Article Text

Abstract
Background Knowledge about and identification of T cell tumor antigens may inform the development of T cell receptor-engineered adoptive cell transfer or personalized cancer vaccine immunotherapy. Here, we review antigen processing and presentation and discuss limitations in tumor antigen prediction approaches.
Methods Original articles covering antigen processing and presentation, epitope discovery, and in silico T cell epitope prediction were reviewed.
Results Natural processing and presentation of antigens is a complex process that involves proteasomal proteolysis of parental proteins, transportation of digested peptides into the endoplasmic reticulum, loading of peptides onto major histocompatibility complex (MHC) class I molecules, and shuttling of peptide:MHC complexes to the cell surface. A number of T cell tumor antigens have been experimentally validated in patients with cancer. Assessment of predicted MHC class I binding and total score for these validated T cell antigens demonstrated a wide range of values, with nearly one-third of validated antigens carrying an IC50 of greater than 500 nM.
Conclusions Antigen processing and presentation is a complex, multistep process. In silico epitope prediction techniques can be a useful tool, but comprehensive experimental testing and validation on a patient-by-patient basis may be required to reliably identify T cell tumor antigens.
- antigen presentation
- antigens
- antigens
- neoplasm
- T-lymphocytes
- immunotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Immunotherapy has revolutionized cancer treatment. At present, various therapeutics for the enhancement or replacement of T cell antitumor immunity have Food and Drug Administration approval for a variety of cancers.1 2 The core principles driving current immunotherapies began to take shape in the late 20th century with the elucidation of the mechanism by which T cells recognize antigens presented by the major histocompatibility complexes (MHC).3 T cell antigens are specific peptide sequences that are recognized by CD8 or CD4 T cells when presented on MHC I or MHC II molecules, respectively. Neoantigens are peptides derived from tumor-specific mutations which have not been previously recognized by the body’s immune system. Experimental evidence supports that tumor antigen recognition by T cells is critical for antitumor immunity and that cancers can evade such immunity by immunodominance, display of immune checkpoints, or immunoediting for loss of specific tumor antigens.4 Some cancers are composed of subclonal tumor cell populations that harbor defects in antigen processing and presentation, suggesting that these cancers may not be curable despite maximal activation of T cell immunity.5 For those cancers without defects in antigen processing and presentation, knowledge of immunogenic T cell antigens may assist in the engineering of immunotherapeutics designed to control and eradicate cancer.6
For a peptide to serve as a natural T cell antigen, the parental protein must first be processed so that its peptides can be presented on an MHC I molecule. Natural processing and presentation of an antigen is a complex and multifactorial process, which is still subject to active research. The available peptide repertoire is constrained by how proteins are cleaved, trimmed, loaded onto MHC, and translocated to the cell surface. Specificity within this process is likely an explanation for why the vast majority of peptides predicted to be high-affinity MHC binders are unable to elicit T cell responses.7 Validated T cell antigens are often tumor-specific as they are derived from expressed tumor-specific mutated genes. An alternative theory for why many peptide candidates do not elicit T cell responses and, thus, are not antigens, is that these mutated peptides are very similar to their non-mutated counterparts, leading to central tolerance and elimination of potentially reactive T cells.
Despite challenges, there has been success in using T cells to target specific antigen–MHC combinations. The cancer testis antigen NY-ESO-1 is one of the best examples.8 As a validated, highly immunogenic antigen, the use of the NY-ESO-1 T cell experimental system has set the stage for an abundance of T cell and cancer biology research over the past two decades.9 Furthermore, its use in clinical trials has led to advances in adoptive cell transfer immunotherapy, including the first successful treatment of a non-melanoma tumor using T cell receptor (TCR)-engineered T cells.9 The successful application of NY-ESO-1-specific T cells suggests that epitope-specific, T cell-based immunotherapy may allow personalized immunotherapy. In this review, we focus on the mechanisms behind natural processing and presentation of T cell antigens and summarize current methods and limitations of antigen prediction and validation.
Biology of antigen presentation
MHC molecule
Class I MHC molecules are polypeptides composed of a polymorphic heavy chain that associates with a constant β2-microglobulin (β2m) subunit, whereas class II MHC molecules are composed of an α and β polypeptide chain. Class I MHC molecules are generally recognized by CD8 cytotoxic T cells, whereas class II MHC molecules are recognized by CD4 T cells. For the purposes of this review, we will only discuss class I MHC molecules and their interactions with CD8 T cells. The mechanisms underlying epitope prediction and discovery for class II MHC molecules, while important, are beyond the scope of this article. The peptide binding groove of an MHC I molecule preferentially binds 8–11mer peptides. The top surface of the groove, where both the MHC protein and bound peptide are exposed, is the portion of the peptide:MHC complex detected by a TCR.10
Humans possess three MHC I genes: human leucocyte antigen A (HLA-A), HLA-B, and HLA-C. These genes encode highly polymorphic proteins, particularly in the peptide interacting region, allowing different HLA molecules to bind different sets of peptides.11 This presents a challenge to the prediction of MHC binding peptides as prediction algorithms must take into account HLA-specific binding preferences.
From proteins to peptides
For a peptide to be presented on an MHC I molecule, it must be processed by the cellular antigen processing machinery. This begins with proteolysis of parental protein precursors. Exogenous proteins that are internalized by the cell through processes such as phagocytosis and endocytosis are processed in the late endosome and primarily presented on MHC II. Endogenous proteins are processed by the proteasome and are presented mainly on MHC I.12
Proteasomes are proteases that break down misfolded, damaged, aberrant, or ubiquitinylated proteins. The core particle of a proteasome is a barrel-shaped structure composed of two outer α-rings and two inner β-rings.13 Variants of the β-subunits are interferon gamma (IFN-γ) inducible and can replace their constitutive counterparts.14 When these are expressed and active, they form a complex known as the immunoproteasome. The constitutive proteasome is capped at each end by a 19S complex that helps proteins into the proteasome lumen by deubiquitinating and unfolding them. The immunoproteasomes are capped by an alternative 11S complex that is also IFN-γ inducible.14 As a result of the different proteolytic subunits and capping structures, the cleavage site specificity differs between constitutive and immunoproteasomes.15
The characterization of proteasomal activity includes two basic approaches. In vitro study involves co-incubation of parental proteins with proteasomes. The resulting peptides are analyzed using mass spectrometry.16 Several proteolysis prediction techniques are trained on in vitro datasets.17 In vivo proteolysis study involves the measurement of peptides that have been eluted from an peptide:MHC complexes and subjected to mass spectrometry.18 A limitation of this strategy is that only a small proportion of cleavage sites can be identified in this manner as most cleaved peptides do not end up being presented on MHC complexes. Furthermore, the influence of other cellular proteases cannot be deconvoluted from pure proteasome function. Several prediction techniques are trained on in vivo datasets and are generally superior at predicting in vivo proteolysis as compared with prediction techniques trained on in vitro datasets.19
Peptide loading onto MHC I
The peptide loading complex (PLC) is a multi-subunit, endoplasmic reticulum (ER) membrane complex which coordinates peptide translocation into the ER, editing, and loading onto MHC I molecules. One of the core components is the transporter associated with antigen processing (TAP), which is made up of the TAP1 and TAP2 subunits. Each subunit contains a transmembrane domain and a nucleotide binding domain which interact to mediate a channel for peptide movement between the cytosol and ER lumen.12 TAP preferentially translocates peptides with 9–16 residues, although longer peptides up to 25–30 amino acids in length can also be translocated with lower efficiency.20
The specificity of TAP has been well studied. On the C-terminal end of the peptide, TAP selects for the presence of an aromatic, hydrophobic, or positively charged terminal amino acid such as phenylalanine, tyrosine, arginine, or leucine.21 On the N-terminal end, the first three residues have significant effects on peptide binding. Aromatic and hydrophobic side chains are favored and an N-terminal arginine is optimal.22 Proteasomal specificity at the C-terminal residue is relatively non-specific and aligns with the restraints of both TAP and MHC I molecules.23 However, the N-terminal ends of TAP-translocated peptides require further processing within the ER to conform to MHC I binding requirements.24 ER aminopeptidase 1 (ERAP1) and ERAP2 are ER luminal components tasked with the intraluminal processing of peptides.25 ERAP1 trims N-terminal amino acids to create 8–11mer peptides capable of fitting within the MHC I binding groove.26 Peptides undergo structural changes when reaching this size, preventing ERAP1 from further trimming.27
Within the PLC, TAP is associated with a number of other proteins. Tapasin interacts with TAP and serves to recruit MHC I–β2m complexes to the PLC.28 Tapasin is further linked to ERp57 and calreticulin, which mediate the folding of newly synthesized MHC molecules.12 Following translocation of peptides by TAP, peptides are released into a molecular basket, confined by two editing complexes composed of tapasin, CRT, ERp57, and MHC I. Peptides are then further edited by ERAP1 and loaded onto MHC I molecules.29 Once loaded onto MHC I, peptides undergo additional processing and quality control mediated by tapasin and TAP binding protein-related protein (TAPBPR).30 TAPBPR associates with empty MHC I molecules, stabilizing their structure. The choice of peptide is based on the stability of the final MHC I–peptide complex, and TAPBPR is released once a high-affinity MHC I–peptide complex is stably achieved.31 A similar mechanism is thought to underlie the function of tapasin in the PLC.29 31 If the affinity for the bound peptide is high and the resulting complex is stable, the MHC I–peptide complex will be transported through the Golgi apparatus and to the cell surface.12 The peptide specificity associated with each step of this process beginning from proteasome proteolysis to MHC I binding is what defines natural processing and presentation of epitopes. For a peptide to be clinically relevant, experimental validation should strive to mimic this process.
Cross presentation
Endogenous proteins are processed through the proteasome pathway and loaded onto MHC I molecules for detection by CD8 T cells, whereas exogenous proteins are processed through the endosomal pathway and presented on MHC II molecules for detection by CD4 T cells. Tumor cells alone are unable to prime an effective, antigen-specific, CD8 cytotoxic T cell response.32 How do CD8 T cells targeting MHC I-restricted antigens become primed and activated against antigens that originated as exogenous proteins?
Cross presentation refers to the presentation of exogenous proteins on MHC I.33 In 1990, Rock and colleagues found that soluble antigens in the extracellular fluid could be internalized, processed, and presented on MHC I molecules in specialized antigen presenting cells (APC).34 One crucial APC is the dendritic cell (DC). Mechanistic studies in mice have demonstrated that the development of antigen-specific T cell responses depends on functional APC.32
DCs can acquire antigens from multiple sources. Dying cells release antigens and they are an important source of antigenic materials.35 Antigens ingested by DC through micropinocytosis, endocytosis, or phagocytosis are efficiently cross-presented.36 While the exact mechanisms are unclear, it is hypothesized that the corresponding downstream intracellular compartments, such as the phagosome or endosome, contain cellular proteins, which are part of the MHC I presentation pathway.33 Additionally, DCs can internalize cytosolic and membrane materials from live cells to prime T cell responses.37 Possible mechanisms for this transfer of antigens includes ingestion of secreted antigens and trogocytosis, where one cell ‘nibbles’ bits off another.38 39
Once a DC has acquired an antigen, processing and presentation occur through either a proteasome-independent vacuolar pathway or a proteasome-dependent cytosolic pathway. To establish a diverse repertoire of T cells that can recognize tumor-presented antigens, an ideal cross-presentation pathway would mimic the natural processing and presentation machinery of a tumor cell. In the vacuolar pathway, proteins are lysed into peptides within the endocytic compartments and then loaded onto MHC I molecules without ever encountering the cytosol.33 40 Therefore, the proteasome and TAP are never encountered, meaning that the repertoire of selected epitopes may differ from those presented on a tumor.41 In the proteasome-dependent pathway, proteins progress through the cytoplasm and ER.42 Several studies have confirmed that exogenous antigens appear in the cytosol.43 Once in the cytosol, the proteins undergo the same process described earlier. Studies have verified that peptides generated by the immunoproteasome from conventional endogenous antigens are identical to immunoproteasome-generated peptides from exogenously delivered proteins.44 Presentation of tumor antigens by APC to naïve T cells in lymphoid tissues then primes T cells for clonal expansion and cytotoxic effector function.
Antigen identification in cancer therapy
Many sources of cancer antigens exist. Epitopes derived from single nucleotide variations (SNVs), insertions/deletions (INDELs), transcript splice variants, gene fusions, and endogenous retroviral elements can all serve as tumor-specific antigens.45 46 Some immunotherapies do not rely on the identification of tumor antigens for use, while other forms of immunotherapy do require knowledge of antigen and possibly HLA type. Immune checkpoint blockade immunotherapy is widely used but induces durable tumor control in only one subset of patients and does not require a priori knowledge of antigens.47 Adoptive transfer of bulk cultured tumor infiltrating lymphocytes (TIL) as an immunotherapeutic approach has produced durable regression of malignancy in a limited subset of patients, and this approach does not rely on the identification of specific tumor antigens.48 Conversely, adoptive cell transfer of TCR-engineered T cells does rely on the identification of a specific antigen target, the HLA restriction, and the TCR specific for this antigen:MHC complex. Autologous T cells are engineered to express TCRs that target antigen:MHC complexes presenting tumor-specific neoantigens.49 Personalized cancer vaccine therapies similarly rely on the identification of tumor-specific neoantigens.50 51 However, the utility of these potential therapies is constrained by the difficulties underlying the discovery of tumor-specific epitopes that elicit T cell responses.52 Here, we review the commonly utilized epitope discovery techniques and key considerations underlying these methods.
Identifying tumor-specific epitopes and epitope-reactive T cells
A general approach to identify neoepitopes and accompany reactive T cells from humans is a reverse immunology approach based on tumor sequencing. A section of surgically resected tumor or metastasis is cultured in media containing cytokines selective for lymphocyte growth, notably IL-2, to expand TIL in vitro. In addition, another portion of the tumor is sent for genome sequencing to identify genetic mutations and the corresponding repertoire of potential neoepitopes. The resulting neoepitopes can be further analyzed in silico to predict the best candidates for MHC I cell surface presentation. Next, T cells are cocultured with APC expressing the antigens of interest and analyzed for activation signals. T cells reactive against antigens can be assessed further via TCR sequencing or expanded as primary effector cells.
Tran and colleagues have successfully applied this strategy to identify T cells reactive against mutant KRAS G12D in a patient with metastatic colorectal cancer.48 Three metastatic lung lesions were resected and used to generate TIL cultures and whole exome sequencing (WES) data. In total, 61 mutations were identified. Cultures of TIL were screened against all identified mutant neoepitopes expressed by the tumor. For each mutation, a minigene encoding the mutation flanked by 12 amino acids on either side was generated and synthesized in tandem to create tandem minigene (TMG) constructs. Five total TMG constructs were made, in vitro transcribed into RNA, and electroporated into autologous DCs to undergo natural processing and presentation of epitopes. TIL cultures were reactive against TMG-1, so the corresponding peptides were synthesized and pulsed onto DCs, followed by coincubation with TIL. Adoptive transfer of four T cell clonotypes that demonstrated antigen-specific reactivity resulted in objective regression of all lung metastases.48 The TMG approach to identify tumor-specific T cell antigens is highly comprehensive as it interrogates responses from all possible neoantigens.
Methodological considerations
Sequencing and mutation calling
The identification of expressed genetic mutations is critical to neoepitope discovery. After tumor and normal tissue samples are sequenced, typically using next-generation sequencing (NGS), the presence of somatic genetic variants is assessed through the application of variant calling pipelines. Comparisons of different pipelines have been summarized.53
In the context of neoepitope prediction/analysis, somatic variants can be classified into SNVs, INDELs, and structural variants. Somatic SNVs are capable of generating immunogenic neoepitopes through amino acid substitutions, and reactive T cells to SNV-derived neoepitopes are detected with higher frequency in cancers with higher mutational burdens.54 55 Less well studied are INDELs, which can lead to frameshift mutations that create neo open reading frames (neo-ORF). Frameshift INDELs lead to higher proportions of neoantigens per mutation compared with SNVs. Additionally, INDELs more commonly result in mutation-specific predicted high-affinity binding neoantigens compared with that of SNV mutations.56Several studies have discovered T cell clones reactive against frameshift INDELs.57 58 While tumors can harbor hundreds of neoantigens, the number of mutant peptides that can be tested in parallel is limited by time and cost. Therefore, prioritization of candidate antigens for experimental validation is important when conducting large-scale experiments.
As the advent of NGS has made the study of genetics-based precision medicine more clinically feasible, investigators must also be aware of its limitations. Study of putative neoantigens to date has primarily focused on the coding region of the genome, given that antigens must be transcribed and translated into protein that can be processed into peptides. The combination of WES and RNA-seq is commonly employed for consistent variant analysis and provides consistent results across different sequencing centers.59 It is worth mentioning that many tumor-rejection antigens have been identified from non-canonical protein sequences derived from retained introns, non-coding regions, alternative ORF, or proteasome-mediated splice variants.60–62 To capture these possibilities, studies may use a combination of WGS, WES, RNA-seq, and mass spectrometry.
Lastly, there are considerations related to the method of tissue sample preservation. Formalin-fixed, paraffin-embedded (FFPE) tissues are an invaluable resource, but the process of sample fixation may modify nucleic acids to cause inaccurate or incomplete capture of information. Studies have shown that FFPE processing of tissue can lead to reduced transcript detection in RNAseq data compared with fresh frozen (FF) samples.63 Currently, the use of FF tumor samples is the gold standard.
Antigen testing
The question of natural processing and presentation is of great concern as most experimental systems permit T cell responses against non-naturally processed and presented epitopes.64 In the study by Tran et al, natural processing and presentation of epitopes was ensured by using TMG constructs electroporated into APCs, meaning that the in vitro transcribed TMG mRNA directly entered the cytosol and was translated into protein, and the protein was processed in a proteasome-dependent manner. Coincubation of APCs with synthetic long (15–30mer) peptides is an alternative strategy for APCs to acquire and present exogenous antigens.65 When designing peptide pulsing experiments, it is important to consider the peptide length. In a comparison of overlapping 24-mer long peptide versus whole protein, Zhang and colleagues found that 24-mer peptides trafficked to both endosomes and cytosol to activate CD8 and CD4 T cells, whereas whole protein (HIV nef, 206 amino acids) was found only in the endosomal compartment and not in the cytosol, activating CD4 but not CD8 T cells.66 Pulsing of minimal epitope or short peptides of length 8–16 may also be misleading as this does not guarantee natural processing by the immunoproteasome, transport by TAP, or trimming by ERAP1. In a study of the TEL-AML1 translocation, an immunogenic minimal epitope did not illicit T cell responses when pulsed into APCs as part of a long peptide, indicating that the minimal epitope was not naturally processed and presented.67 Electroporation of mRNA encoding the full-length protein of interest (mimicking endogenous peptide) into APCs or pulsing APCs with long peptide (mimicking exogenous peptide) that require processing may be the most effective means of ensuring that identified antigens are naturally processed and presented from the parental protein. Small 8–11mer epitopes can then be used directly after narrowing down candidates to confirm the identity of the minimal epitope.
While TCR signaling strength is generally thought to be proportional to peptide–MHC affinity, TCR binding of peptide:MHC complexes does not always result in T cell activation.68 Greater than 15% of T cell clones do not produce antigen-specific responses even after peptide–MHC recognition due to structural mechanisms.68 The existence of naturally processed and presented epitope:MHC complexes that do not elicit T cell activation introduces a wrinkle into methods that attempt to predict antigenic targets based solely on MHC binding and tumor cell antigen processing. Several methods have been developed to directly characterize T cell specificities. Kula and colleagues developed T-scan as a high-throughput, unbiased method for the discovery of antigens on a genome scale. Using this system, T cell pools are used to recognize cells virally transduced with antigen libraries. Functionally recognized cells are then isolated using a reporter for granzyme B activity and sequenced, allowing identification of cognate antigens.69 Studies by Baltimore and colleagues use a similar schema of querying antigen libraries, but use alternative reporter mechanisms of trogocytosis or activation of synthetic signaling molecules to perform high-throughput, unbiased epitope discovery.70 71 The application of these methods to screening of autologous TIL against patient-specific antigen libraries or the screening of candidate TCRs for off-target effects holds promise. Of note, although many T cell clones overlap between tumor and blood, tumors display increased clonality and harbor-enriched clones which are absent or rare in the blood.72 Therefore, TIL should be prioritized over PBMC as the preferred T cell source for experimental validations.
In silico prediction algorithms
The advent of in silico epitope prediction algorithms began with the hypothesis that certain peptide sequence motifs were more likely to bind to an MHC molecule compared with others.73 These motifs were generally based on primary anchor residues that highly influenced binding and secondary anchor positions that were less important for binding.74 For example, positions 2 and 9 serve as anchor residues in HLA-A*02:01 and are most often occupied by small hydrophobic amino acids such as valine, leucine, and isoleucine.75 From these motifs, matrices were developed which assigned a numerical score corresponding to the contribution of each amino acid at each position; values for each amino acid within a peptide were then combined to generate a score for each peptide:MHC combination.76 By design, these models assumed that the contribution of an amino acid at a particular position was independent of its neighboring positions. This may not be true.77
To handle these interactions and to improve predictions over motif-based models, researchers turned to machine learning algorithms. The machine learning algorithms used for T cell epitope prediction are trained on large-scale datasets, resulting in a model that deciphers patterns from a known set of input data and applies them to new data to generate predictions. Datasets used for T cell epitope prediction can be classified as datasets of peptides with known binding affinity to MHC molecules and datasets containing peptides that either bind or do not bind to MHC molecules.78 Models trained on the combination of MHC binding and ligand elution data show superior performance compared with models trained on binding datasets alone.79 An additional benefit of machine learning is that methods can be developed to predict binding for rare HLA alleles using data from well-known HLA alleles (pan-specific predictors).79 This is important because rare HLA alleles often do not have sufficient data to train their own prediction algorithms. The quality of the available datasets determines the performance of the machine learning algorithms. Several databases compile epitope data in a consistent manner to enable proper training of machine learning algorithms.80
Most in silico epitope prediction algorithms aim to predict binding affinities of peptides to MHC I molecules. This is based on the assumption that peptide binding to MHC is the single most selective step in the antigen presentation pathway.77 79 Comparison of a combined proteasome/TAP/MHC affinity predictor with a predictor based on MHC affinity alone determined that the MHC affinity predictor offers higher sensitivity whereas combined predictor offers higher specificity. For a hypothetical protein of 300 amino acids, one would need to functionally test 11 peptides to identify a new T cell-reactive epitope with 90% likelihood when using a combined predictor versus 13 peptides when using a MHC affinity predictor.81 Furthermore, widely used prediction algorithms do not incorporate peptide:MHC complex stability, which is a more important determinant of immunogenicity than binding affinity.82 Incorporation of peptide dissociation data into current algorithms may improve prediction performance.83 Current immune epitope database (IEDB) guidelines recommend using MHC binding predictions alone by default and using prediction scores that incorporate antigen processing as an additional filter if resources require limiting.84
The ways in which researchers use MHC epitope prediction data are varied. Studies of MHC I binding affinity and T cell epitope recognition have shown that predicted MHC binding IC50 of 500 nM or less results in approximately 90% sensitivity for identification of true T cell reactive epitopes.85 Genetic differences between HLA alleles create disparities; some alleles are better able to bind oncogenic mutations while others are predicted to have very few peptide binders with IC50 <500 nM.86 Therefore, the rank of an epitope’s MHC binding affinity in comparison with all other possible epitopes (percentile rank) has been suggested as an alternative. When generating predictions across multiple HLA alleles, percentile ranks are preferred to avoid bias towards certain HLA alleles.87
In the development of a personalized melanoma vaccine, Ott et al utilized NetMHCpan to identify up to 30 patient-specific MHC I-restricted high-affinity binders derived from mutant peptides to include in each vaccine.88 The final list of epitopes was chosen based on a rank list that prioritized neo-ORFs over SNVs, mutations in anchor residues over secondary residues, and low MHC IC50. For each patient, up to 20 putative neoantigens were ultimately incorporated into a personalized vaccine and delivered. All neoantigens had predicted MHC IC50 <500 nM or rank in the top one percentile. Ex vivo analyses demonstrated that 15 of 91 (16%) predicted immunizing peptides-induced CD8 antigen-specific T cell responses across six patients. The total number of predicted epitopes that can be included in each vaccine is limited, but the use of MHC I binding prediction algorithms with astute prioritization criteria generated sufficient specificity to generate T cell reactive epitopes for each patient. Of six vaccinated patients, four were recurrence-free 25 months after vaccination and the remaining two experienced complete tumor regression after treatment with anti-programmed death receptor-1 (PD-1) therapy.88
Assessment of in silico prediction
We utilized the IEDB proteasomal cleavage/TAP transport/MHC class I combined predictor tool to retrospectively generate total prediction scores and MHC binding affinities of a list of known and validated epitopes derived from the Cancer Antigenic Peptide Database (https://caped.icp.ucl.ac.be/).89 The total score is a combined score incorporating proteasomal cleavage, TAP transport, and MHC binding predictions where higher scores represent higher efficiency for MHC presentation.90
Validated tumor-specific neoantigens are summarized in table 1 and validated tumor-associated antigens derived from germ-line or differentiation antigens are summarized in table 2. Tumor-specific and tumor-associated antigens displayed similar MHC IC50 and total score profiles and were combined for further analyses. A significant proportion of epitopes displayed weak predicted binding (IC50 >500 nM, 30.3%) to their respective MHC alleles, with several epitopes exhibiting very weak predicted binding (IC50 >10,000 nM, 9.7%) (figure 1A). One possible explanation for why low-affinity peptides elicit strong T cell responses is that these weakly immunogenic antigens have not been subjected to immunoediting and therefore represent the immunodominant antigen following elimination of more immunogenic targets. When considering discovery of tumor-specific putative neoantigens, nearly all studies utilized a comprehensive querying technique that did not use, or minimally used, epitope prediction algorithms. Methods that heavily incorporated epitope prediction methods were also successful, but rarer in this database. Robbins et al tested the top 55 predicted 9mer or 10mer MHC I binders from a melanoma cell line and demonstrated that the 5th, 18th, 19th, and 39th ranked peptides elicited T cell responses.7 The hit rate of 4/55 and seemingly random rank of peptides validated as antigens raises the question of whether peptide prediction algorithms perform better than random testing. Unfortunately, it is impossible to know how many similar studies failed to recognize successful epitopes. The only solution to achieve maximum sensitivity for detecting antigens is comprehensive screening of all possible neoantigens, which should be used when resources are available.
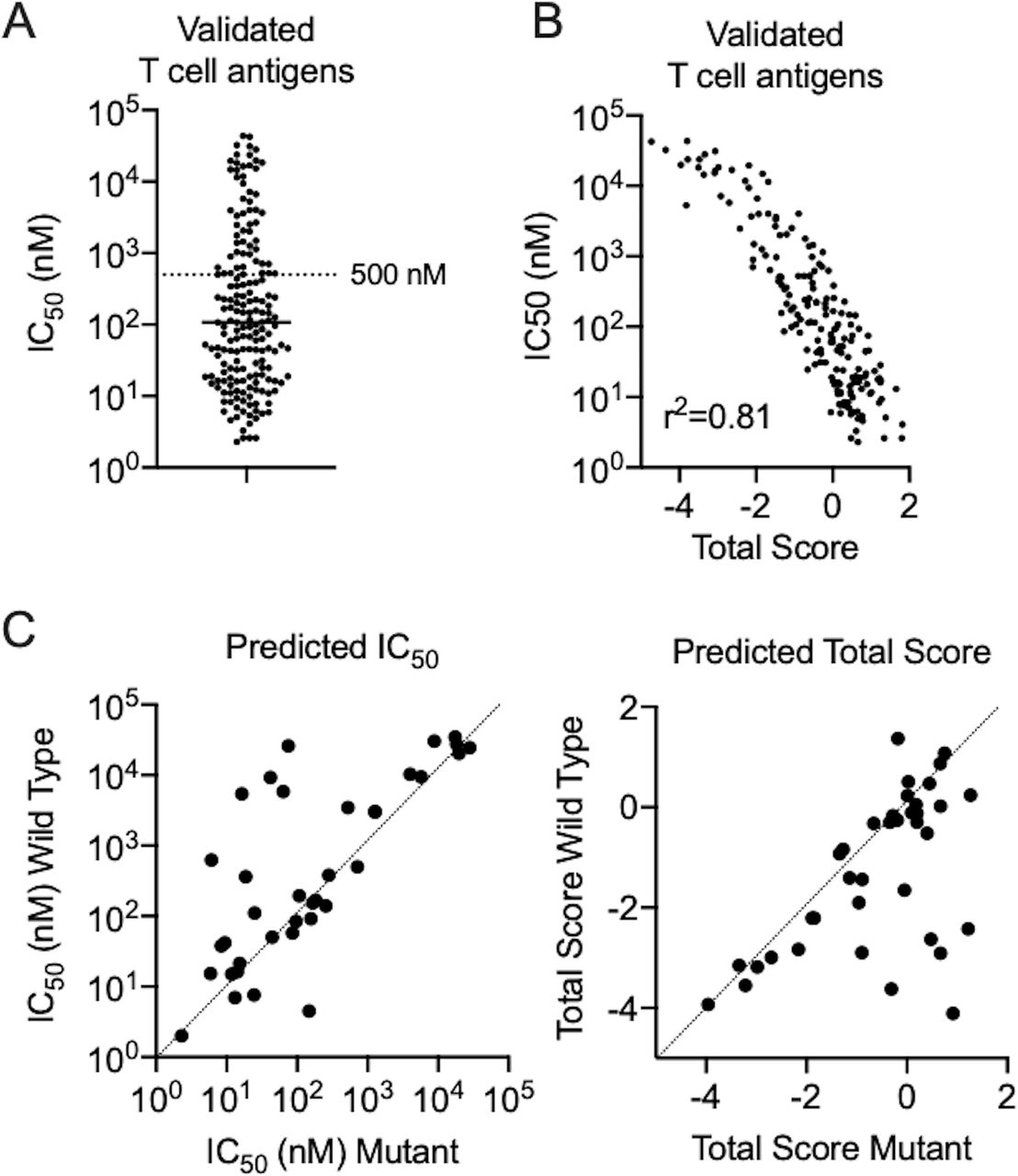
{kind=link}
Performance of in silico predictions in validated T cell antigens. (A) Dot plots of predicted MHC IC50 for a panel of validated T cell antigens. A reference level of IC50 500 nM is provided to delineate a common cut-off score used by researchers. (B) Scatterplot of MHC IC50 and total prediction score showing a linear correlation (Pearson correlation, p<0.001). (C) Scatterplots of predicted MHC IC50 and total score between wild-type and mutant peptides for validated neoepitopes. MHC, major histocompatibility complex.
Table of mutated tumor-specific antigens from the Cancer Antigenic Peptide Database
Table of germ line and differentiation antigens from the Cancer Antigenic Peptide Database
There was high correlation between MHC IC50 and total prediction score in each group (figure 1B), indicating that use of MHC binding predictions alone compared with total prediction scores is likely to generate similar results. When comparing validated neoantigens with their wild-type (WT) counterparts, many neoantigens had predicted MHC I binding affinities and total prediction scores that were similar to that of the WT sequence (figure 1C). Since WT sequences are widely presented throughout the body, central T cell tolerance should have protected the body against auto-immunity. Shaping of T cell repertoires through thymic selection is a critical step in the determination of tolerance on exposure to various antigen:HLA complexes.91 The simplest experimental models of thymic negative selection have demonstrated that negative selection is specific enough that single amino acid substitutions in self-peptides can effectively be recognized as foreign by T cells and serve as neoantigens.92
Why predictions are inconsistent
The stability of the peptide:HLA class I complex may determine presentation to and development of an activated T cell clone to a greater degree than simple peptide binding affinity.93 The algorithms detailed earlier do not incorporate stability as a variable, illustrating one deficiency of current, widely utilized modeling approaches. Weighted inclusion of stability in predicting immunogenic epitopes outperformed predicting based on binding affinity alone, and such parameters may be incorporated into future prediction pipelines.83
Conclusions
The future of immunotherapy will hinge on its ability to achieve durable responses in a large patient population. Given low rates of durable tumor regression observed with immune checkpoint blockade in patients with relapsed malignancy, other T cell-based immunotherapeutic approaches such as personalized cancer vaccines and adoptive transfer of TCR-engineered T cells may be able to improve outcomes. These approaches require the determination of T cell antigens. The discovery of such antigens is largely constrained to the study of few tumor types (mostly melanoma) in the most common HLA alleles. While it seems like bona fide, immunogenic epitopes are the exception and not the rule, new methods of epitope discovery may allow for high-throughput, comprehensive searches of proteins and mutations to the point where personalized cancer immunotherapy is feasible for patients whose tumors harbor intact antigen and processing machinery. However, numerous limitations based on current technologies exist which make reliable identification of a naturally processed and presented antigen difficult. Recent studies have demonstrated proof of concept that epitope discovery can lead to remarkable tumor regression in highly metastatic patients.48 49 88 The development of strategies that allow for efficient and accurate antigen discovery, such as incorporation of peptide:HLA complex stability, is poised to make huge contributions to cancer treatment in the near future as fundamental building blocks of TCR discovery for adoptive T cell transfer and personalized cancer vaccine therapy.
References
Footnotes
Contributors Concept: MYL and CA. Literature reviews: MYL, JWJ, CS, and CA. Computational work: MYL and JWJ. Authorship of manuscript: MYL, JWJ, CS, and CA. Final approval: MYL, JWJ, CS, and CA.
Funding This work was supported by the Intramural Research Program of the NIH, National Institute on Deafness and Other Communication Disorders, project number ZIA-DC00008. This research was also made possible through the NIH Medical Research Scholars Program, a public–private partnership supported jointly by the NIH and contributions to the Foundation for the NIH from the Doris Duke Charitable Foundation (DDCF Grant #2014194), the American Association for Dental Research, the Colgate-Palmolive Company, Genentech, Elsevier, and other private donors.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.