on CD8+ T cells was maintained between day 10 and day 17, the proportion of MHCII+ activated myeloid cells was considerably reduced during this time frame (figure 3C).
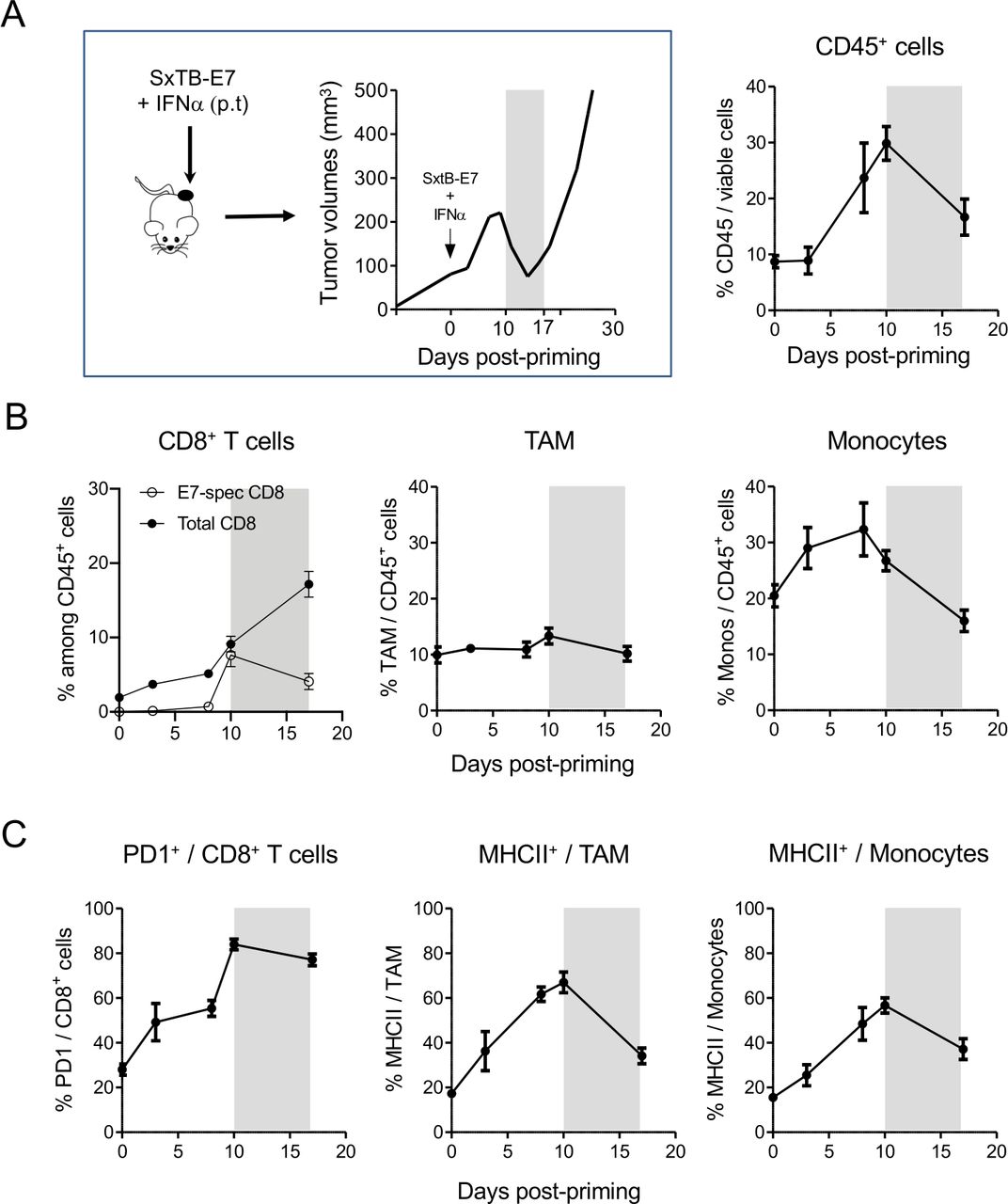
Decrease in immune cell recruitment and activation in the key period preceding tumor relapse. This period, between days 10 and 17 post-priming, is highlighted in gray in all the panels. (A) The proportion of CD45+ cells in tumors of P mice was determined by flow cytometry after dissociation of the tumors at different times during the tumor size evolution. n=20–40 mice from 5 independent experiments. (B) The proportion of total and E7-specific CD8+ T cells, tumor-associated macrophages (TAM) and monocytes among those CD45+ cells for P-treated mice are shown. n=20–40 mice from 5 independent experiments. (C) Tumors from control, PAB-treated or P-treated mice were analyzed by flow cytometry for the expression of PD1 on CD8 T cells and MHC class II on TAM and monocytes. n=6–30 mice from 3 to 5 independent experiments.
All these results show that the suboptimal treatment induced only a transient anti-tumoral immune infiltrate, unable to prevent tumor relapse.
Peri-tumoral IFNα associated with systemic PD1 blockade restores the anti-tumor response
Based on these results, we tested different treatments susceptible to reactivate an anti-tumoral immune response. IFNα has remarkable potential for promoting the activation and recruitment of various immune effector cells, whereas anti-PD1 antibodies are well known to prevent the inhibition of PD1+ T lymphocytes. Separately, both are already used to treat certain types of cancer. We examined to what extent these molecules could prevent tumor relapse after a suboptimal vaccine treatment. Fourteen and 16 days after one peri-tumoral injection of the STxB-E7 vaccine + IFNα that led to tumor regression, two injections of IFNα (5×105 U, p.t.) or anti-PD1 (200 µg i.p.) were performed, separately or together. At day 17, tumors of mice that had received the different treatments were collected and analyzed to determine the potential benefit of such additional treatments. IFNα alone was able to promote an increase in the immune cell infiltrate (figure 4A and online supplemental figure 4A). However, combination with an anti-PD1 was most efficient at recruiting additional CD8+ T cells in the tumors. Indeed, CD8+ T cells represented 37% of CD45+ cells in these treated tumors (figure 4B) compared with 19% or 10% during the relapsing or regressing phase, respectively, when animals were treated with a prime only (figure 3B). Similarly, the proportion of E7-specific CD8 T cells was increased in those tumors (online supplemental figure 4B). Moreover, compared with single therapy with IFNα or anti-PD1 alone, the combination was able to restore the proportion of monocytes found during the regression phase at day 10 (figures 4B and 3B) and to reactivate myeloid cells as shown by MHCII expression (figure 4C). Finally, the association of IFNα and anti-PD1 Ab gave the lowest variability in immune recruitment (online supplemental figure 4C).

Combination of anti-PD1+IFNα is able to restore a well-activated immune infiltrate. Mice received 2 injections of either control (blue circles) or IFNα (light gray circles) or anti-PD1 (dark gray circles) or both (pink circles) in order to prevent tumor relapse at days 14 and 16 post first injection of E7 vaccine+IFNα. (A–C) Measurements were performed at day 17. (A) The proportion of CD45+ cells in tumors after the different treatments was determined by flow cytometry. n=8–15 mice from 3 to 5 independent experiments. Results are expressed as mean±SEM. Tukey’s multiple comparison test. *p<0.05; **p<0.01; ***p<0.001. (B) The proportion of CD8 T cells, tumor-associated macrophages (TAM) and monocytes among that CD45+ are shown. n=20–40 mice from 5 independent experiments. Results are expressed as mean±SEM. Tukey’s multiple comparison test. *p<0.05; **p<0.01; ***p<0.001. (C) The expression of PD1 on CD8 T cells and MHC class II on TAM and monocytes was determined in each condition. n=6–30 mice from 3 to 5 independent experiments. Results are expressed as mean±SEM. Tukey’s multiple comparison test. *p<0.05; **p<0.01; ***p<0.001.
These results show that it is the combination of IFNα + anti-PD1 that was optimal, for the recruitment and the activation of both myeloid and lymphoid cells in the tumors, compared with IFNα or anti-PD1 alone.
Re-activated myeloid and lymphoid cells cooperate to efficiently kill tumor cells
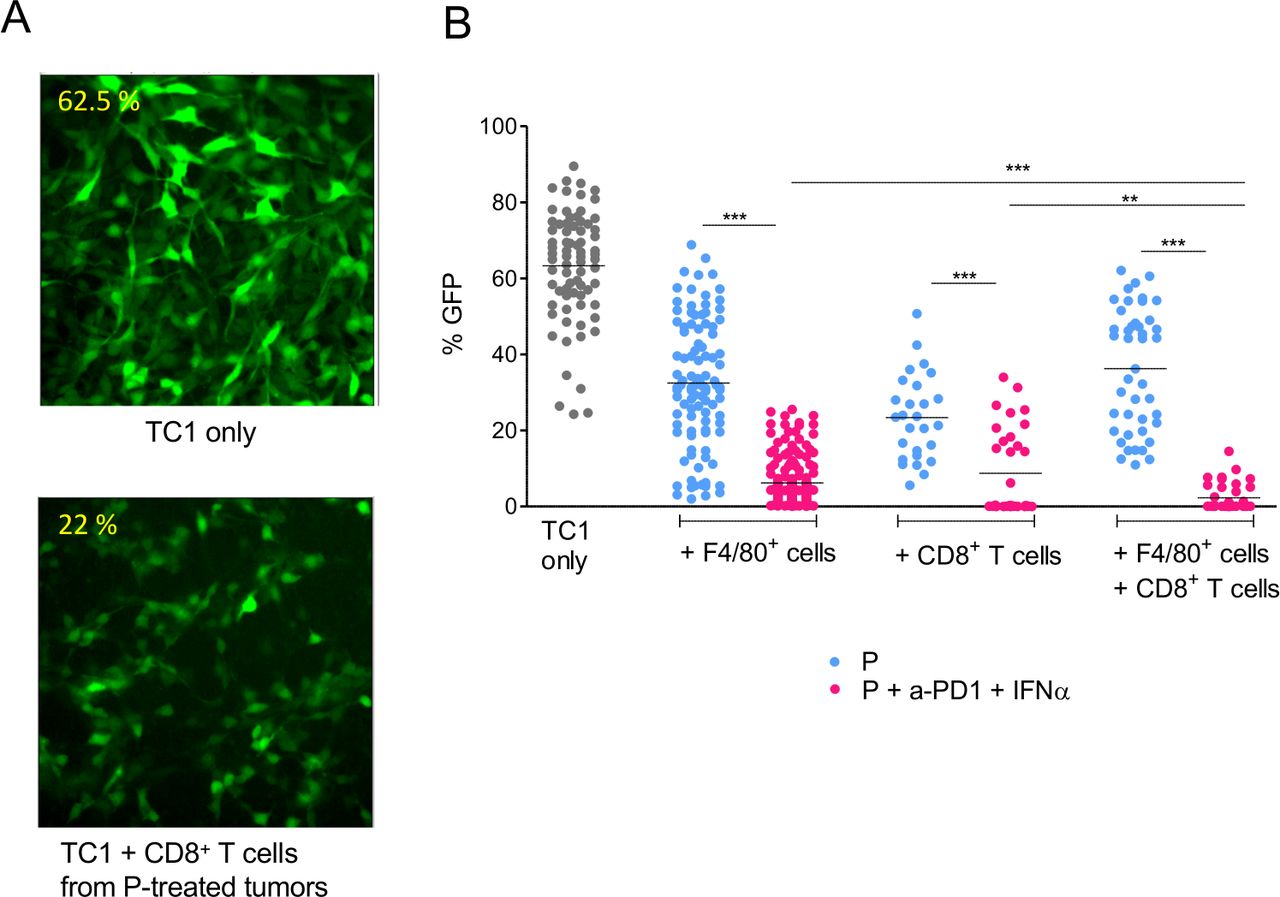
We next examined if the combined anti-PD1+IFNα treatment could increase the cytotoxic activity of CD8+ T cells and myeloid cells. To address this, we purified tumor-infiltrating myeloid (F4/80+ cells) and T cells (CD8+) at day 17 from primed mice, treated or not with anti-PD1 + IFNα. These purified cells were then co-cultured for 24 hours with TC1-GFP+ tumor cells. The GFP fluorescence intensity of the cultures was measured by quantitative imaging in the different conditions. An illustration is shown in figure 5A.
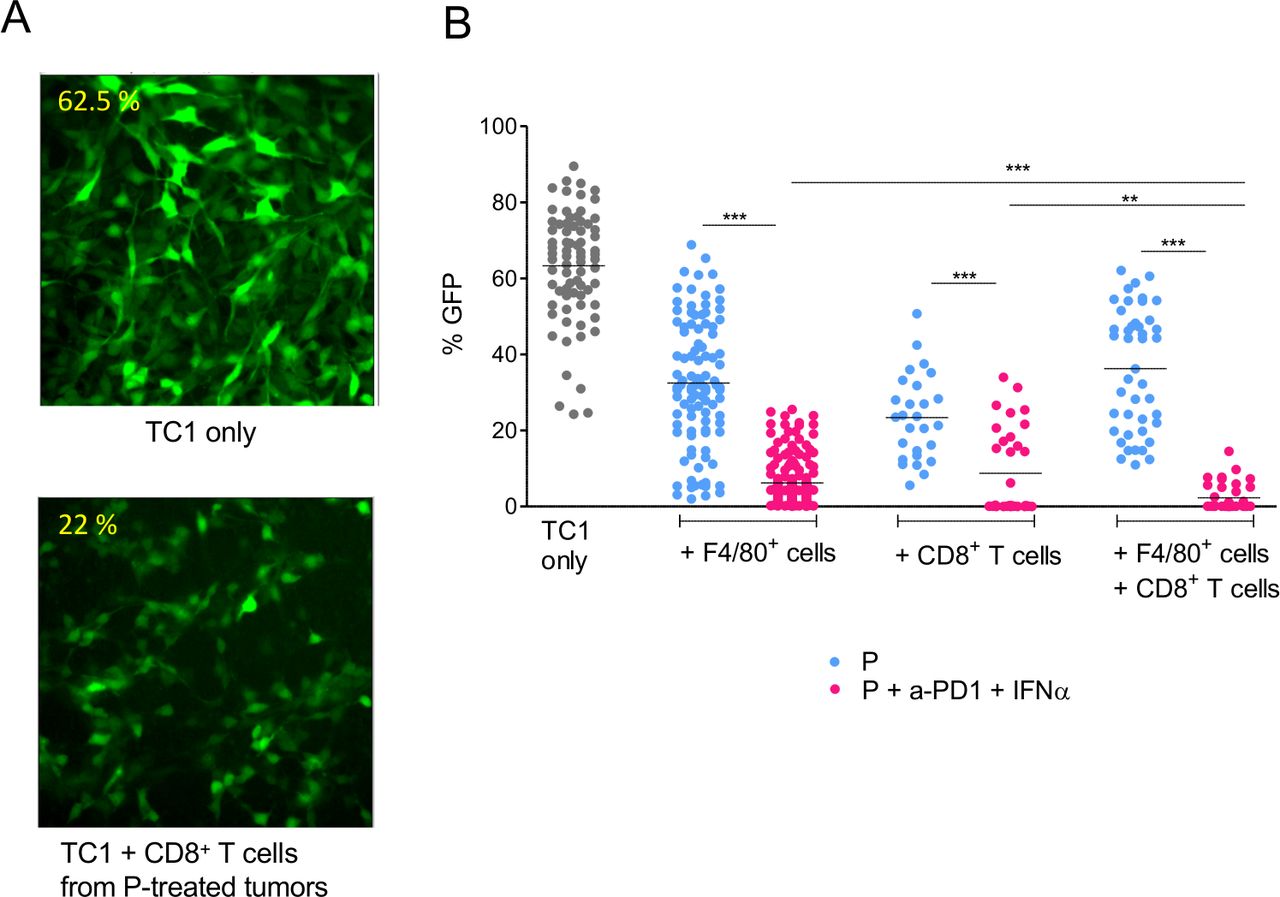
Interaction between myeloid and CD8 T cells for the killing of tumor cells. (A) TC1-GFP+ tumor cells were co-cultured with CD8+ T cells isolated from tumors at day 17. Contrary to control tumors (upper panel), CD8 T cells from P+anti-PD1+IFNα–treated mice (lower panel) were cytotoxic for tumor cells (disappearance of GFP labeling, loss of tumor cells). (B) Quantification of the living cells, from 3 to 5 independent experiments. The apparent fraction of living adherent TC1 cells (fraction of the image occupied by GFP+ cells, measured in 6–17 images per condition) is shown in the various conditions. For each experiment, 3 tumors obtained from 3 mice were pooled to purify enough cells. Student t-test was performed to compare the cytotoxicity of cells purified from P vs P+anti-PD1+IFNα–treated mice for F4/80+ or CD8+ or F4/80++CD8+ T cells conditions. *p<0.05; **p<0.01; ***p<0.001. Tukey’s multiple comparison test was performed to compare the cytotoxicity of F4/80+ or CD8+ or F4/80++CD8+ T purified from P only or P+anti-PD1+IFNα–treated mice.
We observed that TC1-GFP+ cells were more efficiently killed by F4/80+ or CD8+ T cells purified after combined anti-PD1 + IFNα treatment, as detected by the low density of remaining adherent TC1-GFP+ cells, compared with the denser network of TC1-GFP+ cells observed in co-cultures with myeloid cells or CD8+ T cells from P-treated mice (33% vs 7% for myeloid cells and 22% vs 10% for CD8+ T cells purified from P or P + anti-PD1 + IFNα-treated mice, respectively) (figure 5B). Importantly, killing of TC1-GFP+ tumor cells was further improved when both myeloid cells and CD8 T cells were added to the culture of TC1-GFP+ cells. Such a cooperative activity was only seen when immune cells were purified after a combined anti-PD1 + IFNα treatment (3% of GFP+ surface left). Indeed, such a strong cytotoxicity was not observed when myeloid and CD8+ T cells originated from P-treated mice (36% of GFP+ surface left).
Altogether, these data indicate that the combined anti-PD1 + IFNα treatment stimulates the cytotoxicity of myeloid and CD8 T cells, which cooperate functionally to eliminate tumor cells.
Anti-PD1+IFNα, administered as a second-line therapy, prolongs tumor-free survival
Finally, it remained to be established if this combined treatment could prevent tumor relapse. Three injections of anti-PD1 or IFNα or both were performed at day 14, day 16 and day 18 post-priming. IFNα or anti-PD1 Ab injected separately were able to reduce the proportion of mice with relapsing tumors (figure 6A). Indeed, 50% (P + IFNα) and 57% (P + anti-PD1) of the tumors regressed completely in these treated groups of mice, compared with 35% in the P group left untreated (figure 6B). However, the greatest effect was observed with the treatment combining anti-PD1 antibody with IFNα (figure 6A). Indeed, more than 80% of the mice experienced a complete regression of their tumors with this combined treatment (figure 6B). Remarkably, these complete regressions with the combined therapy were sustained over time, as shown by long-term survival for more than 80% of the P+anti-PD1 + IFNα–treated mice, compared with 35%–57% in the primed-only-treated mice or with the single therapies (figure 6C). In addition, we show that the combination of anti-PD1 antibody with IFNα was also efficient for protecting PyMT tumor–bearing mice following a suboptimal treatment with a STING agonist (online supplemental figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Administration of anti-PD1 associated with IFNα in second line induces a long-term tumor-free survival. TC1 tumor–bearing mice primed with the STxB-E7 vaccine received various combinations of treatment to prevent tumor relapse: control, IFNα, anti-PD1 or anti-PD1 + IFNα. (A) Tumor growth curves (individual mice) are shown for each treatment. Dotted lines: days of treatment injections (days 14, 16 and 18). n=13–26 mice from 3 to 5 independent experiments. (B) Proportion of tumors that completely regress at day 30 after priming, after receiving various treatments at days 14, 16 and 18. n=13–26 mice from 3 to 5 independent experiments. (C) Survival of mice that received the different treatments. n=10 mice from 3 independent experiments. Log-rank test. *p<0.05; **p<0.01; ***p<0.001.
Taken together, these results highlight the high efficacy of a combined anti-PD1 and IFNα treatment, administered after an initial tumor regression induced by immunotherapy, in preventing tumor relapse.
Discussion
It is quite common that human cancers initially respond to an anti-tumoral immunotherapy. However, unfortunately, this beneficial effect is frequently followed by a tumor regrowth poorly accessible to a new treatment. Various reasons could explain why this tumor escape occurs: intrinsic alterations of the properties of the cancer cells themselves, modification of the tumor microenvironment or induction of inhibitory molecules such as PD-1 on CD8+ T lymphocytes, which led to a weakened immune response. All these phenomena could be part of the development of a tumor resistance against the immune response, thus leading to a relapse. In order to tackle this issue, we have used in this work a murine model of cancer immunotherapy, in which treatment only induces a transient regression of the tumor. Our main conclusion is that association of anti-PD1 antibody with IFNα is able to prevent the tumor relapse observed in this model. We also demonstrate the importance of a coordinated activation of myeloid cells and CD8+ T cells to fight the cancer cell regrowth.
We have previously shown that a complete tumor regression required a robust immune response.18 We show here that a suboptimal therapeutic intervention leads to a transient immune infiltrate. This strongly suggests that sustaining the immune response over time could prevent tumor relapse. Moreover, anti-tumoral T cells are indeed important to attack tumors, but they cannot be the sole actors. Indeed, their efficacy is maximized when they can cooperate with other cells, in particular with monocytes/macrophages as we have reported earlier.14 18 19 Thus, tumor relapse could be due to an excessive brake exerted on T cells by checkpoint inhibitors and also to a loss of cooperation between T cells and myeloid cells. The suboptimal vaccination protocol used in this study aimed at observing a systematic tumor relapse after an initial vaccination-induced transient regression. At the time where relapses usually start (2 weeks after the vaccination), mice were treated again by systemic anti-PD1 and peri-tumoral IFNα. Such a combined treatment showed a high efficacy for lowering the probability of tumor relapse. Similarly, this combined treatment protected mice with a carcinoma-structured PyMT tumor,19 relapsing after a suboptimal therapy with a STING agonist.
Which key tumor features could explain why a tumor may either completely regress or regrow after a transient regression? To answer this question, we have taken advantage of the difference between mice vaccinated once on day 0 (P) or twice on days 0 and 7 (PAB). Day 10 is a tipping point, when the tumor sizes of the two groups have not yet diverged, but their immune infiltrates start to differ. The comparison of situations P and PAB at day 10 is therefore of major interest. No evidence was observed in terms of immunosuppressive molecules such as IL-10 or TGFβ expression when comparing tumors that will relapse and the ones that will completely regress.
The outcome of these two protocols lay in their respective immune infiltrates. Indeed, PAB-treated tumors, which uniformly regress, harbor more monocytes, and twice more CD8 T cells than P-treated tumors, which are likely to regrow. We expected that intratumoral chemokines would be more abundant in PAB tumors, but unexpectedly this was apparently not the case. Several other features were similar in the two tumor types. In particular, as judged by PD1 and MHC II expression, most CD8+ T cells and monocytes/macrophages were activated in both P-treated and PAB-treated mice. This activation and the abundance of these cells were much larger than that of immune cells found in tumors of mice that had not been vaccinated at all. Thus, the comparison between the P and PAB protocols shows that modest changes in the global immune infiltrate measured at day 10 are nevertheless determinant to allow or not a full tumor regression.
A similar conclusion may be drawn by comparing tumors in mice vaccinated only once, but further treated or not on days 14 and 16 by anti-PD1 or IFNα, or both. We have shown that after a third injection on day 18, the risk of tumor relapse was minimized by the combination, with the isolated treatments having an intermediate effect. The tumor infiltrate was analyzed on day 17, after only two injections, because many tumors would have become too small to be analyzed after three injections. A striking observation (figure 4) is that all treatments (isolated or combined) had an important effect on the abundance of monocytes/macrophages and CD8+ T cells and on their activation status (MHC II expression). It is worth noting that the efficacy of checkpoint inhibitors such as anti-PD1 or anti-CTLA-4 alone is not restricted to their expected effects on T cells. Indeed, it has recently been shown that checkpoint inhibitors can also reshape the myeloid cell compartment,22 which include potentially a direct action on myeloid cells.23 IFNα has an even larger spectrum of action. It has been shown to activate myeloid cells by increasing MHC I, MHC II, CD80 and CD86 expression,24 and to favor the cross-presentation of antigens,25 thus allowing the activation of the adaptive response. In addition, IFN⍺-induced chemokines allow the recruitment/retention of inflammatory monocytes (CCL2, CCL3) and T cells (CXCL10). All these properties could contribute to explain why anti-PD1 alone or IFNα alone were able to increase and activate both cellular effectors and have some anti-relapse efficacy. Thus, the clear anti-tumoral advantage of the combined treatment, in terms of tumor size and survival, cannot be explained with these quantitative features.
Instead, this effect could result from a qualitative difference induced specifically by the anti-PD1 Ab + IFNα combination. Indeed, our results suggested that the combined treatment increased the cytotoxicity of T cells and myeloid cells. Moreover, it was only with the combination that a cooperation between T cells and myeloid cells was observed, resulting with stronger global anti-tumoral cytotoxicity. These results may explain the large proportion of complete tumor regression and the long-term tumor-free survival observed in more than 80% of P + anti-PD1 + IFNα–treated mice.
Conclusions
In conclusion, we have shown that, in order to enhance the anti-tumoral efficacy of checkpoint inhibitors, rather than eliminating myeloid cells whole scale, it makes much more sense to combine these checkpoint inhibitors with treatments aiming at recruiting and activating monocytes locally, as we have done with peri-tumoral IFNα. Other ways of increasing type I IFN could be used, for instance local radiotherapy. This may explain why combining the blockade of CTLA-4 with radiotherapy has shown a clinical benefit associated with a rise in IFNβ secretion in the serum of these patients.26 However, the full production of IFNα/β may be hindered locally by TGFβ as we have reported.9 In this context, the addition of an anti-TGFβ to the local type I IFN inducer plus checkpoint inhibitor would be reasonable.
Acknowledgments
We would like to thank Georges Bismuth for critical review of the manuscript and his helpful suggestions. We are grateful to Agnès Lebon (Cochin Institute, Paris) for providing the IFNα4, and Jean-Pierre Abastado and Bernett Lee (Singapore Immunology Network, Singapore) for the nanostring analysis. We would like to thank Coline Chaput and the staff of the animal facility, as well as the IMAG’IC and CYBIO platforms of the Cochin Institute for their input all along this study. We would like to thank also Eric Tartour (Inserm U970, PARCC, France) for his advice on the production of STxB-E7 vaccine.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
MvG, FR and MT contributed equally.
AT and NB contributed equally.
Contributors NB and AT conceived the study and designed experiments. MT, FR, NB, MvG, HLP and MB performed the experiments. MvG, NB, AT, MT and HLP analyzed the data. NB, MvG and AT wrote the manuscript. MT, FR and NB performed animal experiments. LV and ED prepared IFNα and vaccine, respectively. LJ directed vaccine synthesis.
Funding This research was supported by the Comité de Paris de La Ligue Contre le Cancer (RS17/7536), by a grant from CNRS (PICS 06266 – Projets Internationaux de coopération scientifique) and by the Fondation ARC pour la Recherche sur le Cancer (PJA 2019). MT was granted by the Ministère de l’Enseignement Supérieur, de la Recherche et de l’Innovation and MvG by the Ligue Nationale contre le Cancer. The Johannes team is member of Labex CelTisPhyBio (11-LBX-0038) and Idex Paris Sciences et Lettres (ANR-10-IDEX-0001-02 PSL).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Animal care was performed in compliance with all relevant ethical regulations for animal testing and research of the Federation of European Laboratory Animal Science association. All procedures were approved by the French animal experimentation and ethic committee of Paris Descartes University (CEEA 34, 16-063). Sample sizes were chosen to assure reproducibility of the experiments in accordance with the replacement, reduction, and refinement principles of animal ethics regulation.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as online supplemental information. The authors declare that the data supporting the findings of this study are available within the article and its supplemental information files.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.