Article Text

Statistics from Altmetric.com
- immunotherapy
- programmed cell death 1 receptor
- receptors, immunologic
- translational medical research
- tumor escape
Introduction
Overcoming initial or acquired resistance to programmed death-ligand 1 (PD-(L)1) immune checkpoint inhibitors is a major area of unmet need for many cancers.1 Although the full scope of mechanisms of resistance to these therapies has yet to be determined, different forms of tumor-derived extracellular PD-L1 have been linked to resistance in multiple clinical studies.2–4
Malignant cells produce trans-acting extracellular PD-L1 in three distinct forms. First, tumor cells transcribe and secrete soluble PD-L1 (sPD-L1) splice variants.4 5 Second, enzymes ADAM10 and ADAM17 shed sPD-L1 ectodomain directly from the tumor cell surface.6 7 Both forms of sPD-L1 carry known homodimerization domains and can be detected by ELISA. sPD-L1 can outcompete PD-(L)1 inhibitors, kill CD8+ T cells, and limit the ability of healthy peripheral blood mononuclear cells to kill tumor cells in vitro.6 In a third form, tumors generate extracellular vesicles (EVs) bearing surface PD-L1.8 9 PD-L1-positive EVs (evPD-L1) exhibit similar properties to sPD-L1 in systemic circulation.9 Each type of trans-acting extracellular PD-L1 correlates with poor survival in multiple clinical trials (online supplementary table 1). While broad-spectrum pharmacological inhibitors and genetic manipulation have been shown to reduce release of these forms of PD-L1 in culture or animal models, none are suitable for clinical use. To date, we know of no reported clinical intervention that safely and reliably eliminates any of these forms of immunosuppressive, systemic extracellular PD-L1.
Supplemental material
Therapeutic plasma exchange (TPE) is a procedure in which blood is passed through an apheresis machine separating plasma from cellular components. Removed plasma is discarded and replaced with either colloid (eg, albumin) or crystalloid and colloid solutions. Unlike dialysis, which removes small ions by diffusion, TPE removes plasma-restricted substances like antibodies that are too large for rapid diffusion. On average, each TPE session removes approximately 65%–70% of large non-cellular, plasma-restricted intravascular components.10
It is unknown whether sPD-L1 (approximately 35 kDa) or PD-L1-positive EVs (50–500 nm) are plasma restricted, non-diffusing, and unbound. If so, we hypothesized that TPE could efficiently remove these substances from patient blood. Such an intervention could, if effective, improve response to immunotherapy.
Results
sPD-L1 levels predict overall survival in patients with melanoma
Each form of extracellular PD-L1 acts in trans as a systemic immunosuppressant through PD-1 signaling (figure 1A, online supplementary table 1).4–9 To confirm the clinical impact of plasma sPD-L1, we measured sPD-L1 levels in a retrospective cohort of 276 patients with melanoma. Exploratory analysis of overall survival (OS) determined a working cut-off value of sPD-L1 (≥0.277 ng/mL) and baseline characteristics at the time of entry into study were similar (online supplementary table 2). Patients with high plasma sPD-L1 levels experienced inferior median OS compared with patients with low plasma sPD-L1 levels (figure 1B, 12.1 vs 18.5 months, p=0.005). In comparison to healthy age-matched controls, patients with melanoma exhibited higher mean plasma sPD-L1 (figure 1C, 1.72 ng/mL vs 0.77 ng/mL, p<0.001). In a multivariate Cox proportional hazards analysis, high sPD-L1 prior to treatment predicted worse survival (HR 1.49; 95% CI 1.06 to 2.09; p=0.025) when accounting for advanced age (not significant), sex (not significant), late stage (p=0.002) and high serum LDH (p=0.01; online supplementary table 3).
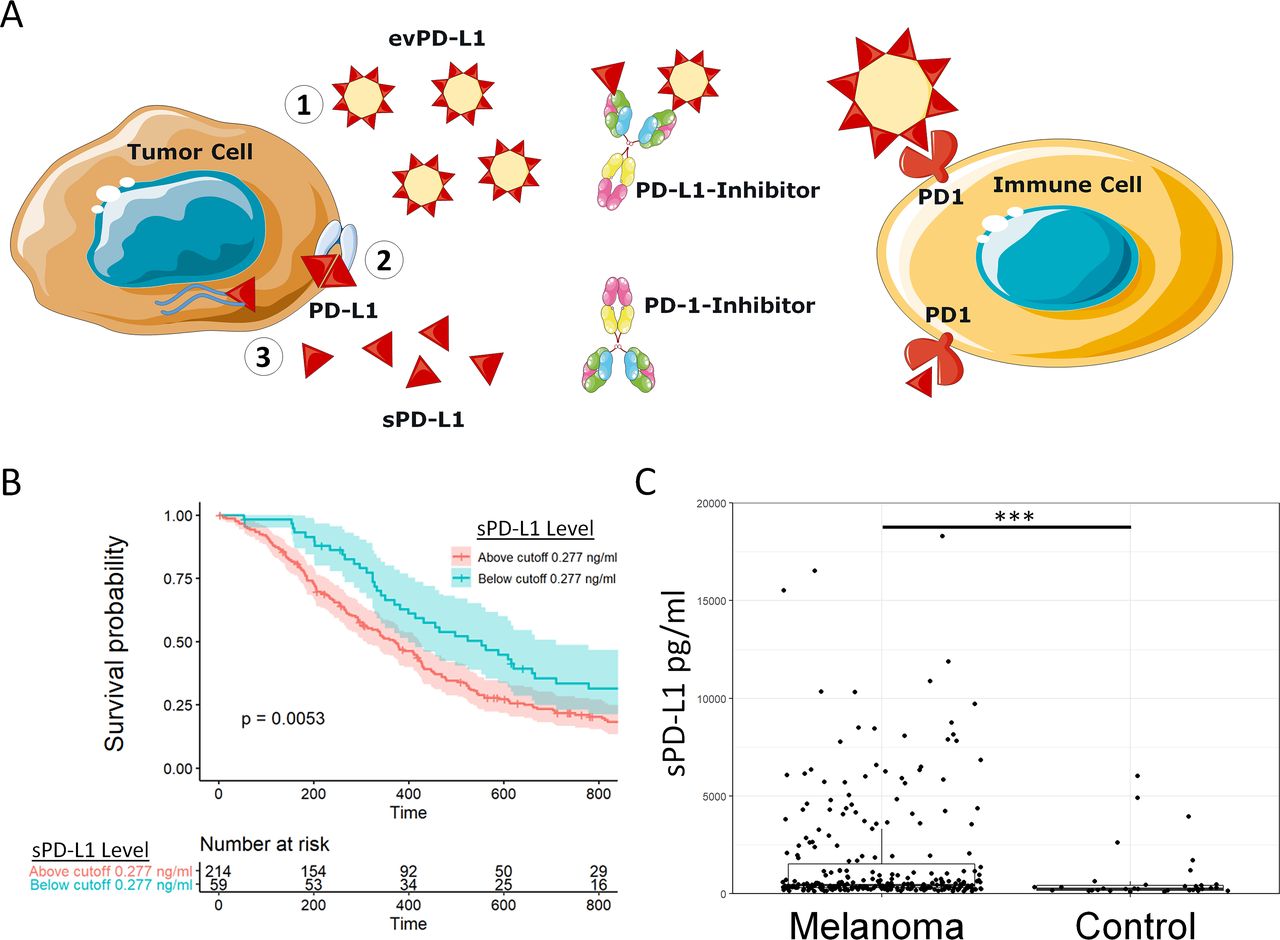
Soluble programmed death-ligand 1 (PD-L1) suppresses antitumor immunity and predicts overall survival in patients with melanoma. (A) A model of three known tumor-derived extracellular PD-L1 forms—(1) evPD-L1, (2) ADAM10/ADAM17-cleaved soluble PD-L1 (sPD-L1) ectodomain, and (3) secreted splice variant sPD-L1—that downregulate antitumor immunity and prevent response to PD-(L)1 inhibition. (B) A Kaplan-Meier plot shows significantly worse overall survival for patients with melanoma exhibiting high (≥0.277 ng/mL) versus low (<0.277 ng/mL) plasma sPD-L1 levels (p=0.005). (C) Patients with melanoma exhibited a higher mean plasma sPD-L1 level (1.72 ng/mL) in comparison to healthy controls (0.773 ng/mL). ***p<0.001.
TPE significantly reduces plasma sPD-L1 levels
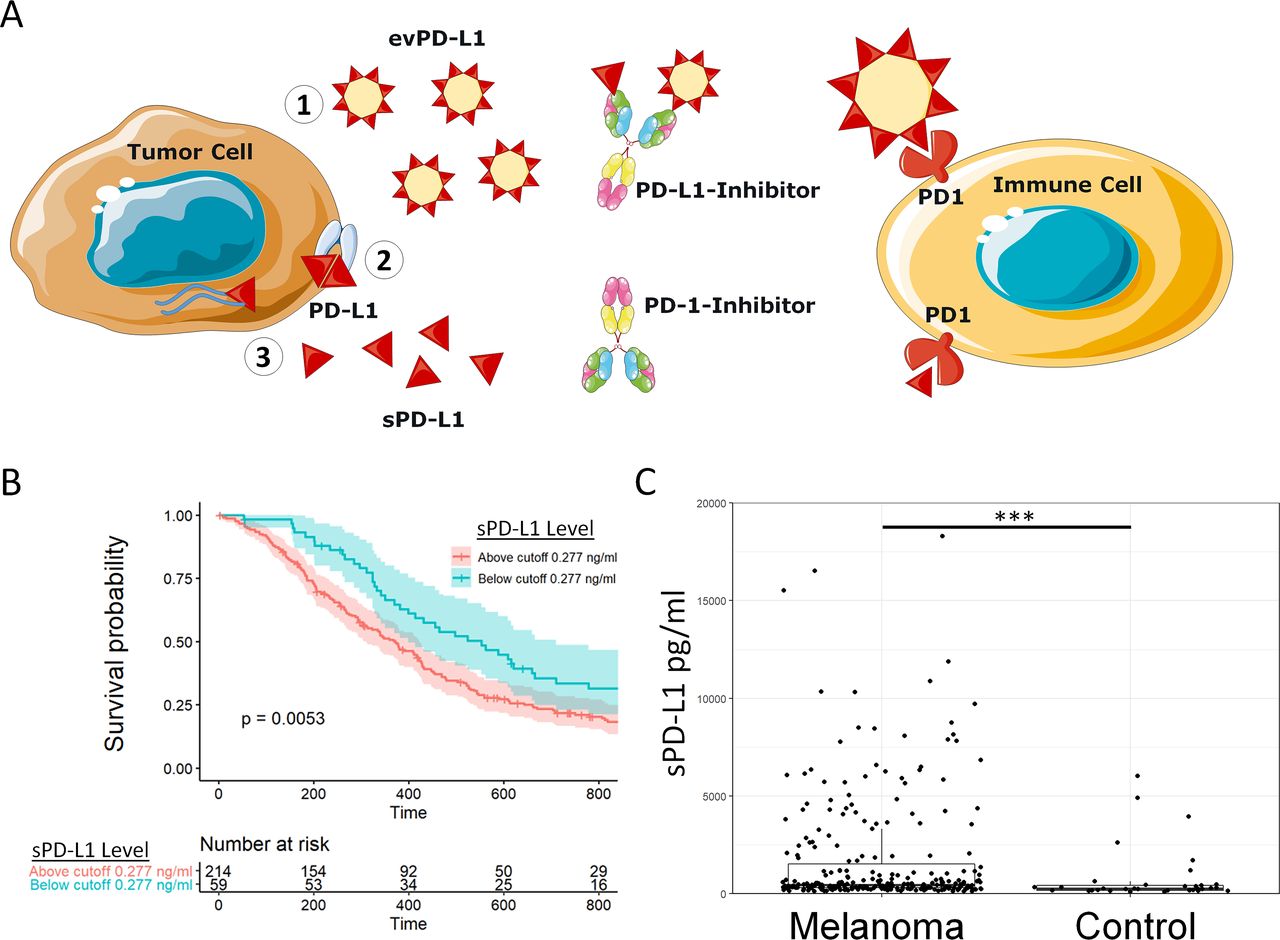
We hypothesized that TPE may remove extracellular PD-L1 in its various forms (figure 2A). To address this question, we prospectively enrolled patients undergoing planned TPE (figure 2B). Twenty-eight patients met inclusion criteria, of which 25 provided informed consent. Baseline patient characteristics are in table 1. One patient was excluded for biotin-containing supplement use as biotin interferes with the established sPD-L1 detection assay. The remaining 24 patients underwent plasma exchange and sample collection before and after the procedure as described. Discarded plasma samples from the TPE device waste bag for each session were also collected. sPD-L1 was measured in each sample and most patients undergoing TPE exhibited sPD-L1 levels above the clinically relevant 0.277 ng/mL cut-off from the retrospective melanoma study.
Patient baseline characteristics

Therapeutic plasma exchange (TPE) significantly reduces plasma soluble programmed death-ligand 1 (sPD-L1) levels. (A) A model of the TPE procedure in which patient plasma is separated and replaced to extract non-cellular substances confined to the plasma. (B) A diagram of the present study in which 24 patients undergo plasma exchange. (C) All plasma levels of sPD-L1 immediately prior to (pre) and after (post) TPE using albumin replacement fluid are plotted. TPE significantly reduced sPD-L1 levels in patient plasma by Wilcoxon signed-rank test (p<0.0001). (D) In a typical timeline, patient sPD-L1 levels are reduced by each successive session of TPE (gray bars). See also table 2, online supplementary figures 2–3.
Most patients undergoing TPE did not have an active cancer diagnosis. Baseline sPD-L1 levels in all patients were compared with matched normal controls and patients with melanoma (online supplementary fig 1), and some patients exhibited sPD-L1 above the clinically significant cut-off level determined in the retrospective melanoma cohort. Patients with high baseline sPD-L1 levels were significantly more anemic than patients with lower baseline sPD-L1 even when controlling for the higher number of female subjects in the high sPD-L1 group (female-only mean Hgb 11.4 vs 14.1, p=0.04; male-only mean Hgb 11.8 vs 14.3, p=0.03). Groups were otherwise similar. TPE significantly reduced plasma sPD-L1 levels in patients receiving albumin-only (ie, no FFP) replacement fluid (figure 2C, p<0.0001). Removed sPD-L1 was detected in matching plasma samples from the TPE procedure waste bag. Each TPE session removed a mean 70.8% of detectable plasma sPD-L1; mean regeneration of sPD-L1 between sessions was 33.8% (table 2). TPE sessions were usually separated by 1–3 days.
Soluble programmed death-ligand 1 (sPD-L1) reduction and regeneration per exchange
A representative individual patient treatment course showing sPD-L1 reduction over four successive TPE sessions is also shown (figure 2D). All individual patient TPE courses, including sessions involving donated human blood products (eg, fresh frozen plasma or FFP), are shown in online supplementary fig 2. Pre-TPE and post-TPE sPD-L1 levels for all sessions are also shown (online supplementary fig 3). TPE significantly reduced plasma sPD-L1 even when sessions requiring donated FFP were included (p<0.0001).
FFP is sometimes given during TPE for patients with increased risk of bleeding. We observed that some patients receiving FFP with low baseline sPD-L1 experienced rapid increases in sPD-L1 levels after TPE, presumably passively acquired from donor plasma as this was not observed in patients receiving albumin replacement alone. sPD-L1 was not detected in the discarded plasma from the procedure for these patients. We observed a mild association between post-FFP infusion rises in sPD-L1 levels and the blood type of the recipient, mainly in patients with O type blood. Individuals with group O− blood are universal recipients of FFP products and universal donors of cellular products due to a lack of ABO group antigens and the presence of preformed anti-A and anti-B antibodies, respectively. Recipients of FFP usually receive a mixture of compatible plasma from multiple donors. To determine whether blood type in FFP donors is associated with FFP sPD-L1 content, we measured sPD-L1 by ELISA in plasma from multiple FFP donors (online supplementary fig 4). O-negative plasma donors showed higher sPD-L1 levels than donors with most other blood types.
TPE efficiently reduces plasma EV levels in vivo
We postulated that TPE may remove PD-L1-positive EVs (evPD-L1) from patient blood. To address this question, we measured total EV levels and evPD-L1 in each sample by flow cytometry. We also determined the impact of TPE on platelet-derived CD61-positive EVs, one of the most abundant EV subpopulations in blood (CD61 is a platelet marker), and ADAM10-positive low density EVs (ADAM10 has been implicated in exosome loading and pathogenesis).11–13
TPE significantly reduced total plasma particle concentration (figure 3A, average 33.5% per exchange, p<0.0001). TPE sessions requiring FFP or other human blood product were excluded from analysis, leaving 44 session pairs. PD-L1-positive (evPD-L1) and ADAM10-positive EVs were significantly reduced by TPE (figure 3B,C, p=0.028 and p<0.0001, respectively) and were detected in waste plasma (data not shown). Each TPE session using albumin-based replacement fluid with pre-TPE levels above one million removed a mean 73.1% of detectable PD-L1-positive EVs from patients (online supplementary table 4). Platelet-derived CD61-positive EVs, while abundant, were not significantly reduced by plasma exchange (figure 3D).

{kind=link}
{kind=link}
{kind=link}
Plasma exchange efficiently reduces total, programmed death-ligand 1 (PD-L1)-positive, and ADAM10-positive extracellular vesicle (EV) levels in vivo. Plasma levels of total EVs immediately prior to (pre) and after (post) therapeutic plasma exchange (TPE) are plotted. TPE significantly reduced (A) total (p<0.0001), (B) PD-L1-positive (p=0.028), and (C) ADAM10-positive (p<0.0001) but not (D) CD61-positive EVs (p=0.94) by Wilcoxon signed-rank test. See also online supplementary figures 5–7 and online supplementary table 4.
Individual patient courses showing total plasma, PD-L1-positive, ADAM10-positive, and CD61-positive EV levels before and after each TPE session are shown in online supplementary fig 5 with exemplary nanoflow plots in online supplementary fig 6. Three successive TPE sessions consistently depleted total, PD-L1-positive, and ADAM10-positive (but not CD61-positive) EVs. These trends were less pronounced when sessions in which patients received donor FFP were included (online supplementary fig 7). In normal control FFP donors, blood type did not correlate with plasma EV concentrations (online supplementary fig 8).
Discussion
Extracellular PD-L1—in the form of splice variant sPD-L1, ADAM10/ADAM17-cleaved sPD-L1 ectodomain, or evPD-L1-positive EVs (evPD-L1)—mediates resistance to PD-(L)1 inhibitors.4–9 These forms are resistant to clinically tested combinations of chemotherapy and immunotherapies in vitro and in animal models, and are associated with poor prognosis in many cancer types. In the present study, we found that TPE reliably reduces sPD-L1 and evPD-L1. This reduction was most pronounced over approximately three consecutive single plasma volume treatment sessions. Given the dramatic reduction in sPD-L1 and evPD-L1, TPE may provide a novel approach to combating these mechanisms of resistance.
While promising, the present study was limited to patients receiving TPE mainly for non-oncological indications over a short time horizon. One patient in the study had melanoma receiving pembrolizumab and exhibited high pre-TPE evPD-L1 that was reduced on treatment. Another patient had a uterine neuroendocrine tumor receiving atezolizumab and exhibited high pre-TPE sPD-L1 that was reduced on treatment. The purpose of TPE in all cases, however, was to blunt paraneoplastic autoimmunity or treat some other coexisting autoimmune disorder—not the underlying malignancy. Neither of these patients experienced improvement in their autoimmunity after TPE. The source of sPD-L1 and evPD-L1 in these cases is uncertain as no assay currently differentiates tumor-derived and non-tumor-derived PD-L1. We observed that these forms of immunosuppressive extracellular PD-L1 exist naturally, although at lower levels in healthy subjects than in patients with cancer, suggesting a potentially beneficial immunoregulatory role. While we observed some regeneration for both sPD-L1 and evPD-L1 between TPE sessions, it is unknown to what degree malignant cells may regenerate and maintain extracellular PD-L1 homeostasis. Relatedly, it is uncertain how other plasma substances removed by TPE may affect response to immunotherapy or, more broadly, cancer immunity overall. Nor is it known at what level sPD-L1 and/or PD-L1-positive EV removal would become clinically relevant. These facets will be tested in future studies.
Immunotherapy resistance is widespread and costly. In most instances, PD-(L)1 inhibitors such as pembrolizumab, nivolumab, atezolizumab, durvalumab, and avelumab are used in situations in which less than half of tumors are expected to respond. Of patients that benefit, many do not experience a sustained durable response. These treatments represent a major investment; the cost of PD-(L)1 checkpoint blockade commonly reaches several hundred thousand dollars over the course of therapy.
To our knowledge, this is the first report of an intervention to achieve consistent, rapid reduction in either sPD-L1 or PD-L1-positive EVs in a clinical setting. TPE is safe and commonly prescribed. Thus, preimmunotherapy TPE may combat immunotherapy resistance. In light of the heavy investment that anti-PD-(L)1 therapy entails, the added cost of TPE in selected patients may be practical.14 While the durability of extracellular PD-L1 reduction in malignancy will be explored in future studies, the present study suggests that this approach warrants further investigation.
Beyond evPD-L1, this is also to our knowledge the first known intervention to reliably deplete EVs in a clinical setting. EVs have been implicated in oncogenesis and metastasis through miRNA carriage and direct protein signaling independent of PD-L1.15 16 Beyond cancer, EVs have also been implicated in autoimmunity,17 aging/neurodegeneration,18 infection,19 obesity,20 and heart disease.21 The selective removal of ADAM10-positive (ie, likely immune-derived) versus CD61-positive (ie, likely platelet-derived) EVs in this study suggests flexible, selective EV depletion may be both possible and expedient in other indications. The present study is only a proof of concept and additional exploratory studies in these areas are necessary.
In summary, TPE reduces extracellular forms of PD-L1 associated with PD-(L)1 checkpoint inhibitor resistance. Future studies will explore the potential role of TPE in improving cancer immunotherapy.
Methods
Retrospective melanoma outcomes study design
In a retrospective analysis, baseline blood samples from 276 patients with melanoma prior to treatment in one of three clinical trials by the North Central Cancer Treatment Group: N057e,22 N0775,23 and N087924 between 2006 and 2014 were tested for sPD-L1. 82% of patients were diagnosed with cutaneous melanoma and none received immunotherapy treatments. Blood from 86 healthy volunteers undergoing blood donation at Mayo Clinic was also tested.
Prospective TPE study design
In an investigator-initiated open-label single-center observational study, adults undergoing TPE were approached from December 2019 through March 2020. In consenting subjects, samples of whole blood immediately prior to TPE and on completion of the procedure were collected in ACD vacutainers (BD). In each case, the first 8 mL of blood was discarded to avoid contamination after which an 8 mL sample was obtained in sequence. Plasma was isolated by centrifugation. A postprocedure blood sample was obtained after completion of the procedure. In addition, matching samples from discarded plasma from the procedure waste bag were collected. Samples were obtained from up to four consecutive procedures for each patient. If a patient underwent fewer than four TPE procedures, samples were obtained from as many procedures as possible.
Patients included were adults able to give consent and undergoing TPE for a variety of hematological, neurological, and renal diseases as indicated by published guidelines from the American Society for Apheresis (ASFA) or according to the medical judgment of the referring physicians.25 Patients taking biotin supplements were excluded from the study due to biotin interference with the sPD-L1 ELISA assay. Procedures were performed using centrifugation-based cell separators, either the Fenwal Amicus (Fresenius KABI USA LLC, Lake Zurich, Illinois, USA) or the Spectra Optia (Terumo BCT, Lakewood, Colorado, USA). For each patient, a single plasma volume was exchanged using either peripheral intravenous (preferred) or central lines for vascular access. For this study, due to the possibility of sPD-L1 or PD-L1-positive EVs present in donor plasma, only TPE sessions using no donor plasma (ie, fresh frozen plasma, FFP) in the replacement fluid were included in calculations. Anticoagulation consisted of either 500 mL of acid citrate dextrose solution A (ACD-A) or 500 mL of ACD-A with 5000 units of unfractionated heparin. Anticoagulant to blood ratios were 1:13 when ACD-A was used and 1:26 when ACD-A/heparin was used. Patients did not receive routine electrolyte replacement but 10 mL of 10% calcium gluconate was administered by slow intravenous push for signs and symptoms of hypocalcemia related to the ACD-A anticoagulant in one patient.
ELISA
ELISA was performed as previously published.26 Both secreted splice variant and shed sPD-L1 are reliably detected by this ELISA. In brief, paired mouse IgG2 monoclonal antibody clones H1A and B11 against extracellular human PD-L1 were utilized in a capture-detection plate assay using biotinylation and HRP-streptavidin detection. This assay is specific for sPD-L1 and does not exhibit cross reactivity to other B7-H homologues, nor to evPD-L1. Concentrations were determined by optical density measurements along a known standard curve of recombinant human PD-L1. ELISAs were performed by team members who were blinded to the identity of the samples.
Flow cytometry
Flow cytometry for EVs was performed as previously published.27 In brief, plasma samples were centrifuged twice at 2000g to deplete platelets. Resultant platelet-free plasma were analyzed using an A60-Micro Plus Nanoscale Flow Cytometer (Apogee FlowSystems) gating for mid-intensity light angle light scatter and markers of interest. Anti-PD-L1 (Genentech: atezolizumab), ADAM10 (R&D Systems: clone 163003), and CD61 (BioLegend: clone VI-PL2) antibodies were conjugated to fluorophores (Life Technologies: Alexa-647, PE phycoerythrin, and Alexa-488) and titrated prior to use. Nanoscale flow cytometer calibration was performed using a standard reference bead mix as previously published. Flow cytometry was performed by team members blinded to the identity of the samples.
Statistical analysis
All statistical analyses were performed using R Statistical Software (R Foundation). Retrospective progression-free survival was analyzed using Kaplan-Meier and Cox proportional hazards modeling. Optimal cut-off values for sPD-L1 levels were determined using the greyzoneSurv package for R. Wilcoxon signed-rank test was used to compare paired pre-TPE and post-TPE patient sample sPD-L1 and EV levels as indicated. Baseline clinical characteristics for the study were compared by Kruskal-Wallis test for continuous variables and Pearson’s χ2 test for discrete variables as indicated. Otherwise, groups were compared by unpaired two-sided Student’s t-test. Figures comprising box plots show quartile values and individual data points. Mean values and 95% CI are indicated in corresponding online supplementary figures and tables. P <0.05 was considered statistically significant. In figures, p values are denoted <0.05 with *, <0.01 with **, and <0.001 with ***.
Acknowledgments
Statistical guidance was provided generously by Nathan Foster of the Mayo Clinic Center for Clinical and Translational Science. Some illustrations were created using Servier Medical Art templates, which are licensed under a Creative Commons Attribution 3.0 Unported License; https://smart.servier.com. Additional illustrations were provided by Mayo Clinic Media Services. The authors thank Daniel Summerfield, MD MS for use of his likeness in Fig 2A.
References
Footnotes
Twitter @JakeOrmeMDPhD
Contributors JO originated hypotheses, designed the study, oversaw experiments, performed analyses and wrote the article. EALE performed retrospective melanoma cohort analysis. FL-M performed nanoflow cytometry. HD and EB oversaw and performed TPE study enrollment, sample collection/processing, and blinding. SMH performed ELISAs. MB, AM, SP, MB, SNM, YY, HD, RD and JLW helped develop hypotheses, provided clinical samples and reagents, and contributed support and oversight.
Funding R21 5R21CA197878-02 Role of Bim and soluble B7-H1 in monitoring T cell responses to anti-PD-1 therapy in melanoma (HD and RD) L30 CA231541-01 Soluble B7H1 as a PD1 Checkpoint “Remote Control” in Cancer (JJO) U10 CA180790 (EE) K12 CA090628 (YY) Richard M. Schulze Family Foundation (HD and RD).
Competing interests Intellectual property has been filed addressing discoveries disclosed in this manuscript. The authors report no other relevant conflicts of interest.
Patient consent for publication Obtained.
Ethics approval All research protocols involving human subjects were approved by Mayo Clinic’s Institutional Review Board and all human subjects gave written informed consent.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. All data will be available for download at the Open Science Framework at https://osf.io/qtskd/.