Article Text

Abstract
Background There are limited treatment options for patients with metastatic or recurrent cervical cancer. Platinum-based chemotherapy plus the anti-vascular endothelial growth factor antibody bevacizumab remains the mainstay of advanced treatment. Pembrolizumab is Food and Drug Agency approved for programmed death ligand 1 (PD-L1) positive cervical cancer with a modest response rate. This is the first study to report the efficacy and safety of the PD-L1 antibody atezolizumab in combination with bevacizumab in advanced cervical cancer.
Methods We report the results from a phase II, open-label, multicenter study (NCT02921269). Patients with advanced cervical cancer were treated with bevacizumab 15 mg/kg intravenous every 3 weeks and atezolizumab 1200 mg intravenous every 3 weeks. The primary objective was to measure the objective response rate (ORR). Secondary endpoints included disease control rate (DCR), progression-free survival (PFS), overall survival (OS), and safety.
Results In the total evaluable population (n=10), zero patients achieved an objective response as assessed by Response Evaluation Criteria In Solid Tumors (RECIST) V.1.1, resulting in a confirmed ORR of 0%. Of note, there were two patients who achieved an unconfirmed PR. The DCR by RECIST V.1.1 was 60% (0% complete response, 0% partial response, 60% stable disease). Median PFS was 2.9 months (95% CI, 1.8 to 6) and median OS was 8.9 months (95% CI, 3.4 to 21.9). Safety results were generally consistent with the known safety profile of both drugs, notably with two high-grade neurologic events.
Conclusions The combination of bevacizumab and atezolizumab did not meet the predefined efficacy endpoint, as addition of bevacizumab to PD-L1 blockade did not appear to enhance the ORR in cervical cancer.
- tumor biomarkers
- programmed cell death 1 receptor
- genital neoplasms
- female
- drug therapy
- combination
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Cervical cancer is the fourth most common cause of cancer-related deaths in women worldwide,1 with an estimated 13 170 cases diagnosed in the USA in 2020.2 Early-stage cervical cancer can be cured in the majority of cases with surgery, while concurrent chemoradiation is the treatment of choice for locally advanced cancer. Unfortunately, there are limited treatment options for patients with metastatic or recurrent cervical cancer. Platinum-based chemotherapy remains the mainstay of treatment for advanced disease3; the addition of the anti-vascular endothelial growth factor (VEGF) antibody bevacizumab to taxane-based chemotherapy was approved by the Food and Drug Agency (FDA) based on data from the randomized phase III GOG 240 study, which found that the addition of bevacizumab increased the objective response rate (ORR) and improved the median overall survival (OS) by 3.5 months.4
More recently, the anti-programmed death protein 1 (anti-PD-1) antibody pembrolizumab has been FDA approved for the treatment of PD-L1-positive, advanced cervical cancer based on the KEYNOTE 158 study.5 PD-1 is a T cell coinhibitory receptor that is expressed by effector T cells on activation,6 where binding of PD-1 to its ligand PD-L1 on tumor cells and tumor-infiltrating immune cells7 8 results in inhibition of T cell proliferation,9 cytokine production,10 11 and decreased T cell survival.12 13 Atezolizumab is a humanized engineered immunoglobulin G1 monoclonal antibody that selectively targets PD-L1 to block its interaction with programmed cell death protein 1 (PD-1) and the costimulatory molecule B7-1 to reinvigorate tumor-specific T cell immunity.14 Atezolizumab monotherapy has not previously been tested in advanced cervical cancer.
Given the modest ORR seen with anti-PD-1 monotherapy,5 15 and minimal activity of other single-agent chemotherapies in the advanced setting,16 there is ongoing interest in testing novel drug combinations. Specifically, there is a strong rationale for combination of PD-L1 blockade with vascular-targeting strategies such as bevacizumab. In addition to its role in angiogenesis, VEGF exerts a number of immunosuppressive effects, including promotion of myeloid-derived suppressor cells.17 18 Data from a diverse set of preclinical solid tumor models and phase I studies in renal cell carcinoma (RCC) and advanced melanoma suggest that VEGF inhibition might enhance the antitumor activity of immune checkpoint blockade by improving T cell infiltration, upregulating major histocompatibility complex class I expression, and reversing myeloid immunosuppression.18–23 Supporting these findings, in the phase II IMmotion150 study of bevacizumab and atezolizumab in recurrent RCC demonstrated a promising response rate and progression-free survival (PFS) compared with atezolizumab alone.24 These findings were echoed in a randomized study in unresectable hepatocellular carcinoma (HCC), in which combination therapy demonstrated a statistically significant improvement in median PFS over atezolizumab monotherapy.25
Following this rationale, in NCI #10010, a phase II, multicenter, single-arm trial sponsored by the National Cancer Institute (NCI), we investigated the combination of atezolizumab and bevacizumab for the treatment of advanced cervical cancer.
Patients and methods
Patients
Patients were eligible if they met the following criteria: had histologically confirmed, recurrent, persistent or metastatic cervical cancer; progression after 1–2 prior therapies in the advanced setting, including at least one containing bevacizumab; had measurable disease based on the revised Response Evaluation Criteria In Solid Tumors (RECIST) guidelines V.1.1; had Eastern Cooperative Oncology Group (ECOG) performance status score of 0–2; and had adequate renal, hepatic, and bone marrow function. Patients with histologies other than squamous cell, adenocarcinoma or adenosquamous carcinoma were excluded. In addition, patients previously treated with anti-PD-1/PD-L1 or CTLA-4 antibodies, history of autoimmune disease, significant vascular or cardiovascular disease, serious or non-healing wounds, active bleeding conditions, or inadequately anticoagulated thromboembolism were ineligible. Patients were enrolled regardless of PD-L1 status. This study was conducted in accordance with the Declaration of Helsinki and the Good Clinical Practice guidelines and is registered at ClinicalTrials.gov (identifier NCT02921269). The study protocol and amendments were approved by the Central Institutional Review Board (IRB) for the NCI and reviewed by the IRB of each participating institute. Written informed consent was obtained from all patients before conducting any study-related procedures.
Study design and treatment
This was a multicenter, open-label, phase II study following Simon two-stage design. Patients were treated with bevacizumab 15 mg/kg intravenous every 3 weeks and atezolizumab 1200 mg intravenous every 3 weeks. Treatment was discontinued at the onset of disease progression, the development of unacceptable toxic effects, or withdrawal from study. The primary objective was to measure the ORR (either partial response (PR) or complete response (CR)) defined by RECIST V.1.1 criteria. Secondary endpoints included disease control rate (DCR), PFS, overall safety (OS), and safety. The DCR was the sum of CR, PR and stable disease (SD).
Assessments
Disease was assessed by means of physical examination, as well as CT or MRI of the abdomen and pelvis within 28 days prior to study treatment. In patients without disease progression, imaging was repeated every 9 weeks for the first year and then every 12 weeks thereafter. Tumor measurements were assessed according to the RECIST V.1.1. Confirmatory scans were required no less than 4 weeks after a documentation of an objective response, but not required in cases of SD. Safety, as assessed according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE V.4.0), was monitored during each cycle. Adverse events were reported until 30 days after the last dose of study treatment had been administered and were summarized for patients who received any therapy and for whom adverse event information was submitted. Guidelines for the management of adverse events were provided by the sponsor and have been published previously. Patients who required discontinuation of one study agent but met criteria to continue the other study agent could continue treatment after discussion with the study chair.
Multiplex immunofluorescence analysis
Primary antibody staining conditions were initially optimized using standard immunohistochemical staining on the Leica Bond RX automated research stainer with DAB detection (Leica Bond Polymer Refine Detection DS9800). Using 4 µm formalin-fixed, paraffin-embedded (FFPE) tissue sections and serial antibody titrations, the optimal antibody concentration was determined followed by transition to a seven-color multiplex assay with equivalency (online supplemental methods). The antibody panel included CD68, FOXP3, PD-L1, CD31, CD8, and a panel of cytokeratin antibodies (PanCK, CK7, Cam5.2). The antibody clone used for PD-L1 staining (E1L3N) has previously demonstrated high concordance with the clinical 22C3 assay.26 Staining for PD-L1 in tumor cells and tumor-associated immune cells was used to calculate combined positive score (CPS), defined as the number of PD-L1 staining cells in the entire tissue divided by the total number of viable tumor cells, multiplied by 100.
Supplemental material
Genomic analyses
DNA samples extracted from FFPE tissue and matched blood/normal tissues were subjected to MSK-IMPACT targeted sequencing and analysis (n=6) as previously described.27 28 Tumor mutational burden (TMB) was calculated by dividing the number of non-synonymous mutations by the total size of the capture panel in Mb.
Statistical analysis
A Simon two-stage phase II design was used to address the primary endpoint of ORR. The unacceptable response rate was 0.15; desirable response rate was 0.40. With an error rate of type I error of 0.1 and type II error of 0.1, 22 patients were needed. With 22 patients, there was a 90% power to show a difference in response rate from 15% to 40%. The trial could continue to stage 2 only if two or more out of 10 patients demonstrated a response in the first stage. Six or more patients out of 22 must have exhibited a response for the study to be positive.
Standard descriptive statistics were used to summarize clinical results. Safety was assessed in patients who received at least one dose each of bevacizumab and atezolizumab. The ORR defined by RECIST V.1.1 was calculated assuming binomial proportions. PFS was defined from the date of start of treatment to the investigator determined date of progression (by RECIST V.1.1) or death, whichever occurred first. OS was defined from the date of start treatment to the death date. PFS and OS were analyzed using the Kaplan-Meier method to estimate values for medians and two-sided 95% CI. Biomarker analyses were not statistically designed in advance, and p values were calculated by Wilcoxon rank-sum test.
Results
Patients
Between July 2017 and October 2017, a total of 11 women were enrolled from five institutions in the USA. Patient characteristics are as described in table 1. As of the December 1, 2019 data cut-off, all patients had been followed to the time of death and no patients were censored. Median age at enrollment was 48 years (range, 31 to 55 years), 82% of patients were White, 64% of patients had an ECOG performance status of 0, and the majority (55%) had squamous cell carcinoma (SCC). Overall, 73% of patients had prior pelvic radiation with concurrent cisplatin, and all patients had been previously treated with a GOG 240 treatment regimen (platinum/paclitaxel/bevacizumab or topotecan/paclitaxel/bevacizumab). All patients discontinued study treatment, most commonly for disease progression (n=9, 82%).
Baseline demographics and patient characteristics
Efficacy
At the time of analysis, all enrolled patients had died. The median number of treatment cycles was 3 (range, 1–14). One patient developed acute neurologic changes 14 days after administration of the first cycle of study drugs and was not evaluable for the primary endpoint (ORR) and thus was replaced for efficacy analysis.
In the total evaluable population (n=10), zero patients achieved a CR or confirmed PR as assessed by RECIST V.1.1, resulting in a confirmed ORR of 0% (figure 1A). Of note, there were two patients who achieved an unconfirmed PR (figure 1A). Both patients achieved a PR by RECIST V.1.1 at the first or second scheduled scan, but had non-target progression at the next interval scan. The DCR by RECIST V.1.1 was 60% (0% CR, 0% PR, 60% SD). Median PFS was 2.9 months (95% CI, 1.8 to 6, figure 1B) and median OS was 8.9 months (95% CI, 3.4 to 21.9, figure 1C). The 1 year OS rate was 36.4% (95% CI, 11.2% to 62.7%).
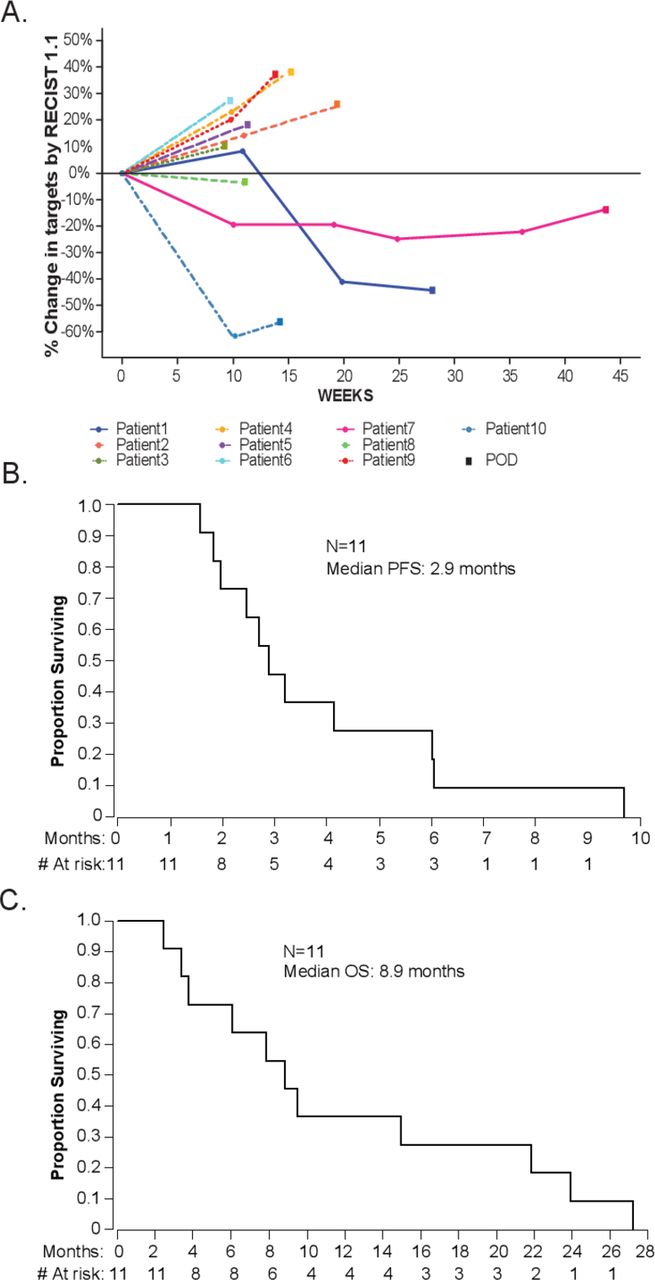
Clinical efficacy of atezolizumab plus bevacizumab in patients with advanced cervical cancer. (A) Spider plot of target lesions assessed per RECIST V.1.1. (B) PFS. (C) OS. OS, overall survival; PFS, progression-free survival; RECIST, Response Evaluation Criteria In Solid Tumors.
Safety
All 11 patients enrolled received at least one dose of atezolizumab plus bevacizumab and therefore were evaluable for toxicity (table 2). The overall rate of grade 3–4 adverse events attributable to study drug was 36.4%. No fatal adverse events attributable to drug were reported. It should be noted that two patients developed grade 3 neurologic events possibly related to study drugs that lead to drug discontinuation. The first patient presented on C1D15 with altered mental status and admitted to the ICU for empiric treatment of encephalopathy and meningitis, possibly attributed to atezolizumab and bevacizumab. Initial microbiology studies were negative, but the patient was treated empirically with both antimicrobials and steroids. The second patient presented on C4D15 with acute hearing loss. She subsequently presented with acute numbness and weakness of both legs and was diagnosed with arachnoiditis by CT scan, attributed to atezolizumab. She was started on high-dose oral (PO) steroids. Several MRIs and a lumbar puncture were completed to further evaluate her condition—all were consistent with inflammatory process with no cellular evidence of malignancy in the cerebrospinal fluid (CSF). There were no other high-grade events attributed to atezolizumab. Grade 3–4 events attributed to bevacizumab were otherwise consistent with published data, including thromboembolic event, gastrointestinal bleeding and anemia.
Toxicity profile and safety summary
The most common adverse events attributed to atezolizumab included fatigue (36%), increased aspartate aminotransferase (27%), nausea (27%), fever (27%), increased alanine aminotransferase (18%), diarrhea (18%) and dyspnea (18%). The most common adverse events attributed to bevacizumab included hypertension (18%) and gastrointestinal fistula (18%).
Tumor microenvironment analysis
All patients underwent pretreatment core biopsy. Eight of 11 patients had adequate tissue for tumor microenvironment analysis. Multiplexed immunohistochemistry was performed using the PerkinElmer Vectra using a panel as previously described (figure 2A,B). Given the paucity of tissue, multiplexed imaging was prioritized; clinically validated PD-L1 staining was not performed. Patients were compared based on clinical benefit, defined as staying on study and receiving therapy beyond 18 weeks (two imaging assessments). There were no significant differences between those patients who derived clinical benefit and those who did not when comparing tumor PD-L1 expression (p=0.663), tumor CD8+ T cell infiltration (p=0.868), or stromal PD-L1 expression (p=0.867), although these results must be interpreted with caution due to the very small sample size (figure 2C). PD-L1 CPS≥1 was observed in 3/8 patients; both of the patients with clinical benefit had CPS>1, with scores of 1.44 and 7.07, respectively (online supplemental table 2). Of interest, the patient with clinical benefit with CPS of 1.44 in addition exhibited strong evidence of angiogenesis in tumor and stroma, as evidenced by the high number of CD31+ cells (figure 2D).

{kind=link}
{kind=link}
Baseline tumor microenvironment and genomic characteristics of the treated patients. (A) Representative multiparameter immunofluorescence images from the patients with SqCC and AdC in CB and no benefit (NB) groups. (B) Heatmaps of the indicated staining parameters performed in tumor and stromal regions. (C) Quantification of the individual indicated immune cell populations in tumor and stroma separated by benefit versus no benefit. (D) Quantification of PD-L1+ and CD31+ cells in tumor and stroma separated by benefit versus no benefit. (E) Targeted genetic sequencing results from the archival tissues from six patients with available data. AdC, adenocarcinoma; CB, clinical benefit; PD-L1, programmed death ligand 1; SqCC, squamous cell carcinoma; TMB, tumor mutational burden.
Tumor genomic analysis
Six of enrolled patients underwent targeted sequencing using the MSK-IMPACT platform28 (figure 2E). Genomic alterations were present in a number of pathways implicated in oncogenesis, including cell cycling (TP53), gene transcription (KMT2D, KMT2A), DNA repair (ATM, BAP1, FANCA, PARP1), growth signaling (ERBB2) and the chromatin remodeling complex (PBRM1). TMB was low, less than 10 mutations per Mb, in all patients. While no strong conclusions could be drawn to distinguish the patients with clinical benefit, one of the patients with benefit exhibited an isolated deep deletion in PBRM1 and BAP1, both of which have been implicated in tumor immunogenicity and response to immune checkpoint blockade.29–31 Conversely, a patient who had progression after only three cycles had a deletion in STK11, which appears to be a marker for PD-1 resistance in patients with non-small cell lung cancer (NSCLC).32 None of the patients had alterations in JAK1 or JAK2, β2 microglobulin, or β-catenin, which have been implicated in primary or acquired resistance to PD-1 blockade in other cancers.33 34
Discussion
This is the first published study to evaluate the clinical activity of an antiangiogenesis agent in combination with an immune checkpoint inhibitor in patients with advanced cervical cancer. The combination of bevacizumab and atezolizumab did not meet the predefined efficacy endpoint with zero confirmed RECIST V.1.1 responses in the 10 evaluable patients. There were two unconfirmed PRs, but these were transient. Overall, the combination of bevacizumab and PD-L1 blockade did not appear to be synergistic in advanced cervical cancer when compared with anti-PD-1 monotherapy5 15 or bevacizumab monotherapy.35 Interestingly, the median OS of 8.9 months was comparable to the survival of 9.4 months observed in the KEYNOTE-158 study and 7.29 months in a phase II study of bevacizumab monotherapy, despite prior bevacizumab exposure in all patients.35
However, these comparisons must be interpreted with caution as the patient population in this study is quite different. First, a significant proportion of patients (45%) had adenocarcinoma or adenosquamous histology, compared with only 6% of patients enrolled in KEYNOTE-158% and 0% in CheckMate 358.5 15 Second, all patients had previously been treated with bevacizumab in combination with chemotherapy in the metastatic setting. Third, only a minority of the enrolled patients (3/8, 37.5%) had positive PD-L1 expression in their tumors.
Positive PD-L1 staining has been reported in 34.4%–96% of cervical cancer samples,36 consistent with the positivity rate in this cohort. It is important to note that in KEYNOTE-158, all of the responses were observed in patients who were PD-L1-positive, while 0% of patients without PD-L1 expression had a response5; this led to the FDA indication of pembrolizumab in PD-L1-positive tumors only. In Checkmate 358, there was a single response in a patient with a PD-L1-negative tumor. The relative paucity of PD-L1-positive tumors in this study is likely secondary to the predominance of patients with adenocarcinoma who were enrolled; multiple studies have demonstrated that SCCs have a much higher rate of PD-L1 positivity (37.8%–54%) when compared with endocervical carcinomas (ECCs, 14%–16.7%).37 38 It is unknown what role, if any, histology may play beyond PD-L1 expression in predicting response to checkpoint blockade, but prior studies suggest that adenocarcinomas may have a different tumor milieu. When compared with SCC, ECC demonstrates lower rates of immune infiltration with CD3+ and CD45+ cells and increased regulatory T cells (Tregs)along with lower chemokine levels.39 Given the rising incidence of cervical adenocarcinoma relative to SCC,40 this presents an opportunity for novel therapeutic combinations beyond single agent PD-1/PD-L1 blockade to improve the outcomes for this group of patients. It is also possible that the staining assay used in our study (E1L3N) could have contributed to relatively low percentages of PD-L1 positivity observed. The E1L3N antibody has been previously compared with the clinical 22C3 assay and was found to be highly concordant in bladder cancer and head and neck squamous carcinoma.26 Similar results were reported in a different study comparing the E1L3N clone to other clones including 22C3 in NSCLC.41 Thus, the potential minor differences in staining sensitivity between the assays likely have a low likelihood of influencing the relative percentages of PD-L1-positive tumors observed in this study.
The safety profile of atezolizumab plus bevacizumab is generally consistent with previously published data for both agents—however, it is notable that 2 of the 11 patients evaluable for safety were diagnosed with high-grade neurologic treatment-related adverse events (TRAEs). Events such as these are associated with bevacizumab42 as well as atezolizumab,43 although the rates appear to be low; retrospective reviews suggest that the true rate of high-grade neurologic events associated with anti-PD-1/PD-L1 blockade is somewhere between 1% and 3.2%.44–46 In regards to the combination, autoimmune encephalitis has been previously reported as a TRAE in the phase Ib study of atezolizumab plus bevacizumab in HCC,47 but no high-grade neurologic TRAEs were noted in the two randomized phase III studies in HCC and RCC.48 49 It will be essential for the FDA to monitor the post marketing safety reporting for this combination to assess the true risk of these devastating TRAEs.
Biomarker studies for the trial were limited by the small sample size and limited amount of tissue material available for analyses, highlighting the challenges of biomarker development in multiinstitutional studies. Two patients with clinical benefit exhibited very different microenvironment profiles, with high levels of CD31, suggestive of vascular predominance, observed in one patient and high levels of PD-L1 observed in another. These findings are hypothesis generating and highlight that responses to immunotherapy/antiangiogenic therapy combinations likely cannot be predicted by unifying tumor microenvironment phenotypes. Similarly, sequencing data were only available for a subset of the evaluable cohort. As discussed above, it is interesting to note that one of the patients who achieved an unconfirmed PR had deletion of PBRM1 and BAP1, which have been postulated to underlie immunogenicity of some tumors. None of the patients had pathogenic alterations in JAK1 or JAK2, β2 microglobulin, or β-catenin, which have been implicated in primary or acquired resistance to PD-1 blockade in other cancers.33 34 The limited nature of targeted sequencing used in this study, however, prevented more detailed characterization of additional alterations potentially involved in immunotherapy resistance, including other components of the IFNγ signaling pathway and major histocompatibility complex (MHC) class 1-encoding genes, which are known to be frequently altered in cervical cancer.50–52
The work here is limited by the size and hypothesis-generating nature of the study. Furthermore, the study unfortunately does not answer the question of whether a combination of antiangiogenic therapy with PD-L1 blockade modifies the tumor microenvironment, as post-treatment biopsies on the study were not possible. Given the published data of KEYNOTE 158, in which there were zero responses to pembrolizumab in patients who were PD-L1 negative, further consideration could be given to combining atezolizumab and bevacizumab in patients with PD-L1-positive tumors. It is also interesting to note that the combination of atezolizumab and bevacizumab has demonstrated robust clinical activity in treatment-naive RCC (ORR 37%)48 and treatment-naive HCC (ORR 27%).53 This is in stark contrast to our study, where all patients were all previously treated with bevacizumab and 1–2 prior lines of therapy in the advanced setting. The heavily pretreated nature of these patients and resistance to bevacizumab likely resulted in a lower clinical benefit rate than we would otherwise expect. Based on this hypothesis, and given the success of combining atezolizumab with bevacizumab and chemotherapy in NSCLC, patients are now being enrolled in BEATcc, a phase III, randomized study to assess the efficacy of atezolizumab administered concurrent to the combination of cisplatin-paclitaxel plus bevacizumab in previously untreated patients with metastatic, recurrent, or persistent cervical cancer.54
Acknowledgments
We thank the patients who participated in this study and the clinical study teams who were involved in data collection and analyses.
References
Footnotes
Contributors CF had full access to all data in the trial and takes responsibility for the integrity of the data and the accuracy of the data analysis. CF, ASC, CA, and DZ contributed to the conception and design of the trial. All authors provided study materials or patients. QZ and AI completed the statistical analyses. ES reviewed the clinical data. DZ reviewed and analyzed the translational data.
Funding The clinical trial was sponsored by the NCI Experimental Therapeutics Clinical Trials Network (ETCTN) (JHU LAO- UM1-CA186691), with funding support provided by Roche/Genentech. The study was supported in part by the MSK Cancer Center Support Grant P30 CA008748. CF and DZ are members of the Parker Institute for Cancer Immunotherapy at MSKCC. DZ is supported by the Ovarian Cancer Research Foundation Liz Tilberis Award, and the Department of Defense Ovarian Cancer Research Academy (OC150111). Clinical outcomes from the study were presented at the 2019 Society for Gynecologic Oncology Annual meeting in Honolulu, Hawaii.
Competing interests DZ reports clinical research support to his institution from Astra Zeneca and Genentech; personal/consultancy fees from Merck, Synlogic Therapeutics, Biomed Valley Discoveries, Trieza Therapeutics, Tesaro, Bristol Myers Squibb, and Agenus; and travel support from Genenetech. These are all outside of the scope of the submitted work. CF reports clinical research support to her institution from Merck, Bristol Myers Squibb and Genentech; personal/consultancy fees from Astra Zeneca, as well as participation in steering committees (compensation waived) for Merck and Genentech. These are outside the scope of the submitted work. AS reports that she is now an employee of Merck and declares Merck stock ownership. ABM reports no conflicts of interest. SF reports clinical research support to his institution from the following: AstraZeneca; Abbisko; Anaeropharma Science; Arrien Pharmaceuticals; BeiGene; BioAtla, LLC; Boehringer Ingelheim; Eli Lilly & Co; Hookipa Biotech GmBH; Huya Bioscience International; IMV, Inc; Innovent Biologics, Co, Ltd; Lyvgen Biopharm, Co, Ltd; MacroGenics; Medivir AB; Millennium Pharmaceuticals, Inc; Nerviano Medical Sciences; NeuPharma, Inc; NIH/NCI; Novartis; OncoMed Pharmaceuticals; Parexel International, LLC; Sellas Life Sciences Group; Soricimed Biopharma, Inc; Tolero Pharmaceuticals. BC reports advisory committees for GSK, Novocure, and Merck. He reports research funding from Clovis. These are all outside the scope of the submitted work. TH reports research funding from Bristol Myers Squibb which is outside the scope of the submitted work.
Patient consent for publication Not required.
Ethics approval The study protocol and amendments for NCI #10010 were approved by the Central Institutional Review Board (IRB) for the NCI and reviewed by the IRB of each participating institute. The study was conducted in accordance with Good Clinical Practice guidelines per the International Conference on Harmonisation. All patients provided written informed consent based on Declaration of Helsinki principles.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Deidentified data will be made available upon request.