Article Text

Abstract
Background MEDI9197 is an intratumorally administered toll-like receptor 7 and 8 agonist. In mice, MEDI9197 modulated antitumor immune responses, inhibited tumor growth and increased survival. This first-time-in-human, phase 1 study evaluated MEDI9197 with or without the programmed cell death ligand-1 (PD-L1) inhibitor durvalumab and/or palliative radiation therapy (RT) for advanced solid tumors.
Patients and methods Eligible patients had at least one cutaneous, subcutaneous, or deep-seated lesion suitable for intratumoral (IT) injection. Dose escalation used a standard 3+3 design. Patients received IT MEDI9197 0.005–0.055 mg with or without RT (part 1), or IT MEDI9197 0.005 or 0.012 mg plus durvalumab 1500 mg intravenous with or without RT (part 3), in 4-week cycles. Primary endpoints were safety and tolerability. Secondary endpoints included pharmacokinetics, pharmacodynamics, and objective response based on Response Evaluation Criteria for Solid Tumors version 1.1. Exploratory endpoints included tumor and peripheral biomarkers that correlate with biological activity or predict response.
Results From November 2015 to March 2018, part 1 enrolled 35 patients and part 3 enrolled 17 patients; five in part 1 and 2 in part 3 received RT. The maximum tolerated dose of MEDI9197 monotherapy was 0.037 mg, with dose-limiting toxicity (DLT) of cytokine release syndrome in two patients (one grade 3, one grade 4) and 0.012 mg in combination with durvalumab 1500 mg with DLT of MEDI9197-related hemorrhagic shock in one patient (grade 5) following liver metastasis rupture after two cycles of MEDI9197. Across parts 1 and 3, the most frequent MEDI9197-related adverse events (AEs) of any grade were fever (56%), fatigue (31%), and nausea (21%). The most frequent MEDI9197-related grade ≥3 events were decreased lymphocytes (15%), neutrophils (10%), and white cell counts (10%). MEDI9197 increased tumoral CD8+ and PD-L1+ cells, inducing type 1 and 2 interferons and Th1 response. There were no objective clinical responses; 10 patients in part 1 and 3 patients in part 3 had stable disease ≥8 weeks.
Conclusion IT MEDI9197 was feasible for subcutaneous/cutaneous lesions but AEs precluded its use in deep-seated lesions. Although no patients responded, MEDI9197 induced systemic and intratumoral immune activation, indicating potential value in combination regimens in other patient populations.
Trial registration number NCT02556463.
- immunotherapy
- drug therapy
- combination
- radioimmunotherapy
- CD8-positive t-lymphocytes
- Th1-Th2 balance
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Agonists of toll-like receptor 7 (TLR7) and TLR8 have shown clinical activity in a range of tumors.1 2 These antitumor effects are thought to result primarily from TLR7-mediated and TLR8-mediated recruitment and activation of myeloid and plasmacytoid dendritic cells, which drive increased antigen presentation and costimulation of CD8 T cells, natural killer (NK) cell activation, and regulatory T cell suppression to enhance tumor cell killing.3–9 However, systemic administration of TLR7 and TLR8 (TLR7/8) agonists may be associated with limiting toxicity such as cytokine release syndrome.10–17 This risk could be overcome using an intratumoral (IT) route of administration, while still promoting an immune response beyond the injected site (ie, an abscopal effect).18
MEDI9197 (formerly 3M-052) is a novel small molecule imidazoquinoline agonist of human TLR7/8, developed for IT administration. The chemical structure includes an 18-carbon chain lipid tail to minimize aqueous solubility, thereby reducing dissemination from the injection site and limiting systemic exposure and potential toxicity.19 In mouse models, IT MEDI9197 was retained in injected tumors with low systemic levels, inhibited tumor growth in injected and uninjected lesions and increased survival.18 19 Treatment modulated the immune response by upregulating genes involved in innate and adaptive immunity, increasing tumor infiltrating CD8+ cells and reducing tumor infiltrating CD4+ T cells.19 In vitro, MEDI9197 enhanced the cytotoxic activity of NK cells and T cells by repolarizing macrophages and shifting immunity from a Th2 toward a Th1 phenotype.19
The ability of TLR agonists to stimulate innate and adaptive immunity may amplify the efficacy of other types of immunotherapy, specifically immune checkpoint inhibitors. Response to inhibitors of programmed cell death 1 (PD-1) or its ligand, programmed cell death ligand-1 (PD-L1), is associated with high levels of tumor infiltrating CD8+ T cells, which are activated by TLR7 agonists.19 20 In mouse models, MEDI9197 combined with a PD-L1 inhibitor showed greater antitumor activity than either agent alone, and an in vitro study showed that MEDI9197 plus the PD-L1 inhibitor durvalumab augmented cytokine production in human dendritic cells and allogeneic T cells.19 Preclinical models suggest increased antitumor activity when TLR7/8 agonists are delivered with radiation therapy (RT),21 which releases tumor-specific antigens. This first-time-in-human study evaluated IT MEDI9197 as monotherapy and in combination with intravenous durvalumab and/or palliative RT in patients with advanced solid tumors.
Patients and methods
Study design
This was a three-part, multicenter, open-label, phase 1 dose-escalation study. The first and third parts enrolled patients with solid tumors; the second part enrolled patients with early stage cutaneous T-cell lymphoma, but was closed early with only one patient recruited and is not described further here.
The primary endpoint was the safety and tolerability of MEDI9197 with or without palliative RT (part 1) or MEDI9197 in combination with durvalumab with or without palliative RT (part 3), as assessed by dose-limiting toxicities, other adverse events (AEs), and determination of the maximum tolerated dose (MTD). Secondary endpoints included pharmacokinetics, pharmacodynamics, and objective response based on Response Evaluation Criteria for Solid Tumors (RECIST) version 1.1. Exploratory endpoints included tumor and peripheral biomarkers that may correlate with biologic activity or identify patients most likely to respond to treatment.
Patients
Eligible patients had locally advanced or metastatic solid tumors and had progressed on, were refractory to, or could not tolerate standard of care therapy, or had no standard treatment options. All patients had at least one cutaneous, subcutaneous, or deep-seated lesion measuring at least 1.5 cm in smallest diameter and at most 5.0 cm in longest diameter, suitable for IT injection. Patients were aged at least 18 years, with Eastern Cooperative Oncology Group performance status 0 or 1, at least one measurable lesion per RECIST version 1.1, adequate organ function, and were not currently using immunosuppressive treatment (with the exception of inhaled, intranasal, intra-articular, and topical steroids). Prior immunotherapy was permitted provided that the last dose was given at least 100 days before starting MEDI9197 for regimens including an anti-CTLA-4 inhibitor, oncolytic virus, or oncolytic peptide, or at least 14 days before starting MEDI9197 for all other immunotherapy regimens (including PD-1/PD-L1 inhibitors). Additional eligibility criteria are provided in online supplemental methods.
Supplemental material
Treatment
Part 1
Patients in part 1 received only local therapy (MEDI9197 alone or with RT). MEDI9197 monotherapy was investigated using a 3+3 dose-escalation design at IT doses of 0.005–0.055 mg (with a starting dose of 0.037 mg) every 4 weeks (Q4W) in patients with cutaneous or subcutaneous lesions. The starting dose for patients with deep-seated lesions was 0.012 mg.
Some patients in the 0.012 mg and 0.037 mg cohorts received palliative stereotactic body RT (6 Gray (Gy)/fraction for 5 days), administered per institutional guidelines to the injected lesion. Additional details are in online supplemental methods.
Part 3
A 3+3 dose escalation design was also used to investigate MEDI9197 0.005 mg and 0.012 mg in combination with durvalumab 1500 mg intravenous Q4W. Durvalumab was administered on day 1 and MEDI9197 on day 3±2. Palliative RT (6 Gy/fraction for 5 days) to the injected lesion was permitted, starting on day 1.
In both parts, treatment continued until disease progression, unacceptable toxicity, administration of other anticancer therapy, or patient refusal.
Study assessments
Patients were monitored regularly for safety throughout the study by physical examination, ECG, assessment of blood hematology, clinical chemistry, urinalysis, and vital signs, and assessment of AEs which were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.03. Disease was assessed at screening, including contrast-enhanced CT imaging of the chest and (in patients with melanoma, breast or lung cancer only) brain MRI or contrast-enhanced CT, then every 8 weeks for 32 weeks, and every 12 weeks thereafter until disease progression, start of new treatment or death. Injected cutaneous lesions assessable by physical examination were measured every week until week 7, at week 9, and every 4 weeks thereafter. Serial blood and tumor samples were collected for pharmacokinetic and pharmacodynamic analyses; full methodology is provided in online supplemental methods.
Statistical analysis
All analyses used the as-treated population, defined as all patients treated with any investigational product, unless otherwise specified. Data were summarized using descriptive statistics. Tumor response was estimated by simple proportions with CIs calculated using the binomial exact method. Statistical evaluation of translational data used a one-way analysis of variance (ANOVA) for comparing peak blood cytokine levels, a repeated measures (mixed model) two-way ANOVA for longitudinal cytokine data, and Student’s t-tests for comparing blood or tumor gene expression at different time points. Statistics were performed using GraphPad Prism or R. P values below 0.05 were considered statistically significant for tumor assessments and adjusted p values (false discovery rate) <0.05 were considered statistically significant for whole blood assessments.
Results
Between November 2015 and March 2018, 35 patients were enrolled in part 1 of the study; 30 received MEDI9197 monotherapy at doses of 0.005–0.055 mg, and 5 received MEDI9197 0.037 mg in combination with RT. Dosing was subcutaneous or cutaneous in 31 patients, and into a deep-seated lesion in 4 patients. Part 3 of the study enrolled 17 patients from June 2017 to February 2018. Fifteen patients received MEDI9197 0.005–0.012 mg (subcutaneous or cutaneous, n=9; deep-seated, n=6) combined with durvalumab, and two received a combination of MEDI9197 0.012 mg (both deep-seated), durvalumab and RT. Baseline demographics and clinical characteristics are shown by study part and by dose cohort in online supplemental tables S1 and S2. Treatment, tumor type and tumor location for each patient are shown in online supplemental table S3. Baseline demographics and clinical characteristics were similar in those who received palliative RT and those who did not. In parts 1 and 3, respectively, the median age of patients was 60 years (range 25–85) and 48 years (range 33–72). Patients were generally heavily pretreated; 32/52 (62%) had received ≥3 lines of systemic therapy.
In parts 1 and 3 of the study, respectively, 34 (97%) and 17 (100%) patients had an AE of any cause, with 24 (69%) and 11 (65%) patients experiencing grade ≥3 events of any cause. The rates of treatment-emergent AEs of any grade and those of grade ≥3 were generally similar across dose cohorts, even in patients who received RT. AEs related to MEDI9197 occurred in 28 (80%) and 16 (94%) patients from parts 1 and 3, respectively, and were grade ≥3 in 14 (40%) and 5 (29%) patients (table 1). Across both parts of the study, the most common MEDI9197-related AEs of any grade were fever (n=29; 56%), fatigue (n=16; 31%), and nausea (n=11; 21%), while the most frequent MEDI9197-related grade ≥3 AEs were decreases in lymphocyte count (n=8; 15%), neutrophil count (n=5; 10%), and white cell count (n=5; 10%).
Treatment-emergent adverse events related to MEDI9197, by study part
In part 1, one patient each from the subcutaneous/cutaneous MEDI9197 0.037 mg and 0.055 mg cohorts had cytokine release syndrome (grade 3 and grade 4, respectively), which was dose-limiting in each case. Both patients recovered. The patient with grade 3 cytokine release syndrome missed the second dose of MEDI9197, but subsequently received more than 10 injections at a reduced dose of 0.012 mg without further problems; MEDI9197 treatment was permanently discontinued in the patient with grade 4 cytokine release syndrome. A third patient enrolled in the subcutaneous/cutaneous MEDI9197 0.037 mg plus RT cohort had grade 2 cytokine release syndrome. The MTD for IT MEDI9197 monotherapy was accordingly determined to be 0.037 mg. No further AEs led to discontinuation of MEDI9197 in part 1, and there were no grade 5 AEs.
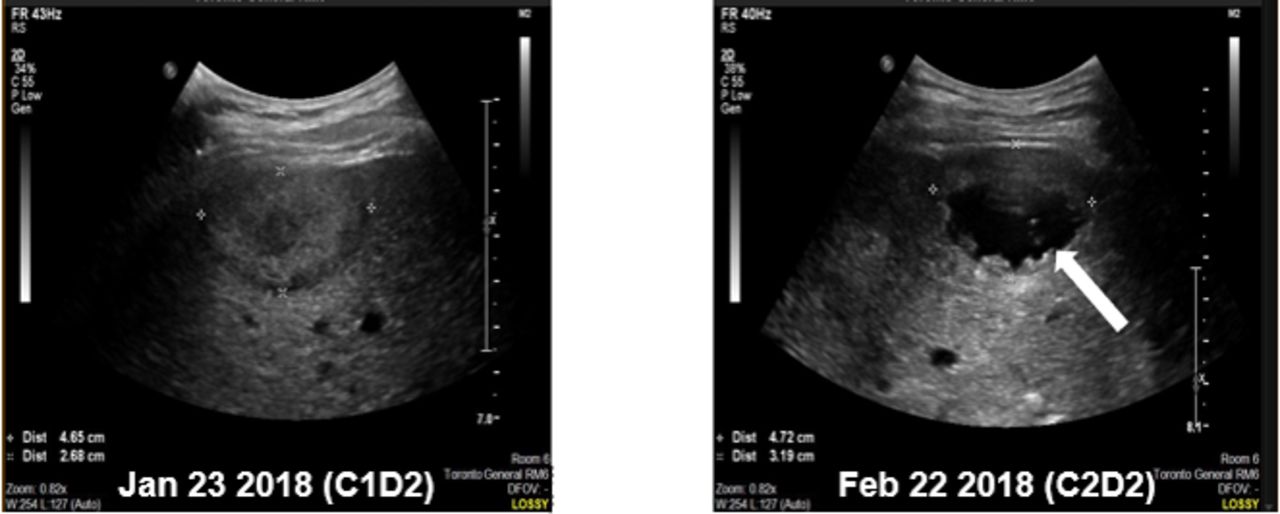
In part 3, 1 patient had a grade 5 AE. This 44-year-old man with anal squamous cell carcinoma received two cycles of MEDI9197 0.012 mg injected into a liver lesion plus durvalumab 1500 mg intravenous; the lesion measured 4.6 cm by 2.7 cm at the time of first injection. He developed sudden-onset, acute right upper quadrant pain on the fifth day of the second cycle, and had a fatal cardiac arrest the next day. An autopsy could not be performed. Images from ultrasound-guided injection on the second day of the second cycle showed a cystic appearance to the injected liver metastasis, which was in the sub-capsular region of the liver and appeared to protrude through the liver contour (figure 1). The injected lesion measured 7.2 cm by 4.4 cm by ultrasound on the day of the second IT injection. The investigator concluded that the death was caused by hemorrhagic shock following rupture of the injected lesion and spontaneous rupture of the liver metastasis, related to MEDI9197 and the injection procedure. This event was considered dose-limiting, and MEDI9197 0.012 mg in combination with durvalumab 1500 mg was the maximum dose administered in part 3. No other AEs in this part of the study resulted in permanent discontinuation of MEDI9197.
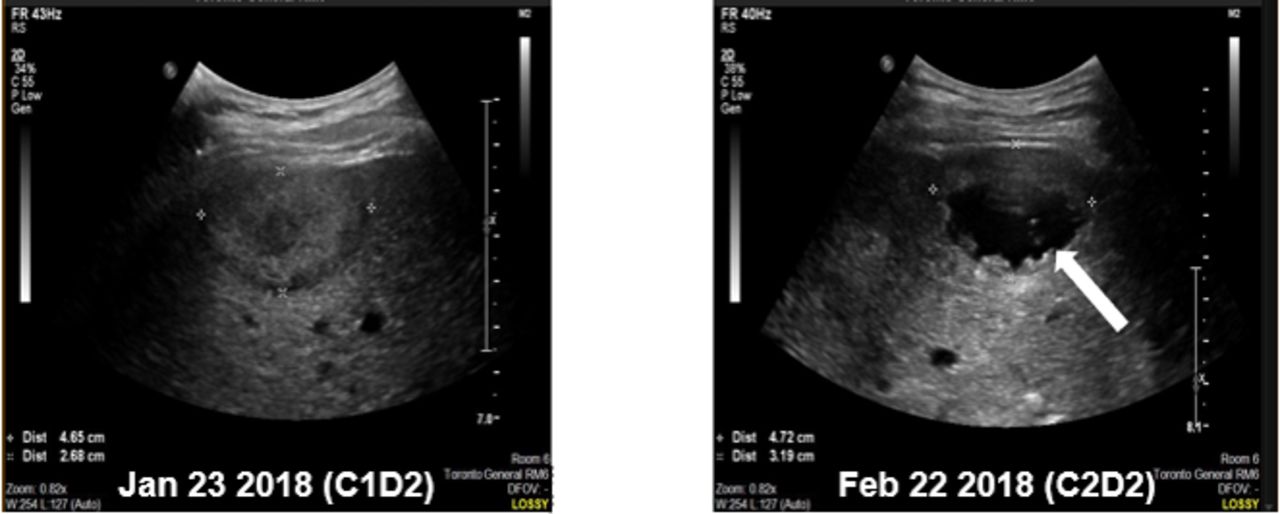
Images from ultrasound-guided injection of MEDI9197 into liver metastases during cycle 1 (A) and cycle 2 (B) in a patient who died of hemorrhagic shock 4 days after receiving his second injection. Note the cystic appearance of the injected lesion (arrow) at cycle 2, day 2.
Another representative patient is shown in online supplemental figure S1A–D. This patient also had metastatic anal squamous cell carcinoma and received MEDI9197 0.037 mg plus 24 Gy RT (4×6 Gy over 3 months). Necrotic changes in the externally visible tumor mass (particularly the injected lesion) suggested a delayed response to MEDI9197 augmented by RT that may have elicited local antitumor immunity.
AEs related to RT and durvalumab are summarized in online supplemental tables 4 and 5.
In patients receiving MEDI9197 monotherapy, peak plasma levels were less than approximately 100 pg/mL, being consistently below the lower limit of quantification in the majority of patients (online supplemental figure S2). Adding RT or durvalumab did not affect peak plasma levels of MEDI9197 (online supplemental figure S3).
Peripheral levels of interferon (IFN)-γ, CXCL10, and CXCL11 increased after the first MEDI9197 monotherapy injection, peaking 18–24 hours post-injection (figure 2A–F), as well as after the second injection (online supplemental figure S4); peak values were unaffected by combining RT or durvalumab with MEDI9197 (online supplemental figure S5). Analysis of whole blood microarray data across all monotherapy doses in subcutaneous or cutaneous lesions demonstrated significant changes in gene expression levels 24 hours (1429 upregulated and 2269 downregulated genes) and 1 week (557 upregulated and 201 downregulated genes) after injection (online supplemental figure S6 and table S6). Increases in Th1 and type 1 IFN gene expression signatures and a transient decrease in CD8A transcript and NK cell signature expression suggested trafficking of T and NK cells (online supplemental figure S7A–D). Similar effects were seen when MEDI9197 was administered in combination with durvalumab (online supplemental figure S7E–H).
Supplemental material

{kind=link}
{kind=link}
Pharmacodynamic effects of intratumoral MEDI9197. Longitudinal plasma cytokine levels demonstrated elevations in (A) interferon (IFN)-γ, (B) CXCL10, and (C) CXCL11 after the first intratumoral injection of MEDI9197 monotherapy (0.005 mg, n=8; 0.012 mg, n=14; 0.037 mg, n=6). One patient treated with MEDI9197 0.037 mg is excluded from these panels due to subsequent receipt of radiation therapy that was not protocol-specified at the time. Peak plasma cytokine levels of individual patients (which occurred 18–24 hours post-injection) are shown for (D) IFN-γ, (E) CXCL10, and (F) CXCL11 (0.005 mg, n=8; 0.012 mg, n=14; 0.037 mg, n=7). Immunohistochemistry staining for (G) CD8+ and (H) programmed cell death ligand-1 (PD-L1)+ cells per mm2 at baseline and day 22 in tumors from patients treated with MEDI9197 0.005 mg, 0.012 mg and 0.037 mg. Quantifications were performed by Definiens quantitative analysis. (I) Relationship of change in CD8 and PD-L1 staining 22 days after treatment with MEDI9197. Closed symbols, subcutaneous/cutaneous lesions. Open symbols, deep-seated lesions. Error bars represent SE of the mean. The lines in (D), (E), and (F) represent the median. *p<0.05 by a repeated measures (mixed model) two-way analysis of variance (ANOVA) (A–C) or by one-way ANOVA (D). Dotted lines at y=1 and x=1 in (I) represent twofold change.
Immunohistochemistry analysis of tumor tissue obtained pretreatment and on-treatment showed evidence of pharmacodynamic effects, with twofold more cells staining positive on-treatment for CD8 or PD-L1 in respectively, 5/19 and 8/18 patients receiving MEDI9197 monotherapy (figure 2G–I), whereas CD8 or PD-L1 staining cells increased by more than twofold in 5/12 and 7/9 patients, respectively, receiving combination treatment (online supplemental figure S8). RNAseq analysis showed 284 significantly differentially expressed genes on-treatment across monotherapy patients with paired tumor biopsies (n=16), including increases in TLR7/8-downstream regulated genes22 and innate and adaptive immune activation signatures in a subset of patients (online supplemental figure S9 and table S6).
Across the study, there were no objective responses to treatment. In parts 1 and 3, respectively, 10 and 3 patients had stable disease ≥8 weeks, thus the disease control rates were 29% (95% CI 14.6 to 46.3) and 18% (95% CI 3.8 to 43.4). Best change in tumor size is shown in online supplemental figure S10 and change from baseline over time is shown in online supplemental figure S11.
Discussion
MEDI9197 monotherapy was generally well tolerated within the dose range of 0.005–0.037 mg. The MTD was 0.037 mg Q4W, with cytokine release syndrome being dose-limiting. The combination of MEDI9197 with durvalumab 1500 mg was not fully evaluated; MEDI9197 dose escalation in part 3 was halted before reaching the planned maximum dose (0.037 mg), after the treatment-related death of a patient from hemorrhagic shock following injection of MEDI9197 0.012 mg into a hepatic lesion. The study was stopped early because of both lack of efficacy and safety concerns about IT injection into deep-seated lesions.
Across the study, most AEs were grade 1 or 2, and the safety profile including fever, chills, influenza-like illness and injection site pain, as well as cytokine release syndrome, was comparable to that of other TLR agonists and consistent with their ability to induce proinflammatory cytokines.10 23–25 The transient leukopenia observed in more than 10% of patients has also been previously reported with systemic imiquimod26 and topical resiquimod.27 28
The local safety profile of MEDI9197 included infrequently observed complications such as swelling, pain, skin ulceration, and hemorrhage. In the patient who died of hemorrhagic shock, analysis indicated that the tumor was located in the sub-capsular region of the liver, which exhibited a dramatic change in texture by ultrasound between cycle 1, day 1 and cycle 2, day 2. A similar local complication—long-lasting delayed skin ulceration—was observed in another patient after administration of MEDI9197+RT. While the safety profile has been acceptable for some immunotherapeutics administered IT in deep-seated lesions, others such as IT CD40 agonists have been associated with severe local AEs.29 This suggests that selection criteria should be more restrictive in future studies of TLR agonists when administered intratumorally in deep-seated lesions. The exact risk of hemorrhage and rupture is unknown; rupture of hepatic lesions is most common in hepatocellular carcinoma, but also occurs rarely in hepatic metastases from other primary tumor types.30–32 Reported risk factors include tumors protruding beyond the original liver contour, exophytic tumors/extrahepatic invasion, tumor size, tumor location within the liver, liver cirrhosis, and hypertension.32–35
Systemic exposure to MEDI9197 was low at all doses evaluated; peak plasma levels of MEDI9197 were approximately 600-fold below the minimal effective concentration (59 ng/mL) that induced cytokines in vitro in human peripheral blood mononuclear cells,19 suggesting that IT MEDI9197 is largely retained in the tumor without extensive systemic dissemination. Nonetheless, pharmacodynamic studies showed rapid systemic and local effects of MEDI9197, consistent with its expected mechanism of action.19 Within the injected tumor, MEDI9197 boosted immune activity in a subset of patients, as indicated by increased numbers of CD8+ T cells and increased adaptive and immune gene signatures. Both type 1 and type 2 IFNs and a Th1 response were induced systemically, indicating activation of both plasmacytoid and conventional dendritic cells. Although these systemic effects suggest the potential for activity in non-injected tumors, no clinical activity was observed. It is possible that the cytokine peaks seen in peripheral blood after IT MEDI9197 injection originated from the tumor rather than representing a systemic response to MEDI9197. Although clinical cytokine release syndrome was reported, we did not see increases in circulating interleukin 6 or tumor necrosis factor-α, which are typically associated with this syndrome. Possible explanations for this include not testing for cytokines at the appropriate time, or that the clinical symptoms reported in the three affected patients were not related to systemic cytokine increase; the exact mechanism triggering those events remains unclear.
Overall, IT MEDI9197 administration in patients with advanced solid tumors, alone or in combination with durvalumab, appeared feasible for subcutaneous/cutaneous lesions but challenging for deep-seated lesions because of the occurrence of one treatment-related death due to hemorrhagic shock after liver metastasis injections. Although no tumor response was observed, MEDI9197 induced local and systemic immune activation, indicating potential value in combination with other drug classes. The absence of synergy between MEDI9197 and durvalumab in patients, as opposed to the synergy observed in preclinical models, is currently unexplained.
Acknowledgments
The authors would like to thank the patients, their families and caregivers, trial nurses, data managers, and all study investigators for their contributions to the study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @zaccoop
Contributors Conception and design: LS, JB, PM, FW, ZC, RK, CF, DSH. Development of methodology: PM, AHR, JW, YW, FW, ZC, CF. Acquisition of data: LS, JB, SG, AM, AJ, PM, JG-O, AHR, AH, RKSW, JW, YW, CM, CF, DSH. Analysis and interpretation of data: LS, JB, SG, AM, PM, JG-O, YW, CM, OH, FW, ZC, RK, CF. Writing, review and/or revision of the manuscript: LS, JB, SG, AM, AJ, PM, JG-O, AHR, AH, RKSW, JW, YW, CM, OH, FW, ZC, RK, CF, DSH.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests LS reports consultant fees for Merck, Pfizer, Celgene, AstraZeneca, MorphoSys, Roche, GeneSeeq, Loxo, Oncorus, Symphogen, Seattle Genetics, GlaxoSmithKline, Voronoi; grant/research funding (for institution) from Novartis, Bristol-Myers Squibb, Pfizer, Boehringer Ingelheim, GlaxoSmithKline, Roche/Genentech, Karyopharm, AstraZeneca, Merck, Celgene, Astellas, Bayer, AbbVie, Amgen, Symphogen, Intensity Therapeutics, Mirati, Shattucks, and Avid. Her spouse reports stocks in Agios. JB reports grant/research funding (for institution) from Merck, Bristol-Myers Squibb, Acerta Pharma, Genentech, Kite/Gilead, Celldex Therapeutics, Celgene. SG reports grant/research funding (for institution) from Bristol-Myers Squibb; personal fees from Bristol-Myers Squibb, Exelixis, Merck, AstraZeneca, and Janssen. AM is the principal investigator of clinical trials for Roche/Genentech, Bristol-Myers Squibb, Merck, MSD, Pfizer, Lytix Pharma, Eisai, AstraZeneca, Chugai, and Tesaro; reports scientific/advisory board fees from GlaxoSmithKline, AstraZeneca, Merck Serono, eTheRNA, Lytix Biopharma, Kyowa, Novartis, Bristol-Myers Squibb, Symphogen, Genmab, Amgen, Biothera, Nektar, Tesaro, OncoSec, Pfizer, Seattle Genetics, AstraZeneca, Servier, Gritstone Oncology, Molecular Partners, Bayer, Partner Therapeutics, Sanofi, Pierre Fabre, Redx Pharma, OSE Immunotherapeutics, Medicxi; speakers’ bureau fees from Roche/Genentech, Bristol-Myers Squibb, Merck, MSD, Merck Serono, AstraZeneca, Amgen, Sanofi, Servier; consultant fees from Roche, Pierre Fabre, Onxeo, Eisai, Bayer, Genticel, Rigontec, Daiichi Sankyo, IMAXIO, Sanofi, BioNTech, Molecular Partners, Pillar Partners, and BPI; patent holder: anti-CD81 (Stanford University); research support from Merus; and research grants (for institution) from Bristol-Myers Squibb, Boehringer Ingelheim, MSD Avenir, and PRTK INCa. AJ reports grant/research funding (for institution) from Bristol-Myers Squibb, Merck Serono, Pfizer, AstraZeneca, Holy Stone Healthcare, Moderna, Iovance, Roche, and Squeeze Therapeutics. PM reports consultant fees from HUYA Bioscience International; leadership roles with Alessa Therapeutics; a patent/royalty/IP with Alessa Therapeutics; honoraria from AtlasMedX, CStone Pharmaceuticals, Xynomic Pharmaceuticals (to institution) McVeigh, Epigene, and Celgene; research funding (to institution) from: Merck, Pfizer, Novartis, GlaxoSmithKline, OncoMed, Celgene, Intellikine, Onconova Therapeutics, Nektar, Sanofi, Merrimack, Roche/Genentech, OncoSec, Bristol-Myers Squibb, Plexxikon, Piramal Life Science, Andes Biotechnologies, Immune Design, and BioMarin. AH reports consultant fees from Amgen, AstraZeneca (to institution), Gritstone Oncology, Incyte (to institution), Lilly, Spectrum Pharmaceuticals (to institution), and Debiopharm Group (to institution); travel/accommodation/expenses from Amgen, Servier, Lilly, AstraZeneca, Roche, and Incyte; other from AbbVie, Agios, Amgen, argenx, Arno Therapeutics, Astex Pharmaceuticals, AstraZeneca, AVEO, Bayer, Blueprint Medicines, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Chugai, Clovis Oncology, Daiichi Sankyo, Debiopharm Group, Eisai, Exelixis, FORMA Therapeutics, GamaMabs Pharma, Genentech, GlaxoSmithKline, H3 Biomedicine, Innate Pharma, Janssen-Cilag, Kyowa, Loxo, Lytix Biopharma, Menarini, Merck, Merrimack, Merus, Millennium Pharmaceuticals, Nanobiotix, Nektar, Octimet, OncoEthix, Onyx, Orion Pharma GmbH, Oryzon Genomics, Pfizer, Pierre Fabre, Roche/Genentech, Sanofi/Aventis, Taiho Pharmaceutical, Tesaro, Xencor, Roche, Servier, Lilly; and honoraria from Merck Serono. JW reports grant/research funding (for institution) from Bristol-Myers Squibb, Merck, Nanobiotix, Mavupharma, and Checkmate Pharmaceuticals; scientific/advisory board fees from Alpine Immune Sciences, Mavupharma, Merck, OncoResponse, and Reflection; consultant fees from AstraZeneca, MolecularMatch, and Nanobiotix; stocks/shares in Checkmate Pharmaceuticals and Reflection; and business ownership of Healios. DSH reports grant/research funding (for institution) from AbbVie, Adaptimmune, Aldi-Norte, Amgen, AstraZeneca, Bayer, Bristol-Myers Squibb, Daiichi Sankyo, Eisai, Fate Therapeutics, Genentech, Genmab, Ignyta, Infinity, Kite, Kyowa, Lilly, Loxo, Merck, Mirati, Mirna, Molecular Templates, Mologen, NCI-CTEP, Novartis, Pfizer, Seattle Genetics, Takeda, Turning Point Therapeutics; travel/accommodations/expenses from Loxo, Mirna, Genmab, AACR, ASCO, and SITC; consultant/advisory fees from Alpha Insights, Amgen, Axiom, Adaptimmune, Baxter, Bayer, Genentech, GLG, Group H, Guidepoint, Infinity, Janssen, Merrimack, Medscape, Numab, Pfizer, Prime Oncology, Seattle Genetics, Takeda, Trieza Therapeutics, WebMD; and other ownership interests with MolecularMatch (Advisor), OncoResponse (Founder), Presagia Inc (Advisor). YW, CM, OH, FW, ZC, RK, and CF are current or former employees of and stockholders in AstraZeneca.
Patient consent for publication All patients provided written, informed consent to their participation in the study.
Ethics approval The study was performed in accordance with ethical principles that have their origin in the Declaration of Helsinki and are consistent with the International Conference on Harmonization/Good Clinical Practice and applicable regulatory requirements. The protocol was approved by Institutional Review Boards (IRBs)/Independent Ethics Committees (IECs) at each center.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. The clinical dataset analyzed during the current study is available at clinicaltrials.gov, https://clinicaltrials.gov/ct2/show/NCT02556463. Any other datasets used and/or analyzed during the current study are available and may be obtained in accordance with AstraZeneca’s data sharing policy, which is described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.