Article Text

Abstract
Background To determine the safety and efficacy of the anti-colony-stimulating factor 1 receptor (anti-CSF1R) monoclonal antibody AMG 820 in combination with pembrolizumab in patients with select solid tumors.
Patients and methods Patients had advanced, refractory mismatch repair-proficient colorectal cancer, pancreatic cancer, or non-small cell lung cancer (NSCLC) with low (<50%) programmed cell death-ligand 1 (PD-L1) expression and were naïve to anti-programmed cell death-1 (PD-1)/PD-L1 or had relapsed/refractory NSCLC after anti-PD-1/PD-L1 treatment with low or high (≥50%) PD-L1 expression; all were anti-CSF1/CSF1R naïve. Patients received 1100 mg or 1400 mg AMG 820 plus 200 mg pembrolizumab intravenously every 3 weeks. The primary endpoints were incidence of dose-limiting toxicities (DLTs) and adverse events (AEs) and objective response rate per immune-related Response Evaluation Criteria in Solid Tumours at the recommended combination dose.
Results Overall, 116 patients received ≥1 dose of AMG 820 plus pembrolizumab (18 at 1400 mg AMG 820; 98 at 1100 mg AMG 820). Most patients (64%) were male; the median age was 64 (range 30–86) years. Seven patients had DLTs (1 at 1400 mg AMG 820; 6 at 1100 mg AMG 820). Almost all patients (99.1%) had AEs, 87.9% with grade ≥3 AEs. The most common AEs were increased aspartate aminotransferase (59.5%), fatigue (48.3%), periorbital/face edema (48.3%), and rash/maculopapular rash (37.1%). The best response was immune-related partial response in 3 patients (3%; duration of response 9.2, 10.0, 12.5 months) and immune-related stable disease in 39 patients (34%). None of the completed phase II cohorts met the predefined threshold for efficacy. Post-treatment there was accumulation of serum colony-stimulating factor 1 (CSF1) and interleukin-34, reduction in CSF1-dependent CD16-expressing monocytes, and increased PD-L1 expression and CD4 and CD8 cell numbers in tumor biopsies.
Conclusions The recommended combination dose of 1100 mg AMG 820 plus 200 mg pembrolizumab had an acceptable safety profile. Although pharmacodynamic effects were observed, antitumor activity was insufficient for further evaluation of this combination in selected patient populations.
Trial registration number NCT02713529
- clinical trials as topic
- drug therapy, combination
- gastrointestinal neoplasms
- immunotherapy
- lung neoplasms
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- clinical trials as topic
- drug therapy, combination
- gastrointestinal neoplasms
- immunotherapy
- lung neoplasms
Background
Immune checkpoint inhibitors that target cytotoxic T-lymphocyte antigen 4 or the programmed cell death-1 (PD-1) and programmed cell death-ligand 1 (PD-L1) axes have been approved for treatment of several cancers.1 Despite durable responses in a subset of patients, many fail to respond or acquire resistance to these agents,2–4 highlighting the need to understand resistance mechanisms and assess combination treatment strategies.
One potential mechanism of resistance to anti-PD-1 therapy is the immunosuppressive activity of tumor-associated macrophages (TAMs), which can prevent T cells from entering the tumor, inhibit intratumoral T cell activation, and directly limit PD-1 blockade by removing anti-PD-1 antibodies from T cells.5–9 TAM recruitment and survival are regulated by binding of colony-stimulating factor 1 (CSF1) to the CSF1 receptor (CSF1R)7 8 10; therefore, inhibition and depletion of TAMs through CSF1R blockade may enhance T cell responses against tumors when combined with PD-1 inhibition.
AMG 820 is an investigational, fully human IgG2 monoclonal antibody directed against human CSF1R that inhibits binding of CSF1 and interleukin (IL)-34 and subsequent ligand-induced receptor activation. In the first-in-human study of AMG 820 in adult patients with relapsed or refractory advanced solid tumors, toxicities were manageable and there was limited evidence of antitumor activity.11
We hypothesized that the combination of AMG 820 with the anti-PD-1 antibody pembrolizumab may overcome the lack of response to anti-PD-1 inhibition and resistance to prior anti-PD-1 therapy, resulting in enhanced antitumor activity. We therefore conducted a phase Ib/II study to determine the safety and efficacy of the combination of AMG 820 and pembrolizumab in patients with select advanced solid tumors that are historically resistant or refractory to PD-1 inhibitors.
Materials and methods
Key eligibility criteria
Eligible patients were ≥18 years old with pathologically documented, advanced colorectal cancer (CRC), pancreatic cancer, or non-small cell lung cancer (NSCLC) refractory to or intolerant of standard treatment. A complete list of eligibility criteria is provided in the online supplementary appendix.
Supplemental material
Patients were grouped by tumor type. Group 1 had advanced mismatch repair-proficient (pMMR) CRC, group 2 had advanced pancreatic cancer, and group 3 had advanced NSCLC and low (<50%) tumor PD-L1 expression. Groups 1‒3 were naïve to anti-PD-1/PD-L1 therapies. Patients in groups 4 and 5 had advanced NSCLC and low (<50%) or high (≥50%) tumor PD-L1 expression, respectively, and either had no response or had relapsed during monotherapy with anti-PD-1/PD-L1 therapies. All groups were naïve to anti-CSF1/CSF1R therapies. PD-L1 expression was assessed by immunohistochemistry using the PD-L1 companion diagnostic test (Agilent, Santa Clara, California, USA).
Study design
This phase Ib/II study was conducted at 15 centers in Australia, Belgium, Canada, Germany, Spain, and the USA. Patients provided a signed informed consent before the start of any study-related procedures.
The phase Ib part of the study had a dose-finding 6+3 design with a planned enrollment of 6–18 patients with CRC, pancreatic cancer, or NSCLC to evaluate the safety of the AMG 820 plus pembrolizumab starting dose. The phase II part enrolled patients in five predefined groups (online supplementary table 1) using a Simon two-stage design to further evaluate safety and to test whether AMG 820 plus pembrolizumab can enhance the antitumor activity of pembrolizumab alone and/or overcome lack of response to pembrolizumab monotherapy.
The starting dose of AMG 820 in phase Ib was 1400 mg every 3 weeks (Q3W), based on results from the first-in-human study.11 Pembrolizumab was administered 200 mg Q3W.12 AMG 820 and pembrolizumab were administered intravenously on the same day, with pembrolizumab administered 15 min after completion of the AMG 820 infusion. If 1400 mg AMG 820 was not tolerated, the dose could be reduced to 1100 mg and then to 800 mg; the pembrolizumab dose remained unchanged. In phase II, patients received the recommended combination dose based on results from the phase Ib part of the study.
Study treatment was discontinued for patients who had a dose-limiting toxicity (DLT; defined in the online supplementary appendix) within the 21-day DLT evaluation period and for those who required >6 weeks from the last dose to recover from DLTs that occurred after the 21-day window. Specific criteria for hepatic toxicity were established based on AMG 820 monotherapy safety data and are detailed in the online supplementary appendix.
Study treatment was withheld for grade ≥3 toxicities or serious adverse events (AEs) unrelated to AMG 820 and/or pembrolizumab until the toxicity resolved to grade 1 or baseline value. AMG 820 could then be administered at the same or a lower dose level. If one study drug was delayed/discontinued, patients could continue to receive the other study drug. Patients continued to receive study drugs until confirmed disease progression (per modified immune-related Response Evaluation Criteria in Solid Tumours (irRECIST)), intolerance to study treatment, clinically significant health deterioration, or withdrawal of consent.
Study endpoints
The primary endpoints were the incidence of DLTs and AEs in phase Ib and objective response rate (ORR; defined as immune-related complete response or immune-related partial response (irPR) rate) per irRECIST in patients treated with the recommended combination dose.
The secondary endpoints included time to response, duration of response, time to progression, overall survival (OS), and progression-free survival (PFS); pharmacokinetics (PK) of AMG 820; and number of CD4, CD8, and CD68 cells in fresh pretreatment biopsies.
The exploratory endpoints were incidence of potential anti-AMG 820 and antipembrolizumab antibodies and biomarker levels before and during treatment.
Study procedures
Assessments included vital signs, complete medical history, physical examination, ECG, laboratory tests, and paired tumor biopsies (if feasible) for PD-L1 and biomarker analysis.
Safety was monitored throughout the study, and AEs were graded according to the Common Terminology Criteria for Adverse Events version 4.0. Events of clinical interest for pembrolizumab (drug-induced liver injury) and AMG 820 (immune-mediated events) were recorded.
Tumor responses per irRECIST were performed by local investigator assessment within 28 days before enrollment, at week 10 (±1 week), and then every 10 weeks (±2 weeks) until confirmed disease progression or start of new anticancer treatment.
Pharmacokinetics
Serum samples for evaluation of AMG 820 PK were collected during cycle 1 on day 1 (predose, end of infusion, 1 hour, and 6 hours) and days 2, 5, 8, and 15; during cycle 2 on day 1 (predose, end of infusion, 2 hours, 4 hours) and days 2, 5, 8, and 15; and during cycle 3 on day 1 (predose and end of infusion). AMG 820 concentrations were measured using an ELISA (lower limit of quantification, 10 ng/mL). Non-compartmental analysis of AMG 820 concentration–time data was conducted using Phoenix WinNonlin v6.3 (Pharsight, Mountain View, California, USA).
Biomarkers
Changes in macrophage populations were evaluated in paired tumor biopsies taken at screening and week 10 (predose in cycle 4). Samples were processed for immunohistochemical analysis of PD-L1, CD4-expressing and CD8-expressing T cells, and CD68-expressing and CD163-expressing macrophages.
Blood samples collected during cycle 1 (predose on day 1 and days 8 and 15) and cycles 2–5 (predose on day 1) were analyzed for circulating biomarkers (CSF1, IL-34) using standard ELISA methods and immune cell populations (lymphocytes (T cells and B cells) and natural killer cells and monocytes with the markers CD4, CD8, CD14, CD16, and CD56) using flow cytometry.
Immunogenicity
A ‘collect-and-hold’ strategy for antibody testing was followed.13 No specific triggers were observed; therefore, anti-AMG 820 and antipembrolizumab antibody testing was not performed.
Statistical methods
The planned sample size was 6–18 patients in phase Ib and ≤185 in phase II. In phase Ib, six patients per cohort provided a 47%–91% probability of observing ≥1 DLT if the true DLT rate was 10%–33%. Sample size considerations for phase II are shown in online supplementary table 1. The Simon two-stage design of groups 1, 2, 3, 4, and 5 provided 80% power if the true ORR was 25%, 20%, 30%, 20%, and 20%, respectively, while maintaining a one-sided, 5% alpha error if the true ORR was ≤10%, ≤5%, ≤15%, ≤5%, and ≤5%, respectively. Enrollment into each group could be stopped early; decisions for early termination were made after reviewing all available data.
Patients who received ≥1 dose of AMG 820 were included in the analysis. Descriptive statistics are provided for demographics, safety, PK, and biomarker data. Kaplan-Meier (KM) estimates of PFS and OS at 6 and 12 months with corresponding 90% CI are provided.
Results
Enrollment, disposition, and baseline characteristics
A total of 116 patients, 15 in phase Ib and 101 in phase II, were enrolled between April 14, 2016 and March 8, 2018; all patients received ≥1 dose of AMG 820 plus pembrolizumab (figure 1).

Flow of patients through the study.
Based on a safety review conducted after 18 patients (6 in phase Ib and 12 in phase II) received ≥1 dose of 1400 mg AMG 820 plus pembrolizumab, the dose of AMG 820 was reduced to 1100 mg; 98 patients received ≥1 dose of 1100 mg AMG 820 plus pembrolizumab.
Baseline patient characteristics categorized by dose of AMG 820 received are summarized in table 1. Most patients (64%) were male, the median age was 64 (range 30‒86) years, and all patients had received ≥1 line of prior therapy.
Baseline demographics and disease characteristics
Exposure to study drugs and AEs
The dose of AMG 820 was reduced to 1100 mg based on safety data from 18 patients who received 1400 mg AMG 820 plus pembrolizumab. Seventeen of these patients had aspartate aminotransferase (AST) elevations (15 with grade 2 and 2 with grade 3) and 12 had alanine aminotransferase (ALT) elevations (6 with grade 2 and 2 with grade 3). Nine patients had periorbital edema (one with grade 3), which was associated with grade 2 uveitis in two patients. Three patients had immune-mediated events (one with fatal pneumonitis) or worsening of previously reported events (two with grade 3 periorbital edema), with onset >6 weeks after the last dose of study treatment. Four patients discontinued from the study, one withdrew consent due to toxicities, and thirteen discontinued due to progressive disease.
The median number of doses received was 2 (range 1–5) with a median treatment duration of 0.7 (range 0.03–2.92) months for patients in the 1400 mg AMG 820 group, and the median number of doses received was 3 (range 1–22) with a median treatment duration of 1.0 (range 0.03–14.4) months for patients in the 1100 mg AMG 820 group.
One of 18 patients who received 1400 mg AMG 820 plus pembrolizumab had DLTs of treatment-related grade 3 immune-related hepatitis and pancreatitis, grade 2 cholecystitis, and grade 4 electrolyte imbalance. Six of 98 patients who received 1,100mg AMG 820 plus pembrolizumab had DLTs: grade 3 maculopapular rash, grade 2 increased lipase, grade 4 epileptic seizure, grade 4 elevated AST, grade 3 elevated AST with grade 1 fatigue that lasted for 147 days and grade 3 generalized rash.
Almost all patients had ≥1 treatment-emergent AE (TEAE), most (88%) had grade ≥3 TEAEs, and approximately half had grade ≥3 treatment-related AEs (table 2). The most frequently reported TEAEs (in ≥30% of patients) were increased AST, fatigue, periorbital/face edema, and rash/maculopapular rash; the most frequently reported grade ≥3 AEs (in ≥10% of patients) were increased AST, rash/maculopapular rash, anemia, increased lipase, and hypophosphatemia (table 3). Approximately two-thirds of patients had treatment-related AEs of clinical interest, the most common of which were increased AST, ALT, amylase, and lipase (online supplementary table 2).
Summary of adverse events
Treatment-emergent adverse events*
Overall, 12% (14 of 116) of patients discontinued AMG 820 and 13% (15 of 116) of patients discontinued pembrolizumab due to TEAEs. Two fatal AEs were considered related to treatment: tumor flare in a female patient who received one dose of combination treatment and pneumonitis in a female patient who received two doses of combination treatment. Four fatal AEs (unrelated to treatment) were due to gastrointestinal hemorrhage (n=1), cardiac arrest (n=1), cerebrovascular accident (n=1), and pulmonary embolism (n=1).
Efficacy
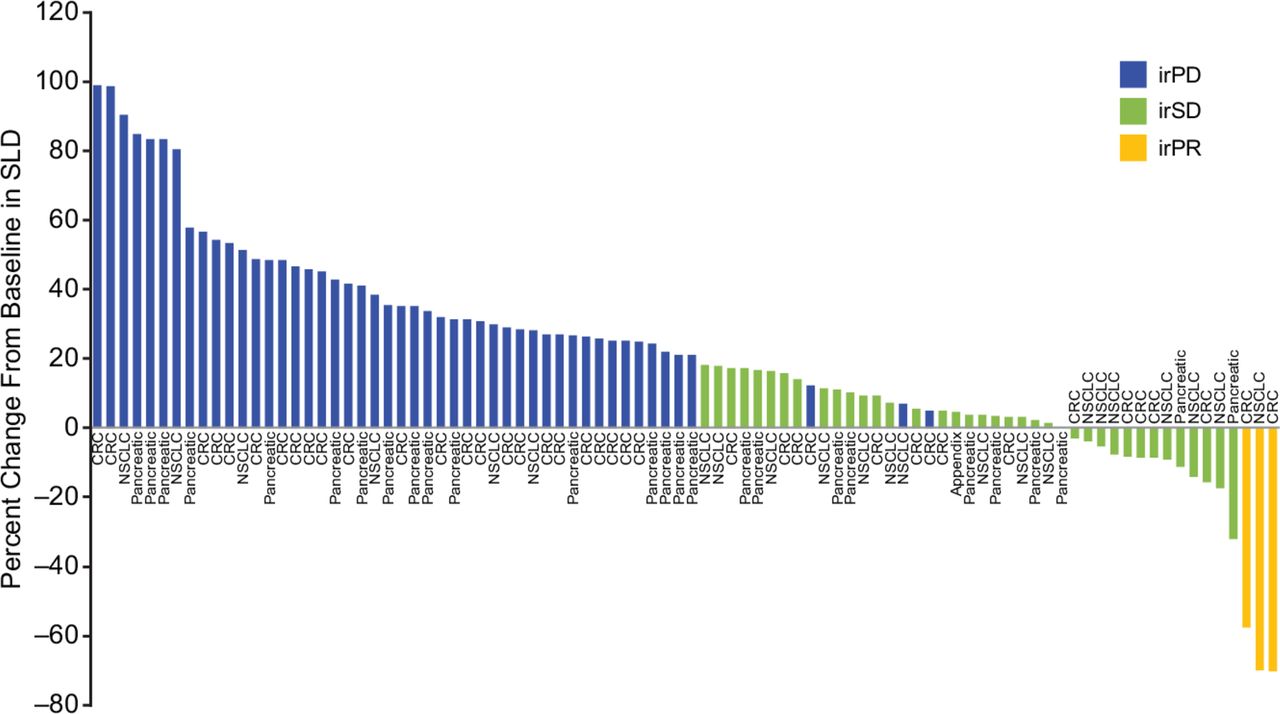
None of the completed phase II cohorts, namely groups 1, 2, and 4, met the predefined threshold for efficacy; enrollment to groups 3 and 5 was stopped early. Of 116 patients evaluable for response, 3 (3%) patients who received 1100 mg AMG 820 plus pembrolizumab had a best response of irPR, including 2 of 41 patients with pMMR metastatic CRC (duration of response, 9.2 and 12.5 months) and 1 of 19 patients with NSCLC who had progressed following prior treatment with nivolumab and had low PD-L1 expression (duration of response, 10.0 months). Thirty-nine (34%) patients had a best response of immune-related stable disease (irSD; table 4). Among patients with a best response of irSD, 13 (2 with pancreatic cancer, 5 with CRC, and 6 with NSCLC) had a decrease in the sum of the longest diameters of lesions compared with baseline (figure 2).

Best response by patient, based on change from baseline in the sum of the longest diameters of lesions. CRC, colorectal cancer; irPD, immune-related progressive disease; irPR, immune-related partial response; irSD, immune-related stable disease; NSCLC, non-small cell lung cancer; SLD, sum of the longest diameter.
Best overall tumor response and time to progression
The median time to progression was longest among patients with NSCLC: 5.4, 5.1, and 7.8 months for groups 3, 4, and 5, respectively. The median time to progression for patients with CRC or pancreatic cancer was 2 months (table 4).
For the overall patient population, the KM median (90% CI) PFS time was 2.1 (1.9–2.3) months and the KM median (90% CI) OS time was 5.3 (3.5–6.7) months. The KM estimate of OS rate (90% CI) at 6 and 12 months was 44% (36–52) and 30% (23–38), respectively.
Pharmacokinetics
PK analyses included 115 patients, 97 patients in the 1100 mg AMG 820 plus pembrolizumab group and 18 patients in the 1400 mg AMG 820 plus pembrolizumab group. Following intravenous Q3W administration of AMG 820 in cycles 1 and 2, AMG 820 exposures appeared to increase in an approximately dose-proportional manner over the dose range of 1100–1400 mg. No marked accumulation of serum AMG 820 (less than twofold) was observed between cycles 1 and 2 (online supplementary figure 1).
Biomarkers
CSF1 (44 patients) and IL-34 (40 patients) accumulated in serum after treatment (figure 3A,B). There was a reduction in CD14+ CD16+ monocytes (CSF1-dependent population) in the peripheral blood of patients (48 patients) at day 8 after dosing (figure 3C).

{kind=link}
{kind=link}
{kind=link}
Levels of CSF1 (A), IL-34 (B), and CD14+ CD16+ monocytes (C) in the peripheral blood of patients before and after dosing with AMG 820 plus pembrolizumab. CSF1, colony-stimulating factor 1; D, day; IL-34, interleukin-34; pre, predose; W, week.
Paired pretreatment and post-treatment tumor biopsies from five patients were available for analysis of immune infiltrate and PD-L1 expression, including three patients who achieved irPRs and two patients who had disease progression. One post-treatment biopsy from a patient with irPR contained tumor tissue, two biopsies contained necrotic tissue, and two contained no tumor tissue. Three paired tumor biopsies, including those from one patient with irPR, showed an increase in PD-L1 expression and doubling of CD4 and CD8 cell numbers after three doses of AMG 820 plus pembrolizumab; there was an increase in CD68 and CD163 macrophages in two patients (online supplementary table 3).
Discussion
In our study, the combination of AMG 820 and pembrolizumab showed preliminary evidence of activity (clinical benefit rate (irPR and irSD) of 36%). However, only three (3%) patients, two with pMMR CRC and one with NSCLC and low expression of PD-L1, achieved an irPR; thus, none of the phase II cohorts met the predefined threshold for efficacy.
Two of the most common AEs reported in our study were increased AST and periorbital/facial edema, both of which are class effects of CSF1R inhibitors. In non-clinical studies of AMG 820, reversible periorbital swelling likely due to increased extracellular matrix was observed (Amgen Inc., data availability statement). In addition, there were reversible increases in serum ALT, AST, and glutamate dehydrogenase in the liver with no evidence of liver injury. Increases in these enzymes result from decreased clearance due to inhibition of macrophages (ie, Kupffer cells).14 In three clinical studies of CSF1R inhibitors, the incidence of all-grade facial edema ranged from 13% to 64% and grade ≥3 increase in AST ranged from 3% to 16%.11 15 16 As demonstrated in our study, the incidences of these toxicities do not appear to substantially differ when a CSF1R and a PD-1/PD-L1 inhibitor are combined. However, the temporal relationship between AMG 820 plus pembrolizumab administration and grade ≥3 elevations in enzymes underscores the importance of safety surveillance. In addition, distinguishing the class effect of CSF1R inhibition as it pertains to normal Kupffer cell function from clinically relevant immune-related toxicities requiring therapeutic intervention, particularly immune-related hepatitis, is especially challenging.
The observed pharmacodynamic effects of AMG 820 plus pembrolizumab were consistent with clinically relevant inhibition of CSF1R and PD-1, including elevation of CSF1R ligand (CSF1 and IL-34) serum concentrations and reduced frequency of CD16+ monocytes. While no decrease in CD68+ or CD163+ intratumoral macrophages was observed following treatment in the limited number of available paired biopsies, this observation may be confounded by the potential inflammatory effects of pembrolizumab treatment, leading to increased monocyte recruitment and macrophage differentiation in the tumor. Notably, in the first-in-human study, AMG 820 monotherapy resulted in increases in CSF1 concentration and reduced the number of skin macrophages.11 There appeared to be no effect of pembrolizumab on AMG 820 exposures, which were similar to those observed when AMG 820 was administered as monotherapy.11
Only 3% of patients who received AMG 820 plus pembrolizumab in our study achieved an irPR. Notably, two of these heavily pretreated patients had pMMR CRC, a disease for which limited responses have been observed with single-agent checkpoint inhibitors.17 18 There were no baseline or on-treatment biomarker results that were predictive of response. In a phase I study of another CSF1R inhibitor, cabiralizumab, in combination with nivolumab, 3 of 31 (10%) patients with advanced pancreatic cancer achieved partial response.19 In a phase I study of durvalumab in combination with pexidartinib in 19 patients with CRC or pancreatic cancer, preliminary results showed a clinical benefit rate of 21% (unconfirmed stable disease in four patients), although this included two PD-1 inhibitor-naïve patients with high-level microsatellite instability CRC.20
Because we were interested in overcoming checkpoint inhibition, we intentionally selected patient populations with tumor types that are known to be resistant to checkpoint inhibition. This study did not investigate whether addition of AMG 820 can enhance responses in PD-1 inhibitor-naïve tumor types known to be responsive to checkpoint inhibition. Furthermore, 90% of patients in this study had received at least two prior lines of therapy for their disease. The effects of prior therapies and its implications in altering the tumor microenvironment are unclear. There are several potential explanations for the lack of efficacy observed in this study. First, there could be the lack of efficient depletion of intratumoral macrophages, either due to the inflammatory response associated with pembrolizumab treatment, as discussed above, or the inherent resistance of some protumorigenic macrophage populations to the effects of CSF1R blockade.21 Second, emerging studies in mice have suggested multiple adaptive resistance mechanisms to CSF1R inhibition, including activation of regulatory T cells and increased recruitment of other suppressive myeloid populations.22–24 These mechanisms of resistance could have been responsible for the lack of efficacy of AMG 820 and may have been exacerbated by the addition of pembrolizumab.
In summary, the recommended combination dose of 1100 mg AMG 820 plus 200 mg pembrolizumab had an acceptable safety profile. Although pharmacodynamic effects of both AMG 820 and pembrolizumab were observed, antitumor activity was insufficient for further evaluation of this combination in the selected patient populations.
Acknowledgments
The authors thank Jackson Egen, PhD, for critical review of the manuscript. Writing support was provided by Kathryn Boorer, PhD, of KB Scientific Communications, and Christopher Nosala, PhD, of Amgen Inc.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @VicMorenoGarcia, @mdmanishshah
Contributors Conception and design: GJ, DN, ER, M-ADS. Patient data collection/acquisition of data: AAR, JMC, VM, MB, ECA, WE, JT, RDH, AR, MAS, AJO, DJ, NL, DPR, KPP, GJ. Analysis and interpretation of data: all authors.
Funding This study was funded by Amgen Inc. Writing support was also funded by Amgen Inc.
Competing interests ARAR reports research support from Amgen and Merck, during the conduct of the study. JMC reports grants from AstraZeneca, Merck, and Tesaro, outside the submitted work; personal fees from Bristol Myers Squibb, outside the submitted work; and other from Roche, outside the submitted work. VM reports personal fees from Bayer, Bristol Myers Squibb, Janssen, and Pieris, outside the submitted work. MB reports grants from Amgen, during the conduct of the study; grants from Ascentage Pharma, AstraZeneca, Bristol Myers Squibb, Merck Sharp & Dohme, Novartis, Oncomed, and Pfizer, outside the submitted work; personal fees from AstraZeneca, outside the submitted work; and non-financial support from AstraZeneca and Merck Sharp & Dohme, outside the submitted work. ECA reports grants from AbbVie, Amcure, Amgen, AstraZeneca, Bayer, BeiGene, Bristol Myers Squibb, Boehringer Ingelheim, CytomX, Eli Lilly, H3, Incyte, Janssen, Kura, Loxo, Macrogenics, Menarini, Merck Serono, Merck, Merus, Millennium, Nanobiotix, Nektar, Novartis, Pfizer, PharmaMar, Principia, PsiOxus, Puma, Rigontec, Roche/Genentech, Sanofi, START, Taiho, and Tesaro, outside the submitted work; and other from AbbVie, Amcure, AstraZeneca, Celgene, Cerulean Pharma, EUSA, GLG, Guidepoint Global, HM Hospitals Group, International Cancer Consultants, Janssen-Cilag, Nanobiotix, Novartis, NPO Foundation Intheos (Investigational Therapeutics in Oncological Sciences), Oncoart Associated, Pfizer, Pierre Fabre, PsiOxus Therapeutics, Roche/Genentech, Seattle Genetics, Servier, and START, outside the submitted work. WE has nothing to disclose. JT reports other funding from Amgen, during the conduct of the study. RDH reports grants from Amgen and Merck, during the conduct of the study. AR reports honoraria for public speaking and advisory board meetings for Roche, Bristol Myers Squibb, Novartis, Pierre Fabre, and Sanofi. MAS reports grants from Merck, during the conduct of the study. AJO reports personal fees from Merck, during the conduct of the study; and personal fees from Array, Bristol Myers Squibb, Novartis, and Pfizer, outside the submitted work. DJ has nothing to disclose. NL reports other from Amgen, during the conduct of the study; personal fees from Innovent Biologics, outside the submitted work; and other from Alexion, ALX Therapeutics, Apexian, Asana BioSciences, Ascentage, BeiGene, Cerulean Pharma, Constellation, CytomX, Formation Biologics, Forty Seven, Ikena, Incyte, InhibRx, Innovent Biologics, Jounce Therapeutics, Livzon, Loxo, MacroGenics, Merck, Northern Biologics, Odonate Therapeutics, Pfizer, Regeneron, Symphogen, and TaiRx, outside the submitted work. DPR reports equity in Acworth Pharmaceuticals and MPM Capital and is an advisor for 28/7 Therapeutics, Gritstone Oncology, Maverick Therapeutics, MPM Capital, and Oncorus, outside the submitted work. ER is an employee and stockholder of Amgen. GJ is an employee and stockholder of Amgen. HW is an employee and stockholder of Amgen. NS is an employee and stockholder of Amgen. M-ADS is an employee of Amgen. DN is an employee and stockholder of Amgen. KPP reports funding to START for conduct of this clinical trial from Amgen; funding to START for conduct of clinical trials from 3D Medicines, AbbVie, ADC Therapeutics, ArQule, Calithera Biosciences, Curegenix, Daiichi Sankyo, EMD Serono, Formation Biologics, Incyte, Jounce Therapeutics, MabSpace Biosciences, MedImmune, Merck, Mersana, OncoMed, Peloton Therapeutics, Regeneron, Sanofi, and Syros Pharmaceuticals, outside the submitted work; and advisory board fees from ArQule, Basilea, and Bayer, outside the submitted work.
Patient consent for publication Not required.
Ethics approval The protocol was approved by the institutional review board at each site. The study was conducted in accordance with the International Council for Harmonisation Good Clinical Practice Guideline and conforms to the provisions of the Declaration of Helsinki.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. Qualified researchers may request data from Amgen clinical studies. Complete details are available at http://www.amgen.com/datasharing.