Article Text

Abstract
To prevent the destruction of tissues owing to excessive and/or inappropriate immune responses, immune cells are under strict check by various regulatory mechanisms at multiple points. Inhibitory coreceptors, including programmed cell death 1 (PD-1) and cytotoxic T lymphocyte antigen 4 (CTLA-4), serve as critical checkpoints in restricting immune responses against self-tissues and tumor cells. Immune checkpoint inhibitors that block PD-1 and CTLA-4 pathways significantly improved the outcomes of patients with diverse cancer types and have revolutionized cancer treatment. However, response rates to such therapies are rather limited, and immune-related adverse events are also observed in a substantial patient population, leading to the urgent need for novel therapeutics with higher efficacy and lower toxicity. In addition to PD-1 and CTLA-4, a variety of stimulatory and inhibitory coreceptors are involved in the regulation of T cell activation. Such coreceptors are listed as potential drug targets, and the competition to develop novel immunotherapies targeting these coreceptors has been very fierce. Among such coreceptors, lymphocyte activation gene-3 (LAG-3) is expected as the foremost target next to PD-1 in the development of cancer therapy, and multiple clinical trials testing the efficacy of LAG-3-targeted therapy are underway. LAG-3 is a type I transmembrane protein with structural similarities to CD4. Accumulating evidence indicates that LAG-3 is an inhibitory coreceptor and plays pivotal roles in autoimmunity, tumor immunity, and anti-infection immunity. In this review, we summarize the current understanding of LAG-3, ranging from its discovery to clinical application.
- adaptive immunity
- immune tolerance
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Immune cells can rapidly activate powerful defense mechanisms when they encounter invading pathogens. However, excessive and/or undesirable immune responses can exert deleterious effects. Immune cells are regulated by various molecules and cells with suppressive functions at multiple checkpoints. Such checkpoints are critical to the development of self-tolerance as the immune cells learn not to attack host cells. However, such checkpoints can be hijacked by tumors and pathogens to escape from the immune system.
Cancer immunotherapies targeting inhibitory coreceptors programmed cell death 1 (PD-1) and cytotoxic T lymphocyte antigen 4 (CTLA-4) significantly improved the outcomes of patients with diverse cancer types, revolutionizing cancer treatment. The success of these therapies verified that inhibitory coreceptors serve as critical checkpoints for immune cells to not attack the tumor cells as well as self-tissues. However, response rates are typically lower and immune-related adverse events (irAEs) are also observed in patients administered with immune checkpoint inhibitors. This is indicative of the continued need to decipher the complex biology of inhibitory coreceptors to increase response rates and prevent such unwanted side effects in patients with cancer.1–5
To date, many stimulatory and inhibitory coreceptors have been identified in addition to PD-1 and CTLA-4. These coreceptors are supposed to control the activation of lymphocytes by regulating the quality and quantity of the antigen receptor signaling to optimize beneficial immune responses while avoiding autoimmunity and excess immune responses. There is an intense competition among pharmaceutical companies to develop novel immunotherapies targeting these coreceptors. Among such coreceptors, lymphocyte activation gene-3 (LAG-3, CD223) is the foremost target next to PD-1, and multiple clinical trials to validate LAG-3-targeted therapies are ongoing.6 7
Molecular characteristics of LAG-3
LAG-3 was identified in 1990 by Triebel and colleagues8 in a screening designed to isolate molecules that were selectively expressed in F5 cells, a CD3-negative interleukin (IL)-2-dependent NK cell line. They screened the cDNA library of F5 cells with a probe obtained by subtraction of the cDNA of F5 cells with the mRNA of Jurkat (T cell leukemia), Laz 388 (Epstein-Barr virus-transformed B lymphoblastoid cell), K562 (erythro-myeloid leukemia), and U937 (cell of histiocytic origin) cells. The LAG-3 cDNA was isolated from 120 cDNA clones that were obtained by the screening.
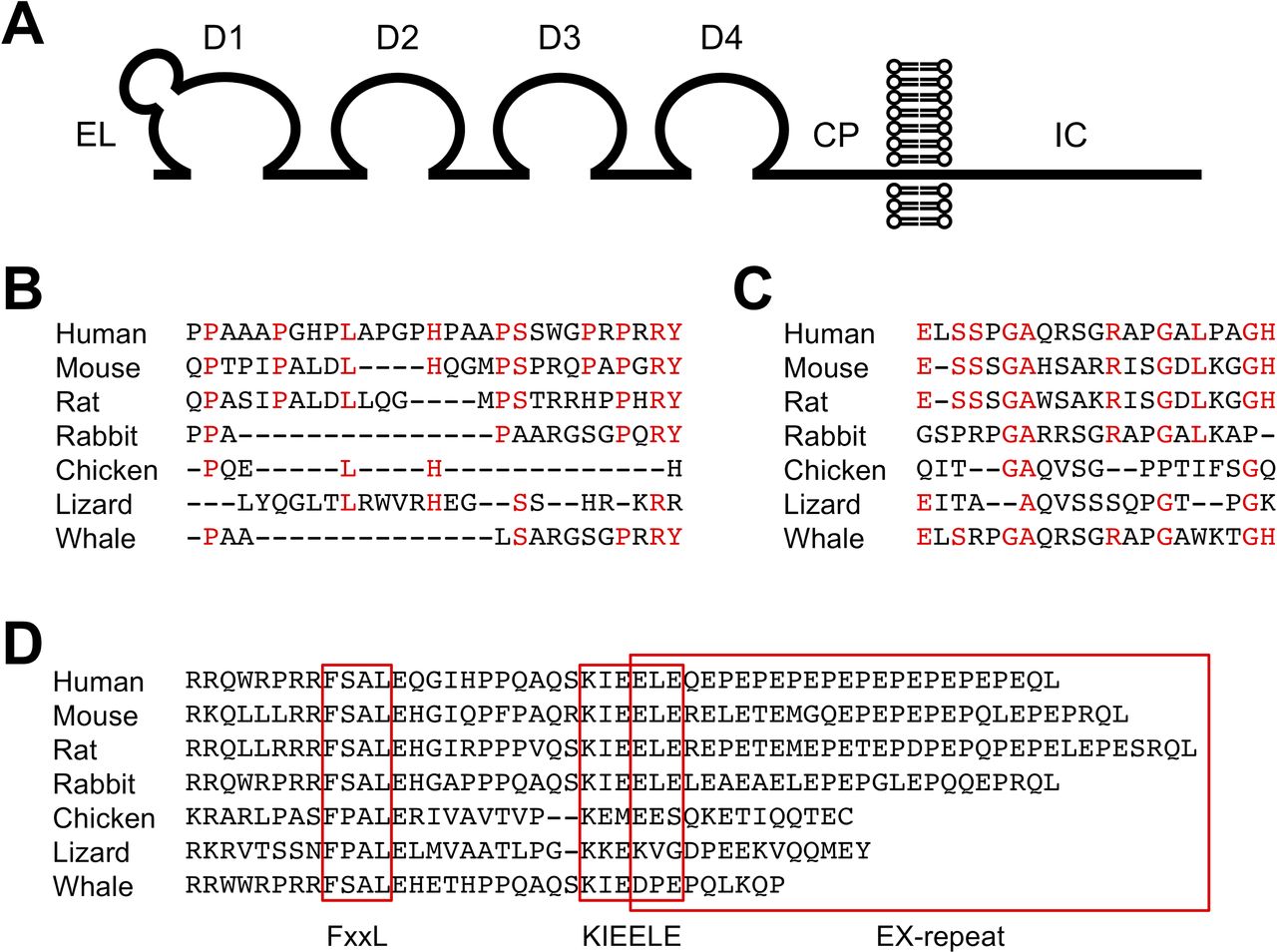
LAG-3 is a type I transmembrane protein with four Ig-like domains termed domain 1 (D1) to domain 4 (D4) (figure 1A). The extracellular region of LAG-3 shares approximately 20% amino acid homology with that of CD4, which is also composed of four Ig-like domains. In addition, the LAG-3 gene is located adjacent to the CD4 gene in most species (eg, on chromosome 12 and 6 in human and mouse, respectively). Hence, it is likely that these genes have evolved by gene duplication. Contrary to the similarity in the extracellular regions, the intracellular regions of LAG-3 and CD4 bear no noticeable similarity. LAG-3 lacks the cysteine motif required for the association with lymphocyte-specific protein tyrosine kinase (Lck) and the palmitoylation site observed in CD4.9–11 The organization of the genomic regions of CD4 and LAG-3 containing exons encoding their extracellular regions is similar, but the genomic organization containing exons encoding their intracellular regions is varied. Thus, CD4 and LAG-3 are closely related but are poised to exhibit divergent functions.
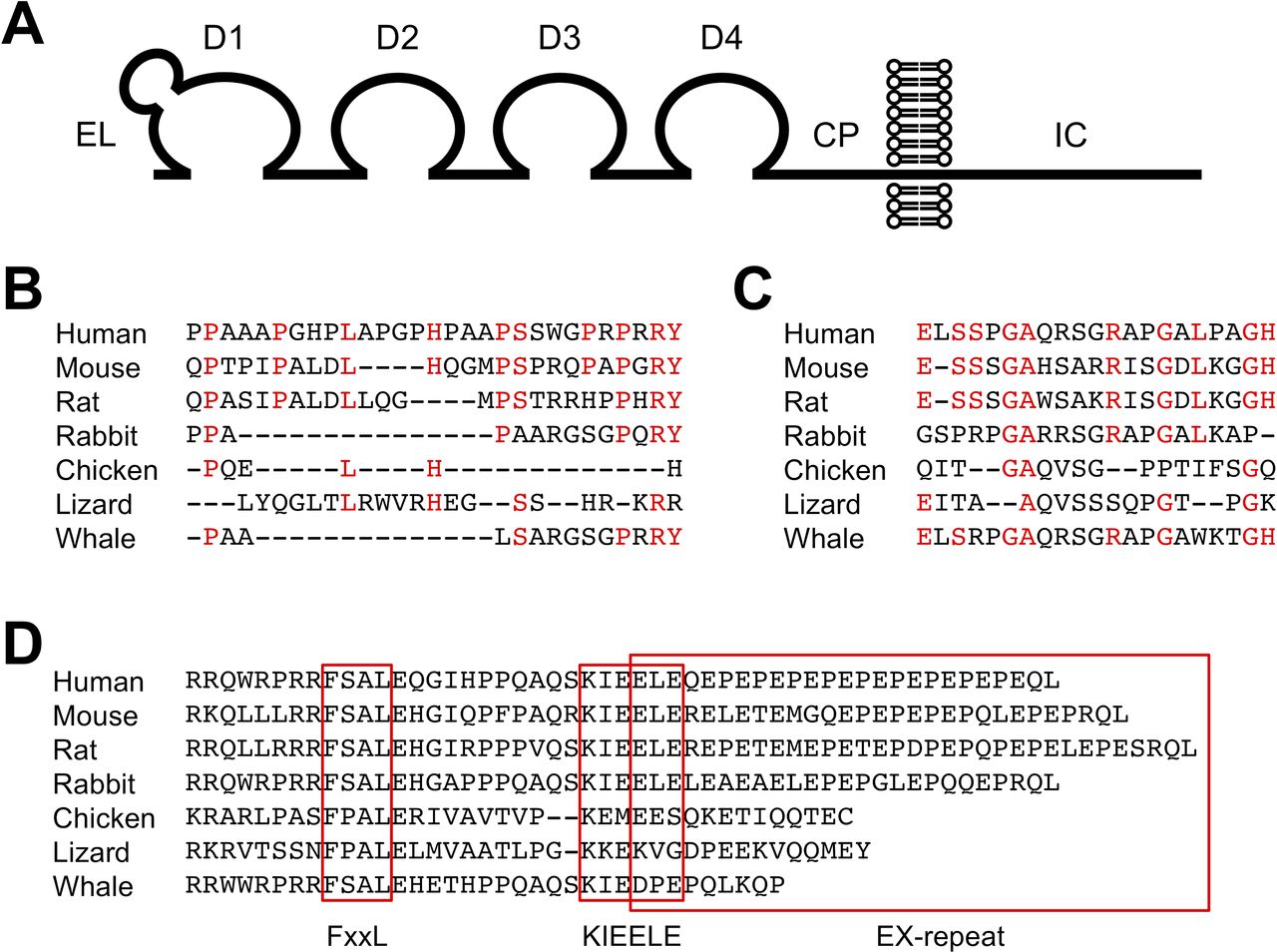
Structure of LAG-3. (A) Schematic representation of LAG-3. CP, connecting peptide; D1-D4, domains 1-4; EL, extra loop; IC, intracellular. (B–D) Alignments of EL, CP, and IC. Amino acid sequences of EL (B), CP (C), and IC (D) are shown for indicated species. Amino acid residues conserved between human and mouse are colored in red for EL (B) and CP (C). Putative FxxL, KIEELE, and EX-repeat are boxed. Amino acid sequences of LAG-3 were retrieved from Ensembl.org.
D1 of LAG-3 consists of nine β-strands that are assigned to the A, B, C, C’, C”, D, E, F, and G strands of the IgV fold. An additional sequence of about 30 amino acids is located between the C and C’ strands, which forms a loop and is termed ‘extra loop’. Although the sequences share low similarity, this loop can be observed in both human and mouse LAG-3 and is reported to engage in the association between LAG-3 and major histocompatibility complex class II (MHCII) (figure 1B).12 13 CD4 does not have an extra loop, and the mechanism of the contribution of the extra loop to the association between LAG-3 and MHCII is currently unknown. LAG-3 has been reported to be highly glycosylated, which is evident by the presence of multiple N-glycosylation sites in D2–D4. Galectin-3 and liver sinusoidal endothelial cell lectin (LSECtin) have been suggested to interact with glycans on LAG-3.14–16
A longer amino acid sequence termed ‘connecting peptide’ is located in the LAG-3 between D4 and the transmembrane region when compared with CD4. Based on the mouse model, Li et al 17 reported that metalloproteinases a disintegrin and metallopeptidase domain (ADAM) 10 and ADAM17 cleaved LAG-3 at CP and released the extracellular region of LAG-3 in soluble form. LAG-3 mutants that can escape from the cleavage by metalloproteinases demonstrate stronger inhibitory effects. Hence, ADAM10 and ADAM17 presumably modulate the inhibitory effect of LAG-3 by regulating the amount of LAG-3 on the cell surface. The homology of the amino acid sequences of the CP between human and mouse is low. Whether human LAG-3 can also be cleaved by these metalloproteinases or not remains to be examined (figure 1C). In addition to the cleavage, soluble LAG-3 comprising D1–D3 can be produced by alternative splicing. Currently, the function of soluble LAG-3 is unknown.7 18
Expression of LAG-3
Like PD-1 and CTLA-4, LAG-3 is not expressed on naive T cells, but its expression can be induced on CD4+ and CD8+ T cells upon antigen stimulation.8 19 As the inhibitory function of LAG-3 strongly correlates with its expression levels on the cell surface,20 the regulation of LAG-3 expression is very critical. Continuous antigen exposure owing to the chronic infection with viruses,21 22 bacteria,23 and parasites24 leads to high and sustained expression of LAG-3 as well as other inhibitory coreceptors on CD4+ and CD8+ T cells. These T cells lose robust effector function and are termed exhausted T cells. LAG-3 blockade has been demonstrated to reinvigorate exhausted T cells and strengthen anti-infection immunity, although to a lesser extent compared with that by PD-1 blockade.22 24–30 Tumor-infiltrating T cells are also persistently exposed to tumor-associated antigens and express high levels of multiple inhibitory coreceptors including LAG-3, resulting in functional exhaustion.31–36 IL-2, IL-7, and IL-12, but not IL-4, IL-6, IL-10, tumor necrosis factor (TNF), and interferon (IFN)-γ, have been reported to augment LAG-3 expression on activated T cells.37 38
LAG-3 expression is also detected on several subsets of CD4+ T cells with suppressive function. Foxp3+ regulatory T (Treg) cells constitutively express LAG-3,39 presumably due to the continuous activation of T cell receptor (TCR) signal by self-antigens, which is required for the differentiation, homeostasis, and suppressive functions of Treg cells.40 Zhang et al 41 demonstrated that LAG-3 on Treg cells limited their proliferation intrinsically. The role of LAG-3 in the effector function of Treg cells is contentious. Huang et al 39 reported that Treg cells from LAG-3-deficient mice inhibited the activation of effector T cells with lower efficiency whereas other studies demonstrated that the suppressive function of LAG-3-deficient Treg cells is comparable.19 42 43 Further studies are required to elucidate the actual role of LAG-3 in association with the Treg cells. LAG-3 is also expressed on CD4+ type 1 T regulatory (Tr1) cells. Although Tr1 cells demonstrate strong immunosuppressive activity by secreting high amount of IL-10, specific cell surface markers that define this population had not been detected until they were observed to express LAG-3 and CD49b.44 In addition, LAG-3-expressing CD4+CD25–Foxp3– T cells that produce IL-10 and transforming growth factor (TGF)-β3 have been proposed to exhibit regulatory function as a distinct cell population.45 ,46 Interestingly, IL-10-producing natural regulatory plasma cells have also been observed to express LAG-3.47 Currently, the roles of LAG-3 in the cell-extrinsic inhibitory function of these non-classical regulatory cells remain unclear and require further investigation.
Multiple transcriptional regulators such as thymocyte selection-associated high mobility group box protein (TOX), nuclear factor of activated T cells (NFAT), nuclear receptor subfamily 4, group A (NR4A), interferon regulatory factor 4, and B lymphocyte-induced maturation protein-1 are known to engage in the generation of exhausted T cells.48–56 Among these, NFAT, NR4A, and TOX have been demonstrated to augment the expression levels of LAG-3, together with other inhibitory coreceptors, when overexpressed in T cells. Early growth response gene 2 (EGR2) is also reported to be a key transcription factor in the induction of LAG-3 expression in CD4+CD25–Foxp3– regulatory T cells.45 On the other hand, T-box expressed in T cells (T-bet) has been reported to repress the expression of LAG-3 and other inhibitory coreceptors and sustain antigen-specific response of CD8+ T cells during chronic infection.57 58 In addition to the transcriptional regulation, the cell surface expression level of LAG-3 is also regulated by subcellular trafficking and proteolytic cleavage.17 59 60
LAG-3 is also expressed on CD3+CD4–CD8– T cells,61 TCRαβCD8αα intraepithelial lymphocytes,62 γδT cells,63 64 and NKT cells.65 Besides, its expression on activated NK cells is reported to be involved in the cytotoxicity against MHCI-negative target cells in mice.8 66 Plasmacytoid dendritic cells67 and activated B cells68 also express LAG-3 on their cell surface. However, the functional roles of LAG-3 in these populations remain poorly understood. Furthermore, LAG-3 is reported to be expressed on neurons and acts as a receptor for α-synuclein fibrils.69
Ligands of LAG-3
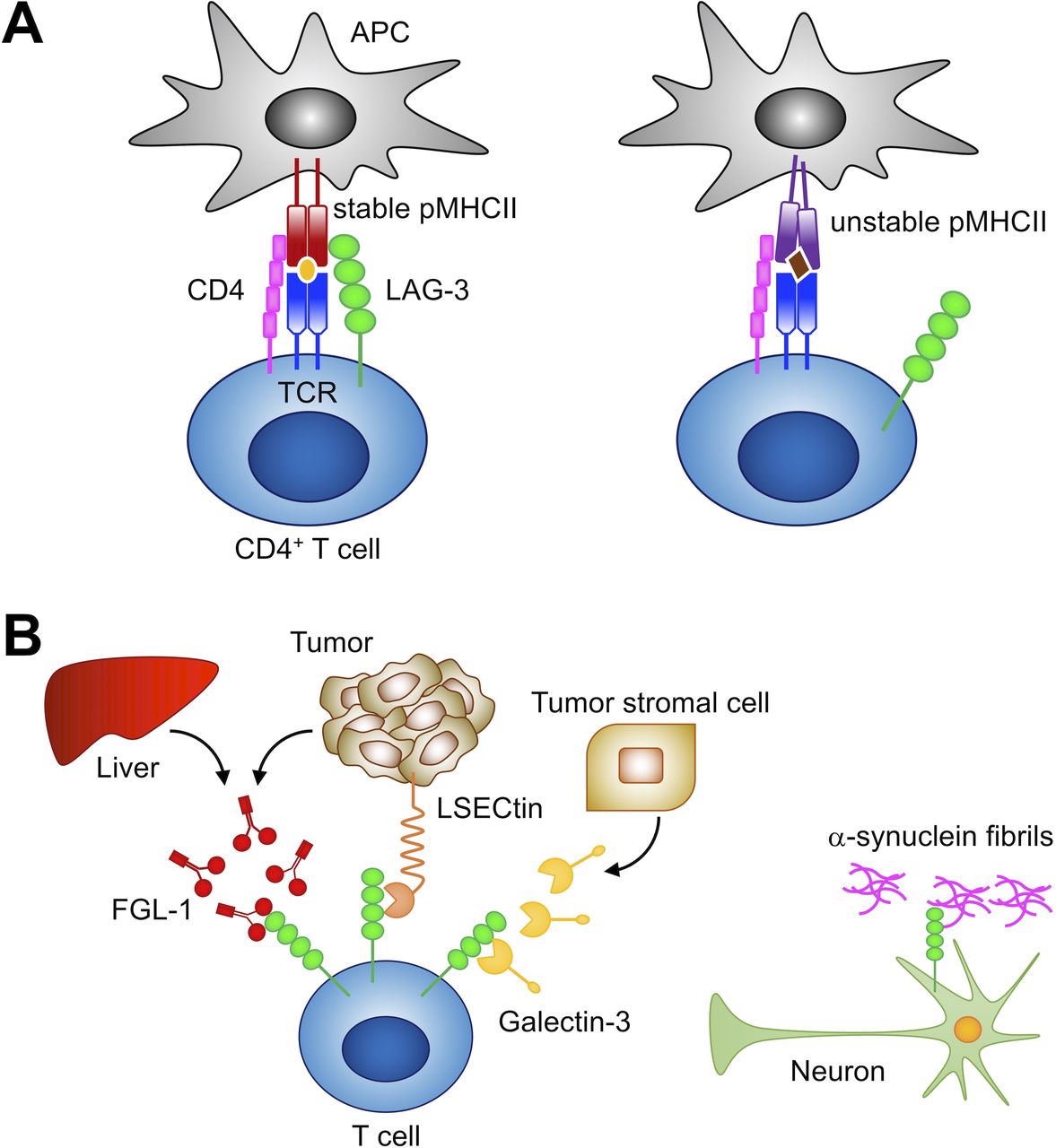
LAG-3 has been proposed to bind to MHCII with higher affinity than CD4 and inhibit T cell activation by interfering with the association of CD4 with MHCII.12 64 70 However, LAG-3 has been demonstrated to inhibit T cell activation by a mechanism that is different from the competitive inhibition of CD4, and the discrepancy between the binding capacity of soluble LAG-3 protein and MHCII expression levels in cells has been recognized, making the actual ligand of LAG-3 elusive.19 71 Recently, MHCII transactivator (CIITA) has been identified as a critical regulator of LAG-3 ligand. CIITA induces the expression of not only MHCII but also MHCII accessory molecules, including CD74 (invariant chain, Ii) and H2-DM. The MHCII accessory molecules contribute to the formation and the cell surface sorting of peptide-MHCII complex (pMHCII) exhibiting stable structural conformation in the conventional pathway of antigen presentation.72 LAG-3 distinguishes the conformation of pMHCII and selectively binds to stable pMHCII. Accordingly, LAG-3 preferentially inhibits the activation of CD4+ T cells that recognize stable pMHCII (figure 2A).71 It has also been demonstrated that LAG-3 does not compete with CD4 for pMHCII binding.71 Instead, LAG-3 inhibits T cell activation by transducing inhibitory signals via the intracellular region, as described in the next section.
{kind=link}
{kind=link}
Ligands of LAG-3. (A) LAG-3 selectively binds to stable pMHCII and inhibits the activation of CD4+ T cells that recognize stable pMHCII. (B) Reported non-MHCII ligands. LAG-3 has been reported to associate with FGL1, LSECtin, galectin-3, and α-synuclein fibrils. FGL1, fibrinogen-like protein 1; LSECtin, liver sinusoidal endothelial cell lectin; MHCII, major histocompatibility complex class II; pMHCII, peptide-MHCII complex; APC, antigen presenting cell; TCR, T cell receptor.
As mentioned, LAG-3 is expressed on exhausted CD8+ T cells in tumors and is a potent therapeutic target for cancer immunotherapy. However, the mechanism by which LAG-3 binding to pMHCII could impact the activation of CD8+ T cells has not been elucidated. Recently, the activation of CD8+ T cells has been demonstrated to be inhibited weakly when antigen-presenting cells express a substantial amount of stable pMHCII in addition to cognate pMHCI, indicating that LAG-3 can also directly suppress CD8+ T cells to a certain extent.71
To date, several molecules other than stable pMHCII have been reported as possible ligands for LAG-3 (figure 2B). As mentioned, galectin-3 and LSECtin have been indicated to interact with glycans on LAG-3. LSECtin is a member of the C-type lectin family and is expressed mainly in the liver.16 73 Xu et al 16 reported that LAG-3 suppressed the IFN-γ production from T cells upon stimulation with anti-CD3 Ab in the presence of LSECtin-expressing melanoma cells. Galectin-3, belonging to the galectin family, is a soluble galactose-binding lectin secreted from various types of tumor cells and tumor stromal cells.74 Kouo et al 15 demonstrated that galectin-3 reduced the frequency of IFN-γ-producing CD8+ T cells upon stimulation with anti-CD3 and anti-CD28 Abs when CD8+ T cells from LAG-3-sufficient and not LAG-3-deficient mice were used. Recently, Wang et al 75 identified fibrinogen-like protein 1 (FGL1), a member of the fibrinogen family, as a potential ligand for LAG-3. FGL1 is secreted from hepatocytes in the liver under normal physiological conditions, whereas some tumor cells can also produce FGL1 at high levels. FGL1 has been demonstrated to reduce the secretion of IL-2 from 3A9 T hybridoma cells expressing LAG-3 upon stimulation with the cognate peptide. Further studies are required to elucidate whether and how these potential ligands independently and/or cooperatively contribute to the inhibitory function of LAG-3.
In addition to the immunoinhibitory role, LAG-3 seems to have a distinct role in the nervous system as well. Mao et al 69 reported that LAG-3 can bind to α-synuclein fibrils, which are associated with the pathogenesis of Parkinson’s disease. The association of α-synuclein fibrils with LAG-3 triggers endocytosis, cell-to-cell transmission, and neurotoxicity of α-synuclein fibrils. In addition to LAG-3, other molecules such as semaphorins and paired Ig-like receptor B are known to exhibit dual roles in immune and nervous systems.76 77 Further studies are expected to demonstrate the similarities and discrepancies in the roles of such coreceptors between immune and nervous systems.
Inhibitory mechanisms of LAG-3
The intracellular region of LAG-3 consisting of approximately 60 amino acid residues lacks a typical inhibitory motif, such as immunoreceptor tyrosine-based inhibitory motif. However, it contains several amino acid sequences that are well conserved over different species of LAG-3 but are not shared with other inhibitory coreceptors. Such sequences include FSAL in the juxtamembrane region, KIEELE in the central region, and 10–15 tandem repeats of glutamate, and favorably but not limited to proline (EX-repeat) in the C-terminal region (figure 1D). As the intracellular region is required for LAG-3 to inhibit T cell activation, it can transduce distinct yet undetermined inhibitory signals via such sequences.
Workman et al 13 reported that the lysine residue of the KIEELE sequence is required for the LAG-3-mediated inhibition of the antigen-dependent activation of 3A9 hybridoma T cells. However, a contradictory report has been published and the mechanisms of the contribution of lysine residue to the LAG-3-mediated inhibition have not been elucidated yet.20 Iouzalen et al 78 identified LAG-3-associated protein (LAP) bound to the EX-repeat of LAG-3 by employing yeast two-hybrid cloning experiment. To date, no follow-up study has been reported and the function of LAP remains elusive.
The inhibitory function of LAG-3 strongly correlates with its expression levels on the cell surface, and the amino acid substitutions and deletions substantially affect the expression levels of LAG-3.20 When the potency of the inhibitory effects of mouse LAG-3 mutants was evaluated relative to their expression levels in the antigen-dependent activation of DO11.10 T hybridoma cells, the substitutions of phenylalanine and leucine in the FSAL sequence to alanine reduced the inhibitory capacity of LAG-3 significantly. In addition, when mutations in the FSAL sequence were combined with the deletion of the EX-repeat, the inhibitory capacity of LAG-3 was completely lost similar to the LAG-3 mutant lacking the entire intracellular region. Interestingly, the deletion of EX-repeat alone does not affect the inhibitory capacity of LAG-3. These results demonstrate that LAG-3 likely transduces two independent inhibitory signals via the FxxL motif and EX-repeat, while the molecular mechanisms of these signals are still unknown.20 Further analyses are expected to delineate the precise molecular mechanisms by which such motifs independently or cooperatively regulate the signaling pathways in T cell activation.
LAG-3 in autoimmunity
Inhibitory coreceptors play critical roles in the establishment and/or maintenance of immune tolerance to self as represented by the spontaneous development of autoimmune diseases in mice deficient for PD-1 and CTLA-4.79–81 In addition, immune checkpoint inhibitors targeting PD-1 and CTLA-4 activate not only tumor-specific T cells but also self-reactive T cells to induce tissue toxicities, termed irAEs.82–84 Unlike PD-1 and CTLA-4, LAG-3 deficiency itself does not cause autoimmunity in non-autoimmune-prone mouse strains. However, genetic deletion or blockade of LAG-3 exacerbates type 1 diabetes (T1D) in non-obese diabetic (NOD) mice, an animal model of T1D.19 85 LAG-3-deficient NOD mice demonstrate accelerated infiltration of autoreactive CD4+ and CD8+ T cells in islets compared with that in the age-matched LAG-3-sufficient NOD mice. In contrast, NOD mice lacking the cell surface expression of LAG-3 on Treg cells exhibit delayed onset and decreased incidence of T1D, which is attributed to the enhanced proliferation and function of Treg cells in the absence of LAG-3.41 Mice with compound deficiency of LAG-3 and PD-1 develop lethal autoimmune myocarditis on BALB/c, C57BL/6, and B10.D2 backgrounds, indicating that LAG-3 acts synergistically with PD-1 to prevent autoimmunity.19 86
In addition, LAG-3 has been reported to mitigate the autoimmune symptoms in experimental autoimmune models. Jha et al 87 reported that LAG-3 deficiency or blockade increased the susceptibility to mercury (Hg)-induced autoimmunity by inhibiting the induction of tolerance to Hg in C57BL/6.H2s mice. In myelin oligodendrocyte glycoprotein (MOG)-induced experimental autoimmune encephalomyelitis (EAE) model, Kadowaki et al 88 demonstrated that LAG-3 blockade abrogated the anti-inflammatory effect of gut environment-induced intraepithelial MOG-specific CD4+ T cells. Kim et al 89 also reported that the ability of in vitro-generated induced Treg (iTreg) cells to rescue Treg-depleted mice from lethal EAE was dependent on the expression of LAG-3 on the iTreg cells.
Given the critical regulatory role of LAG-3 in autoimmunity, LAG-3 has been expected to be a promising therapeutic target in inflammatory and autoimmune diseases. A humanized anti-LAG-3 Ab with antibody-dependent cell cytotoxic activity (GSK2831781) has been developed to treat autoimmune diseases by eliminating LAG-3-expressing T cells that presumably include pathogenic autoreactive T cells.90 In addition, agonistic anti-LAG-3 Ab (IMP761) has been reported to exert immunosuppressive effects both in vitro and in vivo by eliciting the inhibitory function of LAG-3.91
LAG-3 in antitumor immunity
As mentioned, LAG-3 is expressed on exhausted CD4+ and CD8+ tumor-infiltrating T cells that are defective in cytokine production.32–36 LAG-3 is also expressed on Treg cells in the peripheral blood and tumor tissues of patients with melanoma, colorectal cancer, and non-small cell lung cancer.92 93 Such LAG-3-expressing Treg cells produce high levels of immunoregulatory cytokines IL-10 and TGF-β and suppress tumor-specific T cells. Consistently, the levels of LAG-3 expression and infiltration of LAG-3+ cells in tumors have been reported to be associated with tumor progression, poor prognosis, and unfavorable clinical outcomes in various types of human tumors, such as colorectal cancer,94 renal cell carcinoma,95 follicular lymphoma,36 head and neck squamous cell carcinoma (HNSCC),96 non-small cell lung cancer,97 breast cancer,98 and diffuse large B cell lymphoma.99 These results strongly indicate that LAG-3 contributes to immune escape mechanisms in tumors similar to PD-1. Therefore, LAG-3 has been proposed as a promising therapeutic target for cancer immunotherapy, which is also supported by studies using animal models. Tumor growth delayed by anti-LAG-3 Ab has been reported in the mouse models of HNSCC and fibrosarcoma.96 100 Grosso et al 101 demonstrated that the combinatorial therapy incorporating anti-LAG-3 Ab and vaccination with tumor-associated antigen increased the number of activated CD8+ T cells in the tumor and disrupted the tumor parenchyma in the tumor-tolerance model of prostate cancer. However, no substantial difference in the grade of tumor was noted between mice administered with combination therapy or vaccination alone.
LAG-3 acts synergistically with PD-1 to suppress antitumor immunity as well as autoimmunity. In patients with epithelial ovarian cancer, Matsuzaki et al 34 observed that approximately 80% and 50% of LAG-3+ and LAG-3− tumor-infiltrating CD8+ T cells expressed PD-1, respectively. They also reported that the co-blockade of LAG-3 and PD-1 augmented the proliferation and cytokine production of tumor-infiltrating CD8+ T cells upon ex vivo stimulation with the tumor-associated antigen NY-ESO-1. Coexpression of LAG-3 and PD-1 on tumor-infiltrating CD4+ and CD8+ T cells and the strong therapeutic effect of co-blockade or compound genetic deletion of LAG-3 and PD-1 have also been observed in various mouse tumor models, such as B16 melanoma, MC38 colon adenocarcinoma, Sa1N fibrosarcoma, ovalbumin-expressing mouse epithelial ovarian cancer cell (IE9mp1), chronic lymphocytic leukemia derived from Em-TCL1 mice, and recurrent melanoma.86 102–104 Although LAG-3 blockade is expected to activate tumor-specific CD4+ and/or CD8+ T cells in such studies, it may also increase the number of Treg cells, since LAG-3 has been reported to limit the proliferation of Treg cells. According to Goding et al,102 Treg cells are rather reduced by the co-blockade of LAG-3 and PD-1 in the model of recurrence melanoma. Further studies are needed to delineate the exact effect of LAG-3 on the number and function of Treg cells as well as effector T cells.
In most of these studies, anti-LAG-3 Ab resulted in modest therapeutic effects when used as monotherapy but markedly augmented the therapeutic effect of anti-PD-1 Ab. Differences in the inhibitory mechanisms and/or expression profiles of these two molecules most likely explain their functional synergy. Further studies are needed to comprehend the exact mechanisms of the synergistic or additive action of LAG-3 with PD-1.
Clinical application of LAG-3
Based on preclinical observations including those mentioned, agents that block or stimulate LAG-3 functions are expected to provide therapeutic benefits in the treatment of cancer or autoimmune diseases, especially when combined with agents targeting PD-1. To date, at least 13 agents that target LAG-3 have been developed (table 1). Anti-LAG-3 blocking Abs (relatlimab (BMS-986016), Sym022, TSR-033, REGN3767, LAG525, INCAGN2385-101, MK-4280, and BI754111) and antagonistic bispecific Abs (MGD013 (anti-PD-1/LAG-3), FS118 (anti-LAG-3/PD-L1), and XmAb22841 (anti-CTLA-4/LAG-3)) are under clinical trials for various cancers either as monotherapy or in combination primarily with anti-PD-1 or anti-PD-L1 blocking Abs. According to the National Cancer Institute Drug Dictionary (https://www.cancer.gov/publications/dictionaries/cancer-drug), meeting abstracts,105–107 and a published report,108 most of them are supposed to block the interaction between LAG-3 and MHCII, while their effects on the interaction between LAG-3 and reported ligands other than MHCII are not specified.
Summary of LAG-3-targeted drugs under clinical trial
To date, only a few interim reports of combinatorial therapies targeting LAG-3 and PD-1 are available. Future reports are awaited to observe the actual therapeutic efficacy of anti-LAG-3 Abs as monotherapy or the exact additive effects of anti-LAG-3 Abs in the combination therapy targeting PD-1 and LAG-3. In phase I/II study evaluating the safety and efficacy of relatlimab in combination with anti-PD-1 Ab (nivolumab) in patients with advanced melanoma that had progressed during previous anti-PD-1 or anti-PD-L1 immunotherapy (NCT0198609), the combination of relatlimab and nivolumab was well tolerated and the objective response rate (ORR) was 11.5% in 61 patients. ORR was at least 3.5-fold higher in patients with LAG-3 expression in at least 1% of tumor-associated immune cells within the tumor margin (n=33) than that in the patients with less than 1% LAG-3 expression (n=22) (18% and 5%, respectively).109 LAG525 in combination with anti-PD-1 Ab (spartalizumab) exhibited a durable response in 9.9% of patients (n=121) with a variety of solid tumors, including mesothelioma (two of eight patients) and triple-negative breast cancer (two of five patients) in phase I/II study (NCT02460224).110 The precise mechanism of the function of such anti-LAG-3 Abs remains to be investigated. Especially, the examination of target cells in anti-LAG-3 therapy and elucidation of the mechanisms of synergy between anti-LAG-3 and anti-PD-1 therapies have garnered interest and are required for the rational design of anti-LAG-3 therapy with maximum efficacy and minimal adverse effects.
Other LAG-3-targeting agents have also been tested for cancer treatment. IMP321, a soluble recombinant fusion protein comprising the extracellular region of LAG-3 and the Fc region of IgG, has been reported to activate antigen-presenting cells by transducing a reverse signal via MHCII, resulting in the enhanced production of IL-12 and TNF and the upregulation of CD80 and CD86.111 112 IMP321 demonstrated only minimal or modest efficacy as monotherapy or in combination with other therapies in clinical trials conducted so far.113–116 The details of the reverse signal via MHCII remain unknown and require careful investigation.
The development of anti-LAG-3 depleting Ab (GSK2831781) and agonistic Ab (IMP761) as potential therapeutic agents in the treatment of autoimmune diseases has also been reported.90 91 Although these Abs aim to remove or suppress pathogenic T cells, they may also deplete or suppress Treg cells. Further studies elucidating the functions of such Abs together with the biological properties of LAG-3 are expected to help advance their development by providing the rationale for their use.
Conclusions
As checkpoint immunotherapies targeting inhibitory coreceptors PD-1 and CTLA-4 revolutionized cancer treatment, LAG-3 is expected to be a highly promising target in cancer therapies. However, our understanding of LAG-3 is still very limited and many fundamental questions remain unanswered. The signaling mechanism of LAG-3 is unknown and the ligands of LAG-3 are perplexing. LAG-3 is expressed on a variety of cell types. However, the function of LAG-3 and the effect of LAG-3 blockade in each type of cells have not been elucidated. We also need to examine the functional differences, redundancies, and co-operations of LAG-3 and other coreceptors. By elucidating the functional properties of LAG-3 more in detail, we can rationally design LAG-3-targeted therapies for various diseases, such as cancer, autoimmunity, and infection.
Acknowledgments
We thank the lab members for helpful discussions.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 84.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
Footnotes
Contributors TM, DS, IMO, and TO wrote the review.
Funding This work was supported in part by a Grant-in-Aid from the Japan Society for the Promotion of Science (JP18H05417, JP18K19453, JP19H01029, JP19H03423, JP19K16694).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Commissioned; externally peer reviewed.