Article Text

Abstract
Cerebral edema following chimeric antigen receptor (CAR) T-cell therapy can be fatal. ZUMA-2 is a pivotal phase 2, multicenter study evaluating KTE-X19, an autologous anti-CD19 CAR T-cell therapy, in relapsed/refractory mantle cell lymphoma. We describe a 65-year-old patient in ZUMA-2 who developed cerebral edema following CAR T-cell therapy and had complete recovery after multimodality clinical intervention including rabbit antithymocyte globulin (ATG). Biomarker results show early and robust CAR T-cell expansion and related induction of inflammatory cytokines, followed by rapid declines in CAR T-cell and proinflammatory cytokine levels after ATG administration. This clinical profile highlights a potential relevance of ATG in treating severe CAR T-cell-related neurotoxicity.
- case reports
- chimeric antigen
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Chimeric antigen receptor (CAR) T-cell therapies targeting CD19 have demonstrated impressive efficacy in relapsed/refractory B-cell malignancies.1–4 However, neurologic events (NE) are serious toxicities commonly observed with CAR T-cell therapy and remain a therapeutic challenge.5 6 Although low-grade NE are generally reversible, grade 4 NE can be fatal and are typically associated with cytokine release syndrome (CRS), increased blood-brain-barrier permeability, endothelial activation, and poor survival.7–9 CAR T-cell expansion and associated rises in cytokine/chemokine levels are associated with disease remission and treatment-related toxicity.4 10 However, early and rapid expansion of CAR T cells and associated cytokine production have also been correlated with severe NE, including cerebral edema.11 12
ZUMA-2 is a phase 2, multicenter study evaluating KTE-X19, an autologous anti-CD19 CAR T-cell therapy, in patients with relapsed/refractory mantle cell lymphoma (MCL).4 KTE-X19 uses a manufacturing process that removes circulating CD19-expressing cells for use in leukemic indications and MCL.4 13 We present the clinical management of a patient in ZUMA-2 who developed grade 4 NE with cerebral edema that resolved after treatment with multimodality clinical intervention including rabbit antithymocyte globulin (ATG), an immunosuppressant that depletes T lymphocytes in vivo.14
Methods
As previously reported, patients in ZUMA-2 had relapsed/refractory MCL with one to five prior therapies.4 Prior therapy must have included anthracycline or bendamustine-containing chemotherapy, an anti-CD20 monoclonal antibody, and a Bruton tyrosine kinase inhibitor (ibrutinib or acalabrutinib). Induction plus consolidation/maintenance and/or all treatments occurring between sequential complete responses were counted as one regimen. All patients provided written, informed consent, and the protocol was approved by the Institutional Review Board at each site. The ZUMA-2 study was conducted in accordance with the principles of the Declaration of Helsinki. The trial was registered at ClinicalTrials.gov Registry; NCT02601313.
Patients underwent leukapheresis with optional bridging therapy, followed by conditioning chemotherapy (cyclophosphamide 500 mg/m2/day; fludarabine 30 mg/m2/day) on days −5 to −3, as previously reported.4 KTE-X19 was infused at a target dose of 2×106 CAR T cells/kg on day 0.4 CRS was graded per Lee et al.15 National Cancer Institute Common Terminology Criteria for Adverse Events, V.4.03, was used to grade the severity of adverse events, including NE and symptoms of CRS.4 A validated quantitative PCR (qPCR) assay was used to measure the expansion and persistence of anti-CD19 CAR T cells in blood at baseline (prior to conditioning chemotherapy (negative control)) and prior to KTE-X19 administration; and at days 7, 14, 28 and in the long-term follow-up as described in the study protocol.10 11 16–18 The limit of detection of the assay was 1 CAR T cell in 100 000 peripheral mononuclear cells measured in blood. Serum collected at baseline and days 0 (postconditioning and prior to KTE-X19), and at days 3, 7, 14, and 28 was assessed for levels of 44 cytokines, chemokines, circulating angiogenic factors, immune effector molecules, and markers of macrophage activating syndrome using previously reported methods.10 16 17 A qualified flow cytometry assay was performed on immune cells isolated from cerebrospinal fluid (CSF) and other standard immune markers for the identification of T and myeloid lineage cells. All measurements described above were derived from central laboratory assessments. Although the patient was enrolled in ZUMA-2, for comparison in this report with the broader ZUMA-2 population, this patient was excluded from the ZUMA-2 cohort medians and assessments reported herein.
Results
Case report
A 65-year-old man with stage IV relapsed/refractory pleomorphic MCL (ibrutinib-refractory; online supplemental methods) was enrolled. There was no history of prior neurologic disease. Prior to KTE-X19 infusion, the patient had an Eastern Cooperative Oncology Group performance status score of 0, non-bulky disease, a Ki-67 proliferation index of 80%–90%, and baseline lactate dehydrogenase levels of 410 IU/L. An initial MRI of the brain was unremarkable (online supplemental figure S1). Following leukapheresis, the patient received conditioning chemotherapy followed by KTE-X19 infusion. Prior to infusion, prophylactic levetiracetam was initiated and there was no evidence of fever or infection. Serum cytokine levels assessed during and after lymphodepletion were similar to those in other patients in ZUMA-2. KTE-X19 product characteristics were likewise similar to that of other patients. Figure 1 depicts a timeline of clinical events, including imaging assessments and interventions. Online supplemental table S1 provides the level of evidence behind the interventions used. Cytokine levels in serum and CSF and CAR T cell levels in blood and CSF are shown in table 1.
Supplemental material

Timeline of clinical events and interventions in the patient treated with ATG. ALT, alanine aminotransferase; AST, aspartate aminotransferase; ATG, antithymocyte globulin; BID, two times per day; CAR, chimeric antigen receptor; CRS, cytokine release syndrome; FLAIR, fluid-attenuated inversion recovery; N/A, not available.
CAR T-cell levels and serum/CSF analytes by day
Twenty-four hours post-KTE-X19 infusion, the patient developed grade 1 CRS that was treated with tocilizumab 8 mg/kg intravenously. Vancomycin and aztreonam (each 1 g intravenously two times per day) were administered for non-neutropenic fever of 39.1°C. During this interval, the patient had delayed speech and thought processes, with memory deficit, handwriting changes and slight tremor, consistent with grade 2 encephalopathy.
By day 3, the patient presented with marginal leukopenia and a white cell count of 3.7 x 109/L (normal absolute neutrophil count) and CSF cultures were unremarkable, but chest X-ray showed multifocal opacities. e received siltuximab 11 mg/kg intravenously to treat grade 2 CRS, although this was not protocol-specified for CRS management. On day 4, methylprednisolone 500 mg intravenously two times per day was initiated. Some clinical improvements occurred, but the patient’s condition deteriorated, and a second dose of tocilizumab was administered. He was transferred to intensive care unit for worsening aphasia, confusion, and obtundation and was intubated for airway protection. The patient had two tonic-clonic seizures. MRI showed diffuse gyral swelling and an electroencephalogram (EEG) showed triphasic waves and diffuse disturbances. MRI findings confirmed cerebral edema, and the patient was diagnosed with grade 4 encephalopathy. Mannitol 20% (0.25 mg/kg) every 6 hours was initiated along with steroids and cerebral edema management guidelines were followed.19
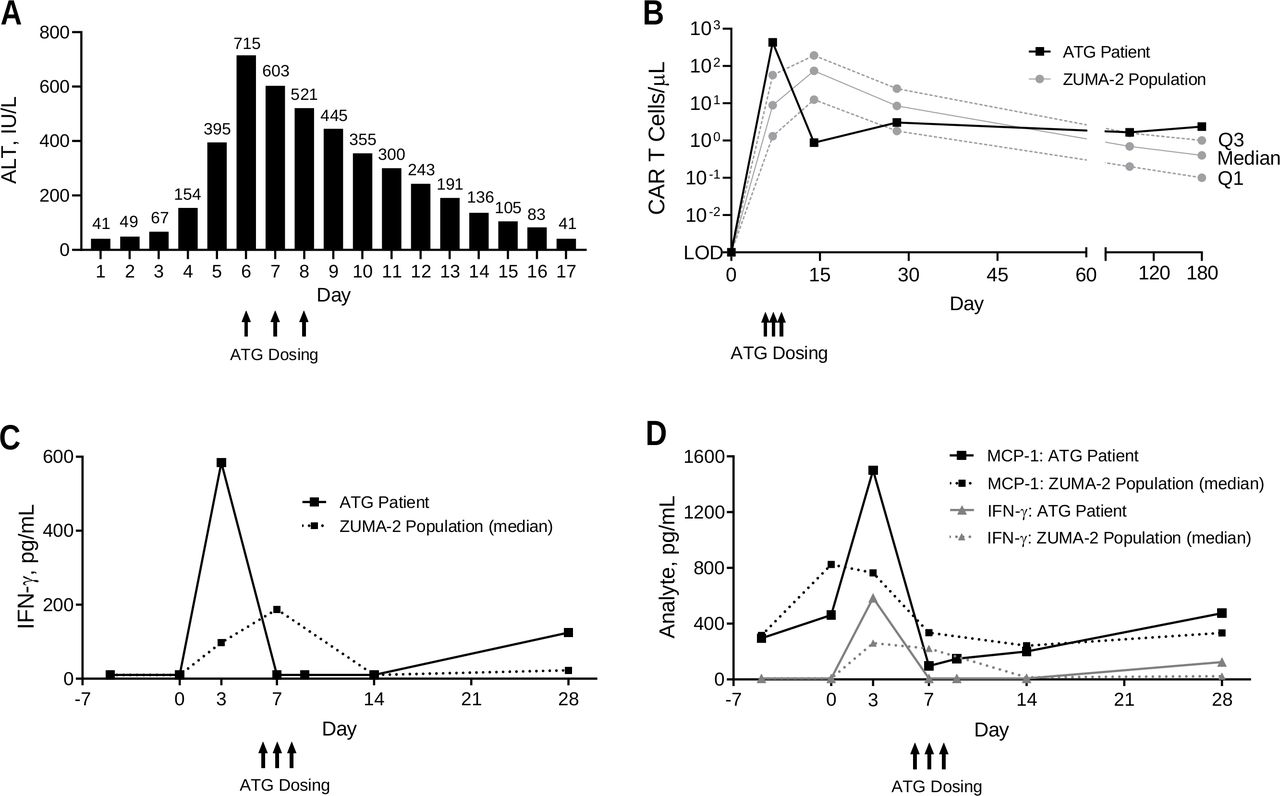
Despite these measures, there was no improvement by day 5, and the patient developed grade 3 transaminase elevation (figure 2A). CSF was clear with an increased opening pressure to 20 cm H2O, elevated CSF protein (6 µg/mm3) and glucose (1.06 µg/mm3) and cytology showing increased white blood cells (24 cells/mm3) and atypical and activated lymphocytes (mainly T cells) admixed with histiocytes and neutrophils (78%). Intrathecal hydrocortisone and ara-C were administered due to worsening neurological symptoms. On day 6, EEG showed bihemispheric cortical dysfunction, and MRI showed cerebral edema and sulcal hyperintensity (online supplemental figure S2). Neurosurgery placed an external ventricular drain to treat the cerebral edema. Prophylactic acyclovir was initiated to mitigate risk of infection. Following no improvement, ATG (2 mg/kg/day intravenous) was administered along with increased methylprednisolone (1 g two times per day) and a third dose of tocilizumab to dampen systemic cytokine storm. Rabbit ATG (Thymoglobulin) and not equine ATG was used due to availability, experience at our center, and the lower dose of ATG required to avoid exacerbation of concomitant CRS. On day 7, an MRI showed stable changes, and a second dose of ATG was given. Reduction in transaminase elevation was noted (figure 2A). A third dose of ATG was given on day 8 after which the transaminase elevation was resolved. On day 11, the patient was extubated, and no further ATG doses were given. Over the next 11 days, he received tapering doses of methylprednisolone with significant clinical improvement. The ventricular drain was removed on day 14 and the encephalopathy was resolved. An MRI on day 20 showed persistent but significantly improved periventricular white matter T2 hyperintensity and resolution of abnormal signal in the dorsal brainstem/thalami (online supplemental figure S3). The patient was discharged to a rehabilitation facility with tapering doses of oral corticosteroids. Two months post-KTE-X19, brain MRI findings were completely resolved (online supplemental figure S4). The patient remains on study, and after 24 months follow-up, he is in complete remission with no persistent neurologic or cognitive deficits.
{kind=link}
{kind=link}
Levels of transaminases, CAR T cells, and cytokines over time. (A) ALT levels in the patient with ATG across the first 17 days post-KTE-X19. (B) Levels of CAR T cells in blood by PCR over the first 6 months following KTE-X19 infusion. In the broader ZUMA-2 population, 66, 65, 59, 65, and 43 patients had data available at days 0 (pre-KTE-X19), 7, 14, 28, and 180, respectively. Levels of IFN-γ (C) and MCP-1 (D) by ELISA over the first 28 days post-KTE-X19. In the broader ZUMA-2 population, 65, 65, 62, 61, 60, and 64 patients had data available at days −4 (pre-conditioning), 0 (pre-KTE-X19), 7, 14, 28, and 180, respectively. ALT, alanine aminotransferase; ATG, anti-thymocyte globulin; CAR, chimeric antigen receptor; ELISA, enzyme-linked immunosorbent assay; IFN-γ, interferon gamma; IL-2, interleukin 2; LOD, limit of detection; MCP-1, monocyte chemoattractant protein 1.
Pharmacokinetics and pharmacodynamics
Peripheral blood CAR T-cell levels measured by qPCR were 431.3 cells/mm3 on day 7 and rapidly declined to <1 cell/mm3 after day 14 (post-ATG; figure 2B). By contrast, median CAR T-cell levels in other patients in the ZUMA-2 cohort with pharmacokinetic data available (N=66) were 8.9 cells/mm3 (IQR 1.3–57) at day 7 and 74.5 cells/mm3 (IQR 12.5–192.7) at day 14. At 24 months, B-cell aplasia was maintained, and low levels of CAR T cells were detectable by qPCR.
Analysis of key serum biomarkers hypothesized to associate with increased risk for CAR T cell-related NE and CRS at baseline (preconditioning) and day 0 (pre-KTE-X19) was unremarkable (table 1).9–11 20–22 Serum C reactive protein, a biomarker of systemic inflammation, was 6.8 ng/mm3 at day 0, compared with a median of 30.5 ng/mm3 (IQR 15.1–63) in the broader cohort. Ferritin, an acute phase reactant, was 606.3 pg/mm3 at day 0 compared with a median of 502.4 pg/mm3 (IQR 273.5–877.7) in the broader cohort.
IL-15, a homeostatic gamma chain cytokine implicated in CAR T-cell expansion and associated with cerebral edema, showed evidence of contributing to the robust expansion observed in this patient.9 22–24 At day 0, IL-15 was 0.0291 pg/mm3 (cohort median, 0.0332 (IQR 0.0254–0.048)) and continued to increase to 0.0561 pg/mm3 (cohort median, 0.0353 (IQR 0.0214–0.0564)) by day 3. Post-ATG levels of IL-15 subsided to 0.0036 pg/mm3 at day 7 (cohort median, 0.0226 (IQR 0.015–0.0386)). Data indicate that rapidly rising IL-15 levels may have contributed to the robust CAR T-cell expansion observed and possibly to the cerebral edema.
IL-2, a homeostatic cytokine produced by activated CAR T cells, was measured in serum at 0.0167 pg/mm3 on day 3 and also likely contributed to the robust CAR T-cell expansion observed. The median at this same time point in the broader cohort was 0.0047 pg/mm3 (IQR 0.0022–0.0100), demonstrating a 3.6-fold increase in this patient relative to others. Post-ATG (day 7), IL-2 was significantly suppressed (below limit of detection in the assay used), indicating a rapid and profound impact of ATG on anti-CD19 CAR T-cell T helper type 1 (Th1) activity. Additional serum biomarker analysis post-KTE-X19 demonstrated excessive CAR T cell and myeloid-related activity that were controlled through the toxicity management strategy reported here. Relative to the broader population, a marked decline between day 3 (post-KTE-X19) and day 7 (post-ATG) in serum interferon (IFN)-γ was observed (figure 2C). In concert with early and elevated CAR T-cell levels, serum IFN-γ, a Th1 cytokine and hallmark of CAR T-cell activity, was 0.5844 pg/mm3 at day 3 and rapidly declined to 0.0075 pg/mm3 (lower limit of detection) at day 7 following ATG administration. By contrast, median IFN-γ in the broader cohort was 0.0978 pg/mm3 (IQR 0.0244–0.2624) at day 3 and 0.1877 pg/mm3 (IQR 0.020–1.2436) at day 7.
Monocyte chemoattractant protein 1 (MCP-1; CCL2), a marker of myeloid-related activity associated with CD19-directed CAR T-cell toxicity, was 1.500 pg/mm3 (above quantitation limit) at day 3 (cohort median, 0.7123 pg/mm3 (IQR 0.5176–1.1424)), indicating the possibility of excessive myeloid activity postinfusion as a contributing factor (figure 2D).7 22 23 At day 7, serum MCP-1 was reduced to 0.0969 pg/mm3 (cohort median, 0.4638 (IQR 0.2714–0.9541)), further demonstrating the impact of ATG in suppressing myeloid-related inflammation.
An analysis of cytokines in CSF taken on day 8 demonstrated elevated levels of IFN-γ-induced protein 10 (CXCL10) and MCP-1 compared with those measured in serum on day 7 as well as measurable levels of intercellular adhesion molecule 1, IL-1 receptor antagonist and IL-2 receptor alpha (table 1, online supplemental file 1). Elevated levels of these cytokines in the CSF are consistent with CAR T-cell activity and myeloid trafficking to the central nervous system (CNS). These results, possibly contributing to the NE, warrant further mechanistic evaluation.
Discussion
Optimal management and pathophysiology of NE associated with CAR T-cell therapy are unclear. We present the first report of a patient with relapsed/refractory MCL treated with anti-CD19 CAR T-cell therapy who developed CAR T-cell therapy-associated grade 4 cerebral edema with concomitant CRS that resolved completely after multimodality clinical intervention including ATG administration. The patient experienced a full recovery with no neurological deficits and achieved a durable response. Considering the serious and dire condition of the patient, multimodality treatments including ventriculostomy were quickly initiated, making it difficult to conclude that ATG administration was solely responsible for the amelioration of patient symptoms. However, the information available and the timeline of events suggest that ATG contributed to the patient’s improvement in neurologic symptoms while simultaneously dampening CAR T-cell levels in blood, cytokine storm, and elevated liver enzymes. Furthermore, pharmacokinetic and pharmacodynamic results indicated that ATG might have contributed to the resolution of cerebral edema, along with other clinical interventions, including corticosteroids, IL-6 or IL-6 receptor blockade with siltuximab or tocilizumab, and a ventriculostomy. Therefore, we believe it is reasonable to suggest that ATG administration, in addition to other clinical interventions, benefited this patient. The clinical profile of this patient, in part, supported revisions in the adverse event management guidelines for NE and CRS in patients treated with CAR T-cell therapy, including the creation of KTE-X19-specific cerebral edema management guidelines (online supplemental table S3), the initiation of corticosteroids for grade 2 NE rather than grade 3, and the initiation of tocilizumab for NE only if occurring in the context of CRS or immune effector cell-associated neurotoxicity syndrome.
Commensurate with clinical observations, peak CAR T-cell expansion at day 7 in this patient was 48 times higher than the median expansion of other patients in ZUMA-2. This rapid and robust expansion alongside exacerbated CAR T-cell activity likely contributed to the severe NE observed. CAR T-cell levels rapidly declined after the administration of ATG to baseline levels, but persisted at low levels over time, with the patient achieving and remaining in complete response 24 months after treatment. These results also support the hypothesis that high levels of CAR T cells and functional activity prior to day 14 may be sufficient for inducing a rapid and durable response.
The rise in several cytokines by day 3 was more pronounced in this patient compared with the broader ZUMA-2 population and rapidly declined by day 7 post-ATG administration. CAR T-cell IL-2 production was approximately fourfold higher relative to the cohort median and promptly downregulated after ATG administration, following dramatic decreases in CAR T cell levels. Serum levels of IFN-γ were sixfold higher relative to the cohort median, indicative of excessive Th1 CAR-related activity and, similar to IL-2 levels, rapidly returned to baseline post-ATG administration. In addition, CXCL10 and MCP-1 were elevated in the CSF on day 8 compared with levels measured in serum on day 7, pointing to an active myeloid inflammatory process in the CNS. Since there was no suspicion of CNS lymphoma, a baseline CSF sample was not collected. Lumbar puncture for CSF on day 3 was unsuccessful, precluding a comparison with CSF cytokines pre-ATG. Although a mechanistic relationship of ATG in resolving cerebral edema post-CAR T-cell therapy could not be established, a broader cytokine dampening induced by ATG and other clinical intervention modalities may have led to an improvement in clinical and laboratory parameters. This case highlights a potential utility of ATG alongside other clinical interventions in managing cerebral edema after anti-CD19 CAR T-cell therapy through rapid control of CAR T-cell levels and immune function. Additional studies are needed to assess the role of ATG in ameliorating NE associated with CAR T-cell therapy before this approach can be used in routine clinical practice.
Acknowledgments
We thank the staff at M.D. Anderson Cancer Center, including Maria Badillo, Javier A. Adachi, Imrana Malik, Luis E. Fayad, and Sudhakar Tummala; and Will Go of Kite, a Gilead Company, for patient management. Medical writing support was provided by Stephanie Morgan, PhD, (Nexus Global Group Science LLC) funded by Kite, A Gilead Company.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors MW designed the ZUMA-2 study in collaboration with Kite, A Gilead Company; MW, PJ, TLC, SEC, AH, and S-PW treated the patient and gathered data. All authors had full access to the data, participated in the analysis and interpretation of the data and manuscript writing and approved the final manuscript.
Funding This study was funded by Kite, A Gilead Company.
Competing interests MW: honoraria from Pharmacyclics, Janssen, AstraZeneca, OMI, and Targeted Oncology; consultancy or advisory role for Pharmacyclics, Celgene, Janssen, AstraZeneca, MoreHealth, Pulse Biosciences, Nobel Insights, and Guidpoint Global; research funding from Pharmacyclics, Janssen, Novartis, Juno, Celgene, Loxo Oncology, VelosBio, and Verastem; expert testimony for AstraZeneca; and travel support from Janssen, Pharmacyclics, Celgene, and OMI. TLC: honoraria from Kite, a Gilead Company; consultancy or advisory role for Kite, a Gilead Company. AH: employment at M.D. Anderson; stock or other ownership in Caris; consultancy or advisory role for Caris and WCG Oncology; research funding from Merck; patents, royalties, or other intellectual property from Celldex. LZ and AVR: employment with Gilead Sciences. JMR: employment with Kite, A Gilead Company; stock or other ownership in Gilead Sciences.
Patient consent for publication Not required.
Ethics approval All patients provided written, informed consent, and the protocol was approved by the Institutional Review Board at each site. The study was conducted in accordance with the principles of the Declaration of Helsinki. The trial was registered at ClinicalTrials.gov (NCT02601313).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Gilead is committed to sharing clinical trial data with external medical experts and scientific researchers in the interest of advancing public health. As such, Gilead shares anonymized individual patient data (IPD) upon request or as required by law and/or regulation. Qualified external researchers may request IPD for studies of Gilead compounds approved in the USA and the European Union with a marketing authorization date on or after 1 January 2014 and are publicly listed on clinicaltrials.gov or the European Union-Clinical Trials Register (EU CTR). For studies of newly approved compounds or indication, the IPD will be available for request 6 months after US Food and Drug Administration (FDA) and European Medicines Agency (EMA) approval. Such requests are at Gilead’s discretion and are dependent on the nature of the request, the merit of the research proposed, availability of the data, and the intended use of the data. If Gilead agrees to the release of clinical data for research purposes, the requestor will be required to sign a data sharing agreement, to ensure protection of patient confidentiality before the release of any data.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.