Article Text

Abstract
Background Minimally invasive radiofrequency ablation (RFA) is used as a first-line treatment option for hepatocellular cancer (HCC) with the weaknesses of incomplete ablation, tumor recurrence, and inferior outcomes. To overcome this limitation, we proposed to develop sunitinib-RFA integrated therapy with a potential of activating anti-HCC immune response.
Methods Using our unique murine model, we developed a novel RFA platform with a modified human cardiac RF generator. Therapeutic efficacy of sunitinib–RFA combined treatment in HCC was tested in this platform. Tumor progression was monitored by MRI; tumor necrosis and apoptosis were detected by H&E and terminal deoxynucleotidyl transferase dUTP nick end labeling; immune reaction was defined by flow cytometry; and signaling molecules were examined with real-time PCR (qPCR), western blot, and immunohistochemical staining.
Results A significantly reduced tumor growth and extended lift span were observed in the mice receiving combined treatment with RFA and sunitinib. This combined treatment significantly increased the frequency of CD8+ T cell, memory CD8+ T cell, and dendritic cells (DCs); decreased the frequency of regulatory T cells; and activated tumor-specific antigen (TSA) immune response in tumor microenvironment. We found that RFA caused PD-1 upregulation in tumor-infiltrated T cells by boosting hepatocyte growth factor (HGF) expression, which was suppressed by sunitinib treatment. We have also demonstrated that sunitinib suppressed VEGF’s effect in enhancing PD-L1 expression in DCs and attenuated heat-sink effect. The results indicate that RFA induced tumor destruction and release of in situ TSAs which can activate a tumoricidal immune response in sunitinib-treated mice, significantly improving anti-HCC therapeutic efficacy.
Conclusions Sunitinib enables RFA-released in situ TSA to ignite an effective anti-tumor immune response by suppressing HGF and VEGF signaling pathways. Sunitinib–RFA as a synergistic therapeutic approach significantly suppresses HCC growth.
- combined modality therapy
- immune tolerance
- liver neoplasms
- lymphocytes
- tumor-infiltrating
- immunomodulation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- combined modality therapy
- immune tolerance
- liver neoplasms
- lymphocytes
- tumor-infiltrating
- immunomodulation
Background
Hepatocellular cancer (HCC) has increased rapidly in the USA over the past three decades and is the second leading cause of cancer-related death worldwide.1 Sorafenib, a receptor tyrosine kinase inhibitor (RTKI), was approved by the Food and Drug Administration (FDA) to treat unresectable HCC in 2008. However, this first systemically administered therapy only increases the median overall survival of patients from 7.9 to 10.7 months.2 Although surgical operation and liver transplantation are potentially curative options, aggressive distant metastasis and scarce donor organs limit their clinical application.3 These facts point to the real need for new therapeutic approaches to treat this lethal disease.4 5
Immunotherapy is revolutionizing HCC treatment.6–8 Monoclonal antibodies (Abs) for programmed cell death protein 1 (PD-1) and its ligand (PD-L1) are able to trigger significant anti-HCC immune response.9 The FDA has approved αPD-1 Abs as a secondary treatment for patients with HCC that is resistant to sorafenib. However, the overall response rate to this therapy is only 14.3%.10 Overcoming tumor-induced immune tolerance in HCC to improve immunotherapy is still an extremely challenging task.11
Radiofrequency ablation (RFA) is designed to destroy tumors by delivering a high-frequency alternating current through an active needle-electrode introduced into neoplastic tissue.12 13 Modern ultrasound, CT and MRI have increased our ability to accurately identify and target hepatic tumors.14 15 Image-guided RFA can be done percutaneously or laparoscopically as a minimally invasive treatment.14 Minimally invasive RFA is used as a first-line treatment option with significant advantages such as lower morbidity, minimized physiologic insult of surrounding tissues, reduced cost, shorter hospitalization time, and intra-procedural visualization for precise targeting.13 16 Studies from Zerbini et al 17 and Mizukoshi et al 12 have demonstrated that RFA monotherapy is able to activate tumor-specific T-cell response, but this effect is not sufficient to control HCC, evidenced by tumor recurrence and inferior outcomes. These findings drive us to think about whether we can develop a novel RFA-integrated therapy which can effectively activate anti-HCC immune response to overcome RFA’s limitation including incomplete ablation, tumor recurrence, and inferior outcomes.
In an effort to develop more powerful immune-based therapeutic strategies against HCC, we developed a unique murine model with immune competent mice via intrasplenic inoculation of oncogenic hepatocytes in combination with administration of carbon tetrachloride (CCl4).18 This approach induces orthotopic HCC tumors arising in the setting of fibrotic liver. In addition, tumor cells in this model specifically express SV40 T antigen (TAg), providing a unique trackable tumor-specific antigen (TSA). The tumors produced in our model have critical features in common with human HCCs.18 Anatomically, the tumors are discrete and multifocal masses in nodular livers; histologically, the tumors display disorganized hepatocyte proliferation, thickened hepatocellular plates, nuclear atypia, and obliterated portal triads; biologically, tumors express embryonic antigens, AFP and GPC3; genetically, the tumors are transformed through the TAg-driven Rb and p53 pathways; immunologically, the tumors induce complete CD8+ T cell–specific tolerance to TSA; therapeutically, the response to αPD-1 Abs monotherapy in our murine model is consistent with results from the subsequent human clinical trials. Using this model, we tested the potential of the different FDA-approved RTKIs in blocking tumor-induced immune tolerance. Although sorafenib monotherapy was found to be superior to sunitinib with significantly less toxicity in the treatment of human patients with HCC,19 we have demonstrated that sunitinib, rather than sorafenib, prevents tumor-induced profound immunotolerance in tumor microenvironment (TME) by significantly suppressing Treg production and PD-1 expression.20 21 Of particular importance, sunitinib at a half clinically equivalent dosage in combination with αPD-1 Abs strongly suppresses HCC growth, associating with the resultant activation of anti-HCC immune response.18 In terms of these findings, we proposed to explore the potential of sunitinib–RFA combined therapy in the treatment of HCC. We hypothesize that sunitinib significantly modulates a profound immunotolerant TME, which allows RFA-released in situ TSA to prime anti-HCC immune response. The resultant anti-HCC immunotoxicity synergizes RFA-induced tumor ablation and sunitinib-mediated chemotherapeutic effect to suppress tumor growth or even destroy the established tumors.
Materials and methods
Unique murine line
Line MTD2 mice served as the source of tumorigenic hepatocytes and were maintained by our laboratory.22 Male C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME) and used as recipient mice for preparation of the HCC tumor model. All experiments involving animals were approved by the Animal Care and Use Committee of University of Missouri. All mice were treated with humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals”.
Intraperitoneal (IP) administration of CCl4 followed by injection of oncogenic hepatocytes to generate the HCC model
CCl4 solution (10% (v/v)) in corn oil (Thermo Scientific, Waltham, MA) was IP injected into C57BL/6 mice twice a week at 8 mL/kg of body weight for 4 weeks.18 Two weeks after the last injection, the mice received inoculation of TAg-transgenic hepatocytes isolated from young male MTD2 mice by intrasplenic (ISPL) injection.18 Briefly, C57BL/6 mice under general anesthesia with isoflurane underwent a 0.5 cm flank incision. Two 10 mm titanium clips were placed between the upper and lower branch of the splenic vasculature on exposed spleen, and the spleen was cut between two clips. Hepatocytes were injected into the lower pole of the spleen and flowed into the liver through the portal vein. After injection, the lower pole of the spleen was removed and the incision was sutured.
Magnetic resonance imaging
Tumor surveillance was conducted with MRI in a small animal imaging center at Harry S. Truman Memorial Veteran’s Hospital.23 24 All MRI scans were obtained on a 7.0 Tesla system (Bruker Biospin, Billerica, MA) with in-plane resolution 0.1 mm and slice thickness 1 mm. Mice were anesthetized with isoflurane inhalation and the vital signs were monitored throughout imaging. Abdominal T2-weighted (T2W) MRI was acquired for tumor volume measurements. Diffusion-weighted (DW) MRI was performed to detect tumor necrosis after RFA treatment.
RFA and sunitinib administration
After anesthesia with isoflurane inhalation, the tumor-bearing mice underwent RFA with an EPT-1000 XPTM cardiac radiofrequency generator (Boston Scientific) which was equipped with a 4 mm cardiac ablation probe (7F/2.33 mm diameter).13 The performance was conducted with the defined conditions: a power output of 80°C for a duration of 60 s.
Sunitinib was orally administered to mice at 20 mg/kg in 0.2 mL of vehicle buffer every other day for 2 weeks by gavage.21
Isolation of liver/tumor-infiltrating leukocytes
Mice were anesthetized with isoflurane, then underwent liver/tumor perfusion with 0.05% collagenase (Gibco, Gaithersburg, MD) in Ca2+-free PBS at a pump speed of 4 mL/min. Livers or tumors were then harvested, smashed, and incubated in 0.04% collagenase in GBSS (Sigma, St. Louis, MO) for 20 min at room temperature with shaking at 240 rpm for the entire digestion period. Samples were filtered through a 250 µm mesh, washed with GBSS (Sigma, St. Louis, MO), and suspended in 15 mL of GBSS plus 18.45 mL 30% Nycodenz solution (Accurate Chemical & Scientific, Westbury, NY), and finally centrifuged at 1400×g for 20 min at room temperature with no brake. Liver or tumor-infiltrating leukocytes enriched in the top layer were collected and washed twice with medium.23
Ex vivo stimulation of liver/tumor-infiltrating leukocytes with peptides
Freshly isolated splenocytes, liver or tumor-infiltrating lymphocytes were suspended and cultured in RPMI 1640 complete cell culture medium (Gibco, Gaithersburg, MD) at 37°C in a 5% CO2 humidified atmosphere. The cells were stimulated with the control or tumor-specific antigen epitope peptides at a dose of 1 µM for 5 hours in the presence or absence of 3 µg/mL of Brefeldin A (Biolegend, San Diego, CA) which prevents cytokine transport.
In vitro stimulation of splenic leukocytes with tumor lysis
Freshly isolated splenic lymphocytes were suspended in RPMI 1640 complete culture medium (Gibco), seeded in 96-well plates pre-coated with 1 µg/mL of anti-CD3, and cultured at 37°C in a 5% CO2 humidified atmosphere. These cells were stimulated with tumor lysates prepared from tumor-bearing mice with different treatments in the presence or absence of recombinant HGF protein (10 µg/mL) (Bio Legend, San Diego, CA), or neutralized anti-HGF antibody (10 µg/mL) (Sino Biological, Beijing, China) for 72 hours, then harvested for the following studies.
In vitro culture of bone marrow–derived dendritic cells (DCs)
Bone marrow–derived DCs were prepared as our previous study.21 Briefly, the bone marrow isolated from femur and tibia of C57BL/6 mice was used to prepare cell suspension. These cells were seeded in six-well plates at a dose of 1×106 cells/well, then received stimulation of GM-CSF (20 ng/mL) in the presence or absence of recombinant VEGF-A protein (10 ng/mL), neutralized anti-mouse VEGF-A antibody (1 µg/mL) (Bio Legend, San Diego, CA), or sunitinib (0.1 µM) (Pfizer, New York City, NY). These cells were cultured at 37°C in a 5% CO2 humidified atmosphere for 7 days, then non-adherent cells were harvested for the following studies.
Flow cytometric analysis
Lymphocytes were isolated from spleen and tumors to get single-cell suspensions. The cells were stained with fluorochrome-labeled antibodies for indicated markers.18 23 25 Stained cells were analyzed with a Fantasia X20 flow cytometer (BD Biosciences, San Jose, CA). Data were analyzed using FlowJo software (Tree Star, Ashland, OR). Staining of intracellular IFN-γ and TNF-α was performed with our protocol as described previously.18 23 25 Staining of FoxP3 was performed with the staining buffer set (Thermo Scientific, Waltham, MA) following the manufacturer’s instruction.
H&E staining, immunohistochemical staining (IHC), and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)
Liver or tumor tissues were fixed with 10% neutral buffered formalin and embedded in paraffin. Tissue sections were processed and stained with H&E as described previously.13 24 To conduct IHC, tissue sections were de-paraffinized with xylene, rehydrated with various grades of ethanol (100%, 95%, 80%, and 70%), antigen unmasked with the provided solution (Vector Laboratories, Burlingame, CA), permeabilized with 0.2% Triton X-100, blocked with serum, then incubated with BLOXALL reagent (Vector Laboratories) to quench endogenous peroxidase. Subsequently, the sections were incubated in succession with primary antibodies, secondary antibody, and DAB substrate at the optimized concentration to develop color. The positive cells were counted in five randomly selected fields in each slide with ImageJ software (National Institutes of Health, Bethesda, MD). TUNEL was performed with TACS 2 TdT-Fluor In Situ Apoptosis Detection Kit (catalog no. 4812-30 K) (Trevigen, Gaithersburg, MD) to detect tumor cell apoptosis according to the manufacturer’s instructions.
Real-time quantitative PCR
Tumor tissues were homogenized by Pellet Pestles (Kontes, Vineland, NJ). Total RNA was extracted with RNeasy Micro kit (Qiagen, Germantown, MD). Reverse transcription of RNA to cDNA was conducted with the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster, CA). qPCR was performed with QuantStudio 3 Detection System (Thermo Fisher Scientific, Waltham, MA) in a 20 µL reaction mixture containing SYBR Green I (Thermo Fisher Scientific) in the following cycle conditions: 95°C for 15 s, 60°C for 15 s, and 72°C for 25 s for total 40 cycles. Expression of different genes was normalized to housekeeping gene 18S rRNA and further analyzed using the 2−ΔΔCT method. All primers were synthesized by IDT (Skokie, Illinois) and their sequences are shown in online supplemental table 1.
Supplemental material
Western blot
Tumor lysate was prepared with lysis protein extraction reagent (Thermo Fisher Scientific) and quantitated with Pierce BCA protein assay kit (Thermo Fisher Scientific). Equal amounts of protein were used to perform western blotting as previously described.18
Statistics
Paired data were analyzed using a two-tailed paired Student’s t-test. A p value of less than 0.05 was considered significant. The comparison of survival curves was analyzed using log-rank (Mantel-Cox) test.
Results
Successful treatment of orthotopic mouse HCC with a modified human cardiac radiofrequency generator
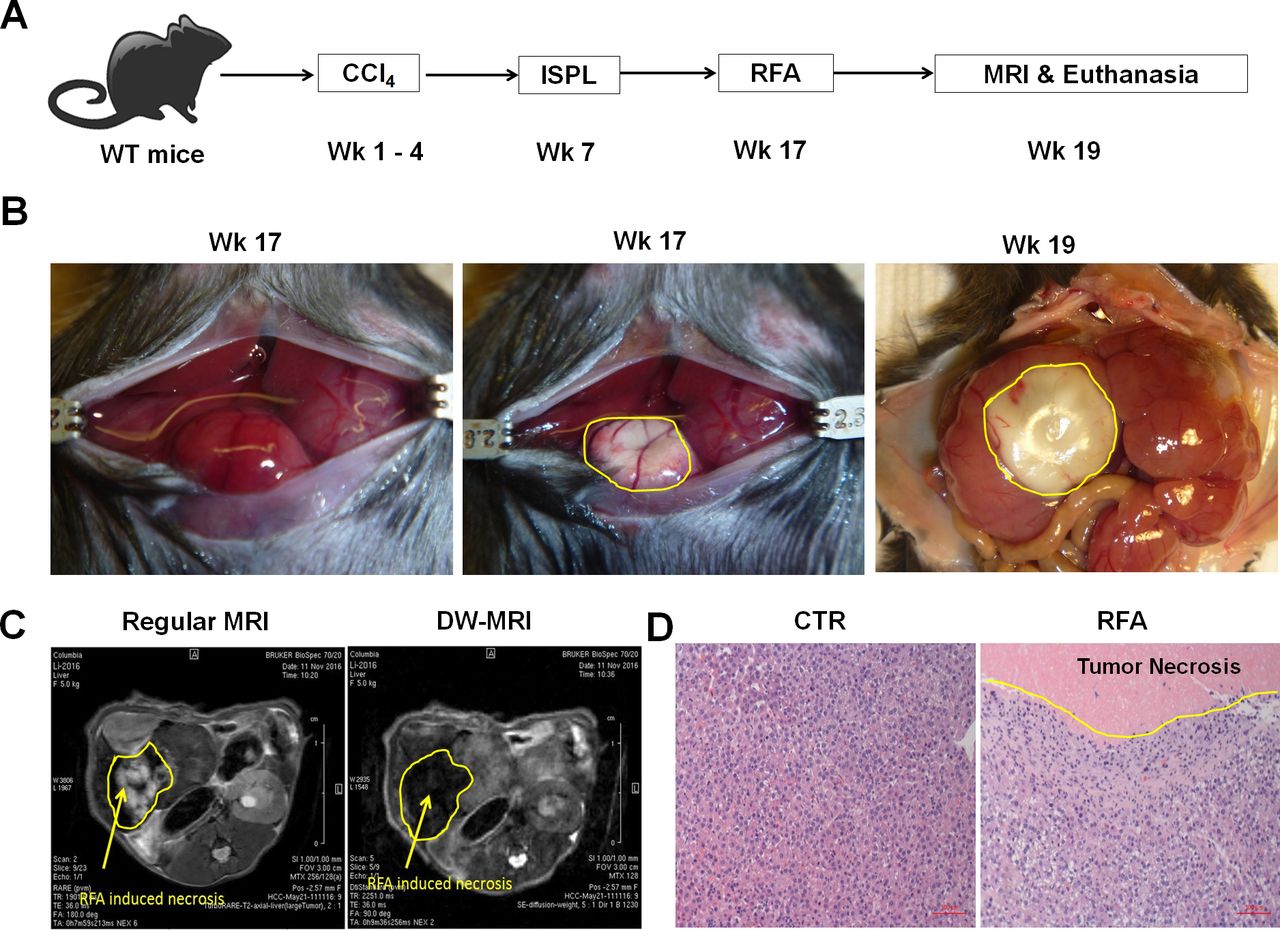
The lack of a RF generator suitable for use in small animals, including mice, critically impedes the capability to study the use of RFA for cancer treatment in murine models. To overcome this limitation, a human EPT-1000 XPTM cardiac radiofrequency generator equipped with a 4 mm ablation electrode was modified and demonstrated successful application in tumor-bearing mice with orthotopic HCC.13 As described in our recent publications,18 23 intrasplenic inoculation of oncogenic hepatocytes in combination with CCl4 injection was used to induce orthotopic HCC tumors in the setting of hepatic fibrosis in wild-type C57BL/6 mice. This model recapitulates the typical features of human HCC and expresses SV40 T antigen (TAg) as a trackable TSA.18 20 23 26 Ten weeks after oncogenic hepatocyte inoculation, these mice developed large tumors and were subsequently treated with RFA. The therapeutic effect was evaluated on week 19 (figure 1A).

Radiofrequency ablation (RFA) of orthotopic hepatocellular cancer (HCC) in a unique and clinically relevant murine model. (A) The schematic representation of orthotopic HCC-bearing mice preparation and RFA treatment. Using our established approach, intrasplenic inoculation of oncogenic hepatocytes in combination with CCl4 injection was used to induce orthotopic HCC tumors in the setting of hepatic fibrosis in wild-type C57BL/6 mice. The resultant HCC-bearing mice received RFA with our modified human RF equipment and optimized parameters. (B) Representative images of RFA-ablated tumors. Left panel: tumors prior to RFA, middle panel: tumor ablation immediately post-RFA, right panel: tumor ablation 2 weeks post-RFA. (C) Regular and DW-MRI for monitoring RFA-induced tumor ablation. Two weeks post-RFA operation, regular MRI (left) and DW-MRI (right) distinguished the RFA-induced tumor necrosis zone with a different pattern from normal HCC tissue. (D) Representative H&E staining of the RFA-ablated tumors. Left panel: tumor section without RFA, right panel: tumor section with RFA. RFA induced the coagulated necrosis of the directly ablated tumor and apoptosis of the surrounding tumor.
Using our previously optimized parameters, we conducted a single RFA in HCC-bearing mice at 80°C for 60 s. These mice were anesthetized with isoflurane inhalation and a 1.5 cm incision was performed to expose the abdominal cavity (left panel in figure 1B). The ablation electrode was placed on the surface of tumors to perform RFA. The RFA-induced tumor ablation was clearly evident at the conclusion of the procedure (middle panel in figure 1B). Two weeks later, the size of ablated tumor could be observed macroscopically (right panel figure 1B). This therapeutic effect was also detected by non-invasive MRI 2 weeks post-RFA ablation. RFA-induced tumor necrosis was revealed as the hyperintensity on T2W-MRI and the hypointensity on DW-MRI (figure 1C). The ablated portion of the tumor and the surrounding tumor were harvested for pathological evaluation. The H&E staining showed that RFA treatment was able to induce coagulation necrosis in the ablated area of the tumor with complete destruction of tumor cells. In addition, RFA also caused tumor apoptosis surrounding RFA-induced necrotic tumor (figure 1D). These results indicate that incomplete RFA is able to directly damage tumors by inducing tumor necrosis and apoptosis.
RFA in combination with sunitinib significantly improves anti-HCC therapeutic efficacy
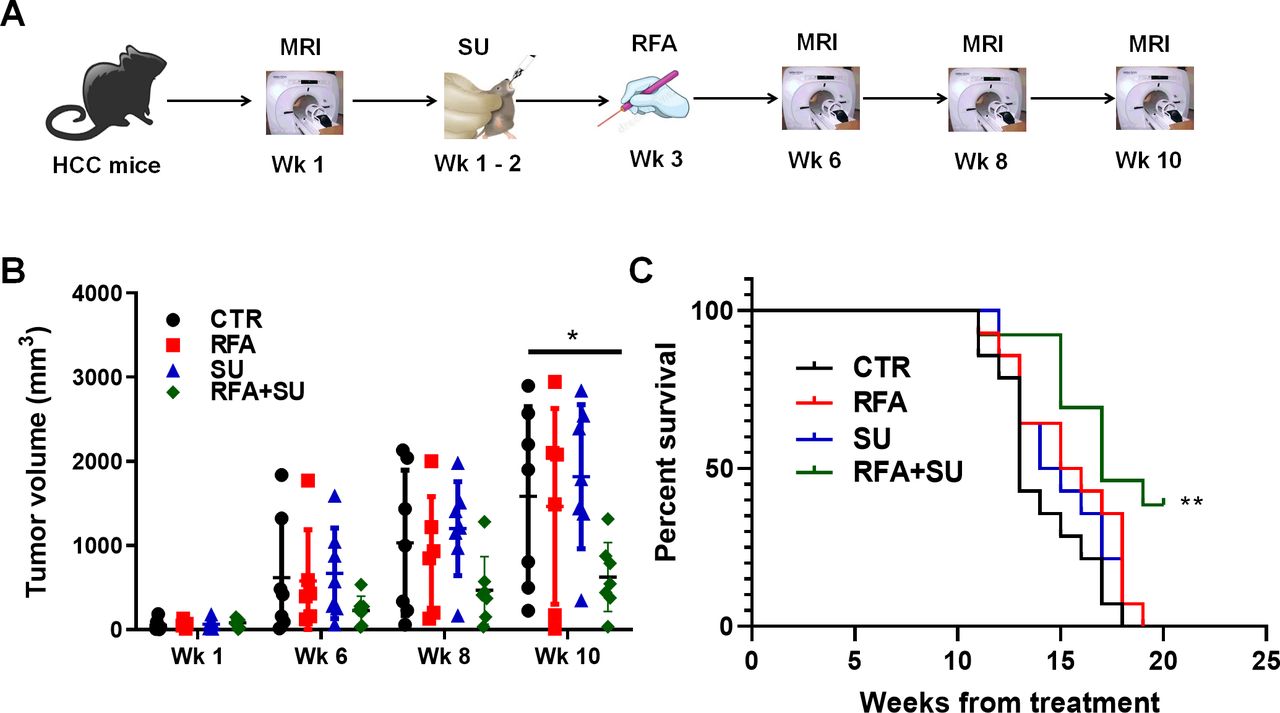
Minimally invasive RFA is widely used in the treatment of small isolated malignancies.13 However, RFA monotherapy often causes incomplete tumor ablation which subsequently results in recurrence, tumor progression, and metastasis. We proposed to improve RFA efficacy by enabling RFA-released in situ TSA to activate an anti-tumor immune response. To achieve this goal, the mice received treatment with sunitinib (SU) prior to RFA, as we previously demonstrated that sunitinib could prevent tumor-induced immunotolerance in HCC.18 To test this therapeutic approach, mice with size-matched tumors were randomly distributed into four groups and received no treatment (CTR), sunitinib monotherapy (SU), RFA monotherapy (RFA), or combinatorial treatment with both (RFA+SU). Tumor progression in each mouse was monitored by MRI (figure 2A). Ten weeks later, significantly slowed tumor growth was detected in the mice receiving the combined treatment compared with control without treatment or monotherapy with SU or RFA (figure 2B). Kaplan-Meier survival analysis revealed that the life span of tumor-bearing mice with the combined treatment was significantly longer than that in control mice without treatment or treated with SU or RFA alone (figure 2C). These results indicate that sunitinib–RFA functions as an effective therapeutic strategy which is superior to each monotherapy, significantly suppressing tumor growth and extending the lifetime of the treated mice.

RFA in combination with sunitinib suppressed tumor growth and extended the life span of tumor-bearing mice. (A) Schematic diagram of the treatments. Mice with size-matched tumors were randomly distributed into four groups and respectively received no treatment, RFA monotherapy, sunitinib monotherapy, or combinational treatment with RFA and sunitinib. The tumor progression was monitored by MRI. (B) Tumor growth over the period of the 3-month treatment. MRI was used to monitor tumor growth, and ImageJ was used to quantitate tumor size. n=7 for the control mice without treatment, n=6 for the mice with RFA monotherapy, n=7 for the mice with sunitinib (SU) monotherapy, n=7 for the mice with the combinational treatment. Error bars represent means±SD. Statistical analysis was performed by Student t-test (Prism8). *p<0.05 indicates statistical significance. (C) Kaplan-Meier survival analysis. The number of survival mice in each group was counted every day. n=14 in the control mice without treatment, n=14 for the mice with RFA monotherapy, n=14 for the mice with sunitinib monotherapy, n=13 for the mice with combinational treatment. Statistical analysis was performed by log-rank (Mantel-Cox) test (Prism8), **p<0.01.
Sunitinib synergizes with RFA to improve anti-HCC immune response in the setting of HCC
Sunitinib has the capacity to significantly improve RFA therapeutic efficacy. We have previously demonstrated that RFA resulted in apoptotic tumor death,13 and that tumor-induced immunotolerance was suppressed by sunitinib.18 25 Thus, we hypothesized that the improved therapeutic efficacy of the combinatorial treatment was attributed to immune activation. To test this hypothesis, we first assessed the change of intrahepatic immunity in each mouse. Two weeks after RFA treatment, the mice in each group were euthanized to isolate splenocytes and tumor-infiltrating lymphocytes (TILs). Flow cytometric assay detected a significantly increased frequency of tumor-infiltrating CD8+ T cells in the mice receiving combined treatment compared with the control mice without treatment or receiving each monotherapy (figure 3A,B). Correspondingly, the obvious increase in the absolute number of CD8+ T cells, but not CD4+ T cells, was detected (online supplemental figure 1). This increase was confirmed by IHC staining for CD8 in tumor tissue section (figure 3C,D)and was also observed in splenocytes (online supplemental figure 2). In response to the combined treatment, we also detected a significantly increased frequency of memory CD8+ T cells (CD44+CD62L−) in splenocytes compared with control mice without treatment or receiving each monotherapy (figure 3E,F). In contrast, we observed that the combined treatment markedly reduced the frequency of tumor-infiltrating FoxP3+ Tregs compared with that in control mice without treatment or with RFA monotherapy (figure 3G,H). Also, a mild effect was observed in splenocytes (online supplemental figure 2). In addition, compared with each monotherapy, the combined treatment was found to induce a significant increase in the frequency of tumor-infiltrating DCs (CD11b+CD11c+) (figure 3I,J). These data suggest that the combination of sunitinib and RFA is able to activate immune response which is associated with the increase of effector CD8+ T cells and DCs, and decrease of immune suppressive Tregs in the TME.
Supplemental material
Supplemental material
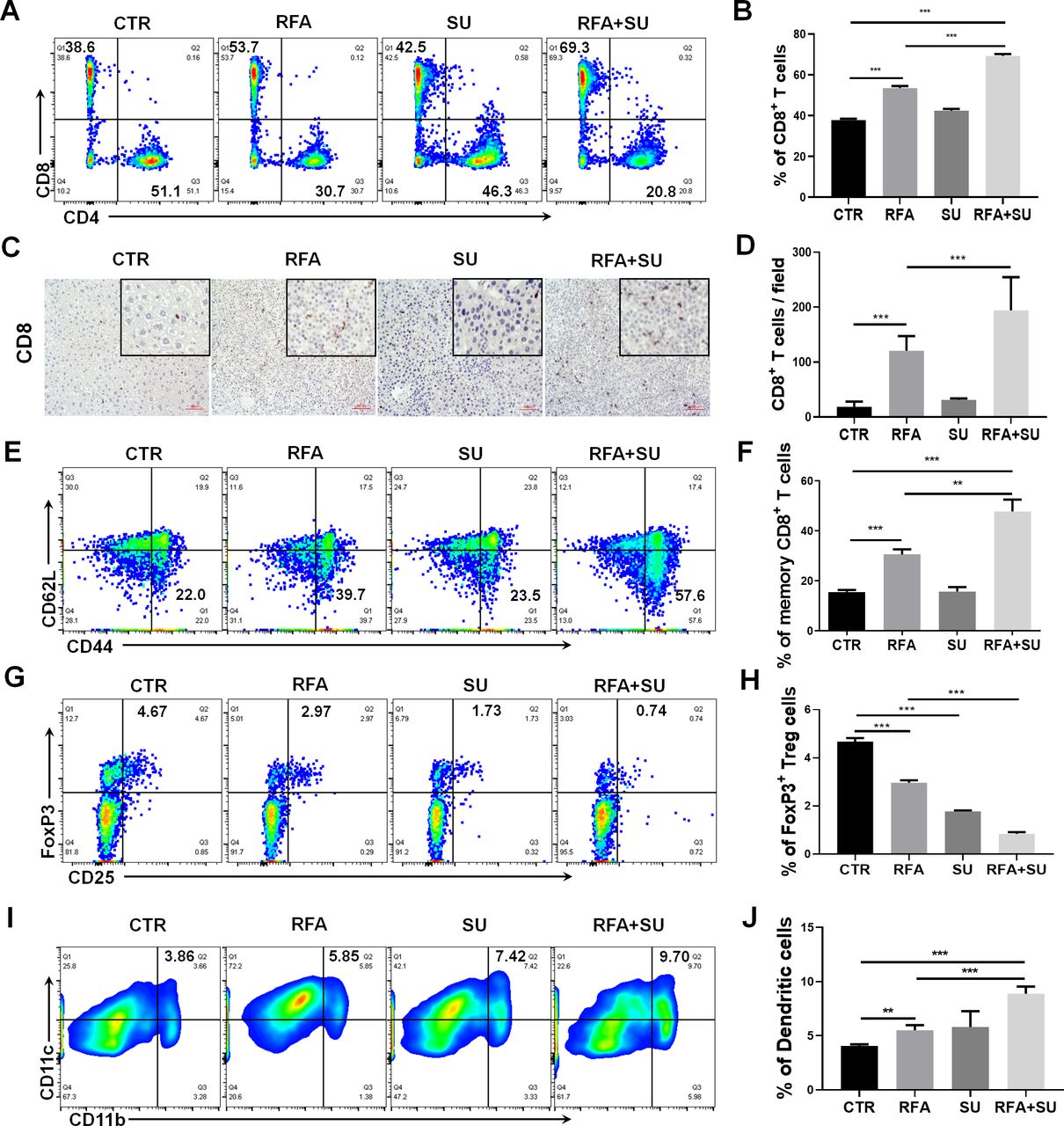
Sunitinib in combination with RFA changed the profile of tumor-infiltrating immune cells in hepatocellular cancer–bearing mice. Mice with size-matched tumors were randomly distributed into four groups and respectively received no treatment (CTR), RFA monotherapy (RFA), sunitinib monotherapy (SU), and combinational treatment with RFA and sunitinib (RFA+SU) as described in figure 2A. Two weeks post-RFA treatment, tumor-infiltrating leukocytes were isolated and used to conduct flow cytometry. (A) Representative flow cytometric assay to show the frequency of tumor-infiltrating CD8+ T cells in each mouse with the different treatments. (B) The cumulative frequency of tumor-infiltrating CD8+ T cells in each group, n=4, ***p<0.001, error bars represent means±SD. (C) Representative immunohistochemistry (IHC) to show the tumor-infiltrating CD8+ T cells. (D) The cumulative frequency of tumor-infiltrating CD8+ T cells detected by IHC in each group, n=4, ***p<0.001, error bars represent means±SD. (E) Representative flow cytometric assay to show the frequency of memory CD8+ T cells in spleen. (F) The cumulative frequency of memory CD8+ T cells in spleen in different groups of mice. n=4, **p<0.01, ***p<0.001, error bars represent means±SD. (G) Representative flow cytometric assay to show the frequency of tumor-infiltrating FoxP3+ Treg cells in different groups of mice. (H) The cumulative frequency of tumor infiltrating FoxP3+ Treg cells in different groups of mice. n=4, ***p<0.001, error bars represent means±SD. (I) Representative flow cytometric assay to show the frequency of tumor-infiltrating dendritic cells (DCs) (CD11b+ CD11c+) in each mouse with the different treatments. (J) The cumulative frequency of tumor-infiltrating DCs in each group, n=4, **p<0.01, ***p<0.001, error bars represent means±SD. Statistical analysis was performed by Student t-test.
Sunitinib enables RFA-released in situ tumor antigens to activate TSA immune response
RFA-induced in situ tumor apoptosis creates an antigen source for the induction of antitumor immunity. In our unique murine HCC model, TAg is specifically expressed by tumors as a trackable TSA, which allows us to investigate TSA immunity in mice in response to the different treatments. As described in figure 3, the mice were randomly exposed to each monotherapy or combined treatment. Two weeks after RFA, the TILs were isolated in each mouse and in vitro stimulated with TSA epitopes including TAg epitope I and IV. Influenza epitope peptide was used as a non-specific control. Five hours post-stimulation, flow cytometric assay detected 3% of IFN-γ-producing CD8+ TILs in the mice with combined treatment. This proportion is significantly higher than IFN-γ-producing CD8+ TILs cells in control mice without treatment (1%), SU-treated mice (1.5%), and RFA-treated mice (1.5%) (figure 4A,B). This increase was also observed in splenocytes, but not as much as it in TILs (online supplemental figure 3). Similarly, a significant increase in the frequency of TNF-α-producing CD8+ T cells in TILs (2.46%) (figure 4C,D)and splenocytes (1.3%) (online supplemental figure 3) was detected in the mice with combined treatment. Also, ELISA detected the increased production of serum IFN-γ (figure 4E), TNF-α (figure 4F), and interleukin-2 (IL-2) (figure 4G) in the mice with combined treatment. This increase was also reflected in mRNA level which was detected by qPCR (figure 4H–J). These results suggest that sunitinib treatment enables RFA-released in situ antigens to activate TSA immune response.
Supplemental material
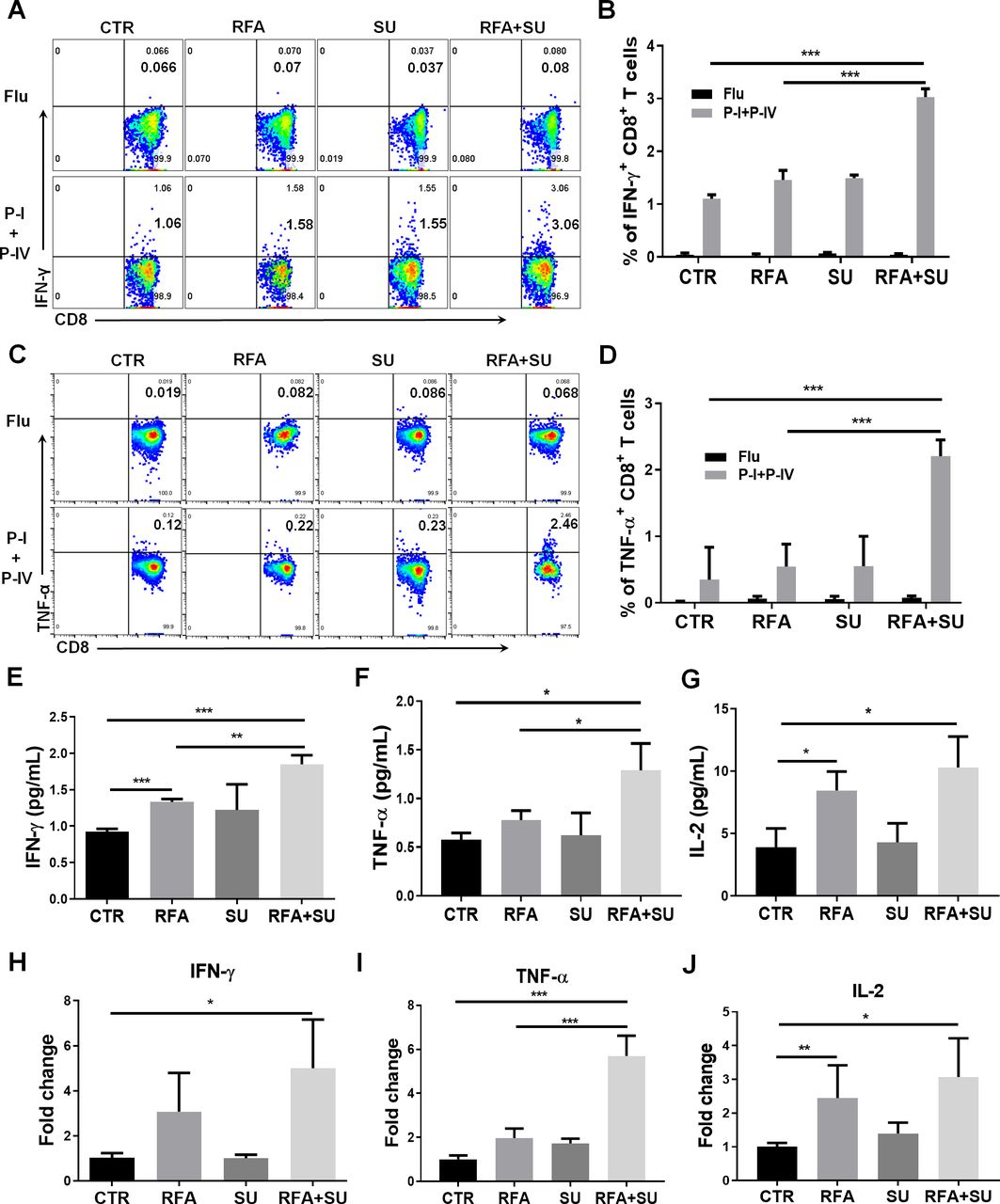
Sunitinib in combination with RFA activated tumor-specific antigen (TSA) immune response. As described in figure 3, mice with size-matched tumors were randomly distributed into four groups and received the indicated treatments. Two weeks post-RFA, the blood and tumors were harvested for the following studies. (A) Representative flow cytometry to show the frequency of IFN-γ-producing CD8+ T cells in tumor-infiltrating lymphocytes (TILs) in response to TSA stimulation. TILs were isolated and received stimulation of TSAs with TAg epitopes I and IV. Five hours after stimulation, the IFN-γ-producing CD8+ T cells were evaluated by flow cytometry. (B) Cumulative frequency of IFN-γ-producing CD8+ T cells in TILs in response to the stimulation with TSA epitopes I and IV, n=4, ***p<0.001, error bars represent means±SD. (C) Representative flow cytometry to show the frequency of TNF-α-producing CD8+ T cells in TILs in response to TSA stimulation. (D) Cumulative frequency of TNF-α-producing CD8+ T cells in TILs in response to the stimulation with TSA epitopes I and IV, n=4, ***p<0.001, error bars represent means±SD. The IFN-γ (E), TNF-α (F), and IL-2 (G) production in the blood. The serum was isolated from the harvested blood in each mouse. Each cytokine production was measured with ELISA. The average levels of IFN-γ, TNF-α, and IL-2 in each group are presented. n=3, **p<0.01, ***p<0.001, error bars represent means±SD. The IFN-γ (H), TNF-α (I), and IL-2 (J) mRNA expression in tumors. Part of tumor tissues was used to isolate total RNAs. The mRNA expression in the tumors of each mouse was measured by qPCR. n=4, *p<0.05, error bars represent means±SD. Statistical analysis was performed by Student t-test.
Sunitinib represses RFA induced PD-1 and PD-L1 upregulation
We previously demonstrated that tumor-driven PD-1 upregulation is implicated in the exhaustion of effector CD8+ T cells in the setting of HCC.18 Therefore, we investigated if sunitinib would allow RFA-released in situ antigen to activate anti-HCC immune response by suppressing PD-1 expression. In this regard, we isolated the TILs from the mice receiving either monotherapy or combined treatment with sunitinib and RFA. Unexpectedly, RFA treatment caused an additional increase in the expression of PD-1 in tumor-infiltrating CD8+ (figure 5A,B) and CD4+ (figure 5C,D) T cells. This increase in the proportion of PD-1+CD8+ T cells and PD-1+CD4+ T cells was dramatically repressed by sunitinib treatment (figure 5A–D). A similar effect was also detected in the splenocytes in the mice with the different treatments (online supplemental figure 4). Sunitinib-mediated repression in PD-1 expression may explain why the combined treatment enables RFA-released in situ antigens to activate antitumor immune response in the context of HCC. Consistently, qPCR detection demonstrated that RFA further increased the mRNA expression of both PD-1 and PD-L1 in the tumors compared with that in the control mice without treatment, and sunitinib treatment significantly repressed this increase with the resultant significantly lower level of PD-1 and PD-L1 in mice with combinational treatment than that in mice with mono-RFA treatment (figure 5E,F). These results suggest that sunitinib treatment represses RFA-induced additional upregulation of PD-1 and PD-L1, enabling the activation of effector CD4+ and CD8+ T cells.
Supplemental material

Sunitinib (SU) suppressed RFA–induced PD-1 upregulation. As described in figure 4, mice with size-matched tumors were randomly distributed into four groups and received the indicated treatments. Two weeks post-RFA, tumors were harvested from each mouse. Part of the tumor tissues was used to isolate tumor-infiltrating lymphocytes for flow cytometric assay. (A–B) Representative and cumulated proportion of CD8+ T cells expressing PD-1. (C–D) Representative and cumulated proportion of tumor-infiltrating CD4+ T cells expressing PD-1. The results showed that RFA promoted PD-1 production in both tumor-infiltrating CD4+ and CD8+ T cells which can be suppressed by SU. n=4, ***p<0.001, error bars represent means±SD. (E–F) qPCR detected the mRNA expression of PD-1 and PD-L1 in the tumors in different groups of mice. n=4, **p<0.01, ***p<0.001, error bars represent means±SD. Statistical analysis was performed by Student t-test.
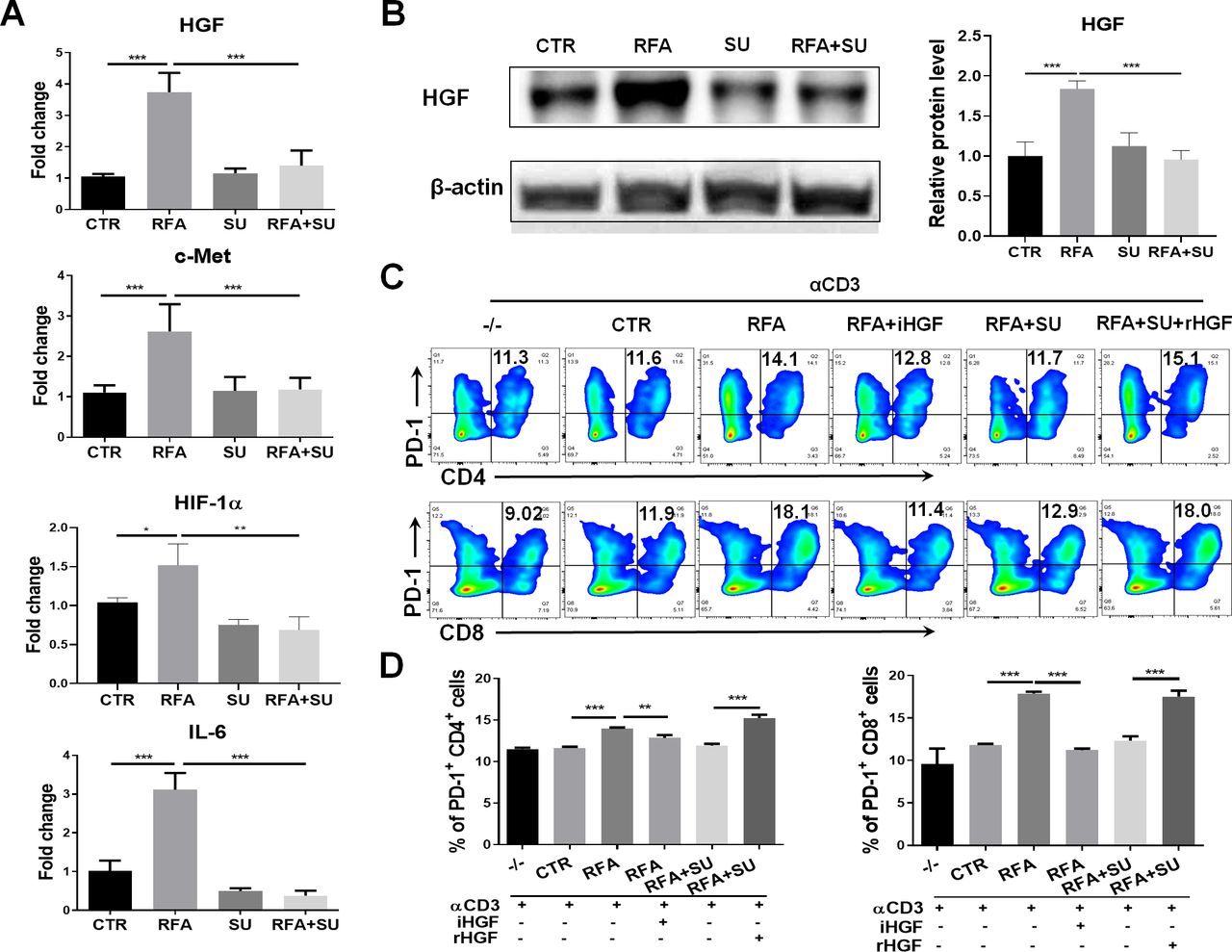
Sunitinib represses PD-1 upregulation induced by the tumors and RFA via hepatocyte growth factor (HGF) signaling
Similar to the clinical findings, RFA treatment caused upregulation of PD-1 and PD-L1 in the HCC-bearing mice. In an effort to elucidate how sunitinib administration suppresses PD-1 expression in RFA-treated tumor-bearing mice, we detected the expression of the different signal molecules associating with tumor-induced immune suppression. As shown in figure 6A, qPCR detected the increased mRNA expression of HGF, c-Met, HIF-1α, and IL-6 in tumors in RFA-treated mice; sunitinib administration effectively repressed this increase. Western blotting detected a similar expressional pattern for HGF at a protein level (figure 6B). These results indicated that HGF was closely associated with tumor growth and sunitinib treatment. To examine if HGF was a critical factor contributing to sunitinib-mediated suppression of PD-1 expression, splenocytes prepared in wild-type mice were seeded in anti-CD3 Abs-coated plate at a dose of 1 µg/mL, then stimulated with tumor lysate extracted from four groups of mice. Seventy-two hours later, the cells were harvested and assayed with flow cytometry. The results indicated that the lysate from RFA-treated tumors significantly increased PD-1 expression in CD4+ and CD8+ T cells, and this increase was neutralized by addition of anti-HGF antibody (iHGF). In contrast, the lysate from the mice with the combined treatment failed to prompt PD-1 expression in the CD4+ and CD8+ T cells, but addition of a recombinant HGF (rHGF) restored this effect (figure 6C,D). These results indicate that HGF is a critical factor in tumor-induced upregulation of PD-1 and sunitinib-mediated suppression of PD-1 in effector CD4+ and CD8+ T cells in HCC-bearing mice.27 28
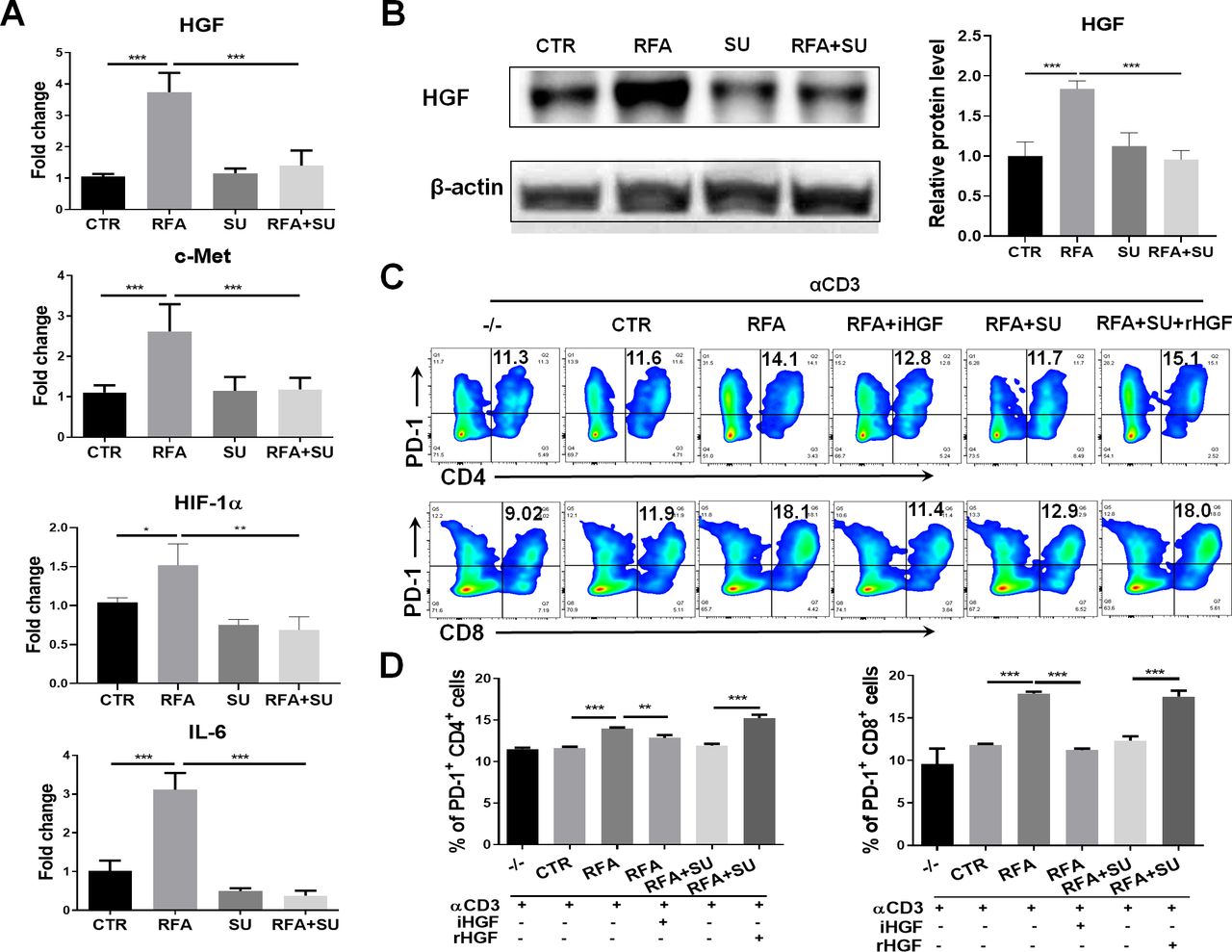
Sunitinib (SU) treatment repressed RFA–induced PD-1 upregulation via HGF signaling. Mice with size-matched tumors were randomly distributed into four groups and received the indicated treatments as described in figure 4. Two weeks post-RFA, tumors were harvested from each mouse and used for the following studies. (A) The gene expression of HGF, c-Met, HIF-1, and IL-6 in the tumors. Total RNAs were extracted from part of tumors in the different groups of mice. qPCR was used to detect the mRNA expression of HGF, c-Met, HIF-1, and IL-6. n=4, *p<0.05, **p<0.01, ***p<0.001, error bars represent means±SD. (B) HGF protein expression in the tumors. Tumor lysate was prepared and used to detect HGF protein expression by western blotting. The protein quantification assay was conducted with ImageJ. n=4, ***p<0.001, error bars represent means±SD. (C) Expression of PD-1 in CD4+ and CD8+ T cells in response to the different stimulation. The splenocytes from wild-type mice were stimulated with the basal level anti-CD3 Ab and the tumor lysate from the mice with the different treatments. Flow cytometry was used to detect PD-1 expression in CD4+ and CD8+ T cells. iHGF, HGF inhibitor; rHGF, recombinant HGF; TL-CTR, tumor lysate from the control mice without treatment; TL-RFA, tumor lysate from the mice with RFA monotherapy; TL-RFA+SU, tumor lysate from the mice with combined treatment with SU and RFA. (D) Average frequencies of PD-1+CD4+ T cells and PD-1+CD8+ T cells. n=3, **p<0.01, ***p<0.001, error bars represent means±SD. Statistical analysis was performed by Student t-test.
Sunitinib blocks VEGF signaling pathways to activate DCs and suppress vascularization
Sunitinib specifically blocks multiple tyrosine kinase receptors including VEGFR that is an immune suppressive factor impacting different immune cells.18 20 21 29–33 As described in figure 2, we treated size-matched tumor-bearing mice with no treatment, RFA, SU, or both. One week after RFA treatment, the mice in each group were euthanized. We observed that RF-induced coagulative tumor necrosis was significantly larger in size in the combined treatment compared with RFA monotherapy (top panel in figure 7A). This effect was further confirmed by direct measurement with calipers. The depth and diameter of the necrotic tumor were evaluated and were found to be significantly greater in the combined treatment mice (lower panel in figure 7A). Consistently, the combined treatment induced more tumor apoptosis detected by TUNEL assay (left panel in figure 7B). The width of apoptotic tumor zone in the combined treatment mice was about twofold of that in mice receiving RFA monotherapy (right panel in figure 7B). qPCR assay detected the increased mRNA expression levels of VEGFA, VEGFR1, and VEGFR2 in RFA-treated mice compared with control mice without any treatment. This increase was significantly suppressed after receiving sunitinib treatment (figure 7C). The impact of RFA and sunitinib on tumor angiogenesis was also confirmed in the protein expression levels of CD31 and VEGFR2 by IHC staining (figure 7D). The results in each mouse under different treatments are very consistent, which were reflected by accumulated data analysis with ImageJ (figure 7E). Recent studies indicate that VEGF appears to diminish host immunity by modulating DCs.34 By treating GM-CSF-stimulated bone marrow cells with sunitinib, VEGF, anti-VEGF antibody, or the indicated combination, we demonstrated that sunitinib suppresses VEGF signaling to promote DC differentiation and suppress DC’s PD-L1 expression (figure 7F–H). This effect is consistent with the finding in vivo where sunitinib induced DC increase in RFA-treated mice (figure 3I,J). Together, sunitinib treatment suppresses vascularization and also activates DCs by blocking VEGF signaling pathways.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Sunitinib (SU) blocks VEGF signaling pathways to activate dendritic cells (DCs), suppress vascularization, and enhance radiofrequency-mediated tumor ablation. Mice with size-matched tumors were randomly exposed to no treatment (control), SU, RFA, or both. One week post-RFA, the mice were euthanized for the following studies. (A) SU treatment enlarged RFA-induced tumor ablation. Upper panel: representative macroscopic examination of the damaged tumors; lower panel: the accumulated depth and the diameter of RFA-induced tumor necrosis zone. n=7 in RFA group, n=6 in RFA+SU group, *p<0.05, error bars represent means±SD. (B) RFA-induced tumor apoptosis. Representative images to show RFA-induced tumor cell apoptosis in the mice treated with RFA or SU/RFA. Green color shows the apoptotic area surrounding the ablated tumor zone; blue color shows the DAPI-stained nuclei; red lines show the width of apoptotic tumors in the different mice. Accumulated width of apoptotic tumors in the indicated mice is shown. n=4, *p<0.05, error bars represent means±SD. (C) mRNA expression of VEGF signaling molecules. qPCR detected the mRNA expression of VEGF, VEGFR1, and VEGFR2 in the tumors from the mice under the different treatments. n=4, *p<0.05, **p<0.01, ***p<0.001, error bars represent means±SD. (D) Protein expression of VEGFR2 and CD31. Representative images of immunohistochemistry staining for VEGFR2 and CD31 in the tumors in the mice with the indicated treatments. (E) Accumulative results to show the numbers of VEGFR2-positive and CD31-positive cells in each field. n=5, *p<0.05, error bars represent means±SD. Statistical analysis was performed by Student t-test. (F) Representative flow cytometric assay for DC differentiation and PD-L1 expression. Bone marrow cells in C57BL/6 mice were stimulated with GM-CSFs in the presence of SU, VEGF, anti-VEGF antibodies, or indicated combination. Seven days later, the suspended cells were harvested. After staining for CD11b, CD11c, and PD-L1, the cells underwent flow cytometric assay. (G) Mean frequencies of CD11b+CD11c+ DCs. n=3, **p<0.01, ***p<0.001, error bars represent means±SD. (H) Mean frequencies of PD-L1+ DCs. n=3, ***p<0.001, error bars represent means±SD. Statistical analysis was performed by Student t-test.
Discussion
Leveraging our unique mouse model and a modified human EPT-1000 XPTM cardiac RF generator, we have successfully established a murine RFA platform to improve this first-line HCC therapy by overcoming its intrinsic limitations. We treated tumor-bearing mice with sunitinib to suppress HCC-induced immunotolerance. The changed TME allows RFA-released in situ tumor antigen to prime anti-HCC immunity. The activated immunological tumor death synergizes sunitinib-mediated chemotherapeutic function and RF-induced tumor ablation, significantly improving therapeutic efficacy. Thus, the combination of sunitinib and RFA is emerging as an effective therapeutic strategy offering a potential cure for patients with either small or large HCC. This therapeutic approach can be quickly translated into clinical application, as both sunitinib and RFA are FDA approved and are readily available cancer therapies.
Development of an effective sunitinib/RFA combination therapy is an important contribution to the field of HCC treatment. Both sunitinib and sorafenib are a small molecular inhibitor of multiple tyrosine kinases,18 20 displaying similar drug profiles and overlapping targets. They initially received FDA approval for the treatment of advanced renal cell cancer (RCC). In 2008, the FDA granted approval of sorafenib as a first systemically administrated therapy for unresectable HCC.19 Preclinical and clinical studies from us and others revealed that sunitinib as a HCC chemotherapy drug was not superior to sorafenib; however, sunitinib shows a very strong immune modulatory effect which makes it a preferable chemotherapeutic agent to use in combination with immunotherapy.18 21 25 Image-guided RFA offers a reliable and reproducible modality to treat hepatic lesions with minimal collateral damage to the surrounding hepatic parenchyma.13 However, RFA is most efficacious in treating smaller tumors (≤2 cm), particularly when an ablation margin of ≥4–5 mm can be obtained. RFA has diminishing utility in large tumors, resulting in reduced 3-year and 5-year overall survival rates when compared with surgical resection. Multimodal approaches to combine RFA with other standard approaches have become a subject of recent research interest. Therefore, we proposed that treating HCC-bearing mice with sunitinib followed by RFA was a rational approach. As expected, a markedly improved synergistic outcome was observed, evidenced by significantly reduced tumor growth and extended lifetime observed in the treated HCC-bearing mice (figure 2). This effect is associated with activation of a robust antitumor adaptive immune reaction induced by RFA-released in situ tumor antigens (figures 3 and 4), which were not detected in HCC-bearing mice receiving each monotherapy. These results revealed the potential of sunitinib treatment in preventing tumor-induced immunotolerance in the setting of HCC. The results also supported our previous finding in which we demonstrated that sunitinib in combination with anti-PD-1 antibody primed antitumor immune response to improve anti-HCC efficacy.18 In addition to immune activation, sunitinib treatment significantly enhanced RFA-mediated coagulated tumor necrosis and peripheral tumor apoptosis, as the size of ablated tumors in sunitinib-treated mice was significantly larger than that in the control mice only receiving RFA monotherapy (figure 7), suggesting that sunitinib-mediated angiogenesis suppression enhances RFA’s tumor destruction by attenuating heat-sink effect.35 In conclusion, the novel therapeutic approach of sunitinib in combination with RFA overcomes each monotherapy’s weakness and generates a synergistic therapeutic effect against HCC.
In the present study, we detected the corresponding expression suppression of HGF/c-Met and PD-1 in HCC-bearing mice undergoing sunitinib/RFA treatment. Previously, we demonstrated that HCC growth induced exhaustion of tumor-infiltrating CD8+ T cells by enhancing its expression of PD-1.18 However, we do not know what tumor-intrinsic factors contribute to this increase. By treating the HCC-bearing mice with sunitinib alone, RFA alone, or a conjunction of both, we demonstrated that RFA induced additional PD-1 expression in tumor-infiltrating effector CD8+ T and CD4+ T cells (figure 5) by enhancing production of HGF (figure 6), and this effect was blocked by sunitinib treatment. Similarly, HGF inhibitor blocked PD-1 expression in effector CD8+ T and CD4+ T cells induced by the tumor lysate in RFA-treated tumors. Recombinant HGF restored the capacity of tumor lysate from sunitinib/RFA-treated mice to enhance PD-1 expression in effector CD8+ T and CD4+ T cells (figure 6). In addition, the mono-RFA treatment caused the increased production of c-Met, the receptor of HGF,27 28 and its downstream targets of HIF-1α and IL-6 (figure 6). The increase of these molecules was obviously suppressed by sunitinib treatment (figure 6). Consistent with these results, a previous clinical study in human patients with ovarian clear cell cancer demonstrated that sunitinib exerts therapeutic effect by modulating IL6–STAT3–HIF signaling pathway.36 Similarly, another group has demonstrated that modulating HIF function is an important mechanism to dampen the tumor-promoting inflammatory response and promote tumor growth through direct growth-promoting cytokine production.37 Together, these results indicate that sunitinib treatment blocks PD-1 upregulation through HGF/c-Met/HIF signaling pathway.
We demonstrated that the combinatorial sunitinib/RFA approach modulates anti-tumor immune response in the setting of HCC by exerting multiple functions. Sunitinib treatment significantly reduced tumor-infiltrating FoxP3+ Tregs and increased tumor-infiltrating effector CD8+ T cells (figure 3). This phenomenon was also found in our previous preclinical studies with the same HCC murine model18 21 and others’ clinical studies in patients with RCC.38 We have also demonstrated that sunitinib-mediated suppression of VEGF promoted DC differentiation and reduced its PD-L1 expression (figure 3I,J and figure 7F,G). This may function as another mechanism to underpin sunitinib-activated anti-tumor immunity. This conclusion is supported by the previous studies which have demonstrated the critical role of VEGF/VEGFR in tumor-induced tolerance by widely modulating different immune cells including DCs.33 In response to sunitinib treatment, we detected the increased frequency of central memory CD8+ T cells expressing CD44high and CD62Llow in RFA-treated HCC-bearing mice (figure 3) as well as the significantly reduced PD-1 expression in effector CD8+ T and CD4+ T cells (figure 6). In vivo studies demonstrated that sunitinib treatment enables RFA-released in situ tumor antigens to activate TAS CD8+ T cell by stimulating cytotoxic cytokines production (figure 4). Taken together, these results suggest that sunitinib/RFA therapeutic strategy enables antigen-specific CD8+ T cells to undergo an activation process and mount an effective CD8+ T cell response which is associated with activation of DCs by suppressing VEGF signaling pathways.39
In this study, TAgs are specifically expressed in tumors in our unique model as a trackable TSA. TAg has been well characterized and relevant tools including transgenic mice with TCR for TAg epitope-I and TAg-expressing cells have been developed. This provides an ideal opportunity to evaluate immunotherapeutic approaches to HCC. Still naturally occurring tumor-associated antigens (TAAs) associated with human HCC would provide even more relevant antigenic targets. We have detected the significant upregulation of AFP and GPC3; both antigens are HCC-associated antigens.18 This provides an opportunity to use our model to evaluate the immune response to antigens that occur naturally in human HCCs.
In summary, the current work provides insight into the mechanisms by which sunitinib/RFA combination improves therapeutic outcome against HCC. Sunitinib treatment abolishes tumor-induced immunotolerance. The educated TME allows RFA-released in situ tumor antigen to ignite immunological anti-tumor immune response. In addition, we elucidate sunitinib-mediated molecular regulation of T-cell exhaustion by repressing PD-1 expression via HGF/c-Met and VEGF/VEGFR signaling pathways.
Acknowledgments
The authors thank VA-BIC in the Harry S. Truman Memorial Veterans’ Hospital for the assistance in conducting MRI to monitor tumor growth.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors XQ: study design; acquisition of data; analysis and interpretation of data; statistical analysis. MY: study design; acquisition of data; analysis and interpretation of data; statistical analysis. LM: performance of MRI for tumor detection. MS: critical revision of the manuscript for important intellectual content. DA: study design; critical revision of the manuscript for important intellectual content. JTK: study design; critical revision of the manuscript for important intellectual content. JB: study design; critical revision of the manuscript for important intellectual content. KC: critical revision of the manuscript for important intellectual content. KFS-O: study concept and design; analysis and interpretation of data; drafting of the manuscript; critical revision of the manuscript for important intellectual content; obtained funding; study supervision. ETK: study design; obtained funding; critical revision of the manuscript for important intellectual content. GL: study concept and design; analysis and interpretation of data; drafting of the manuscript; critical revision of the manuscript for important intellectual content; obtained funding; study supervision.
Funding Grant support: VA merit Award I01 BX004065-1 (ETK, KFS-O); R01CA208396 (Mark Kester, GL, KFS-O).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All mice used in these studies were housed, managed, and cared for in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, 2011), and all animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Missouri (Columbia, MO).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement The data in the current study are available from the corresponding author on reasonable request.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.